
Herpesvirus gB: A Finely Tuned Fusion Machine


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Viral Entry and Membrane Fusion
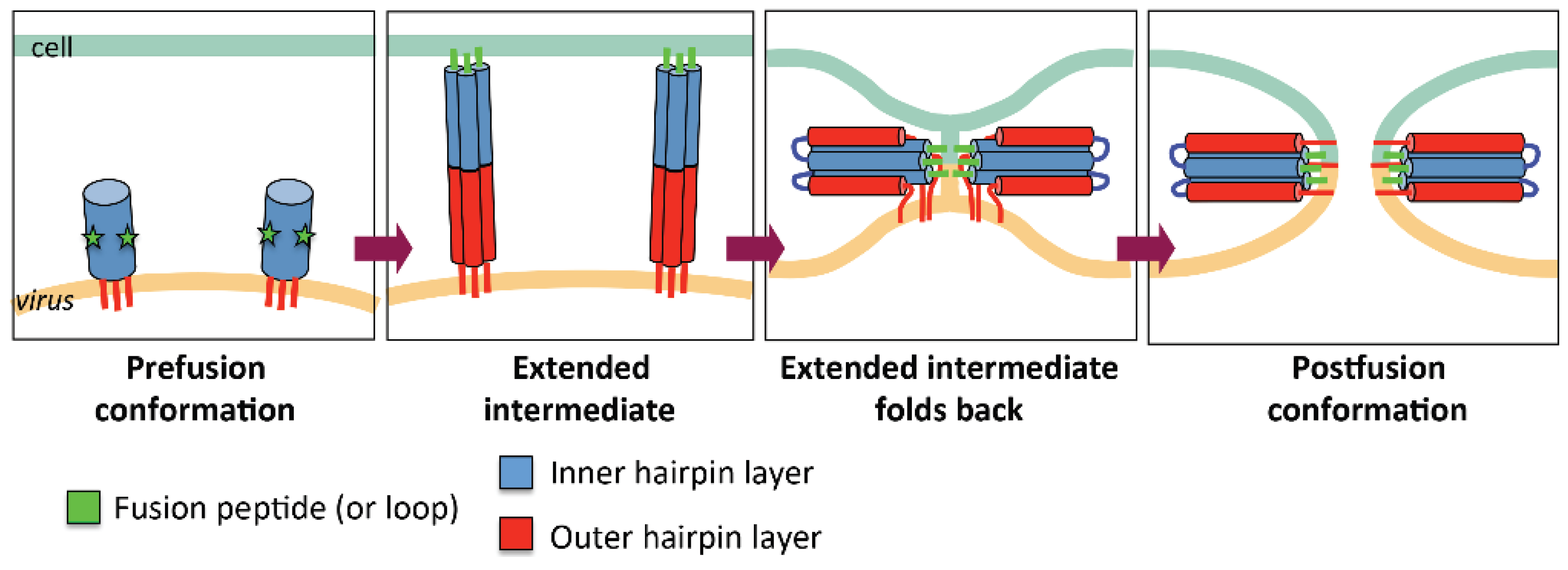
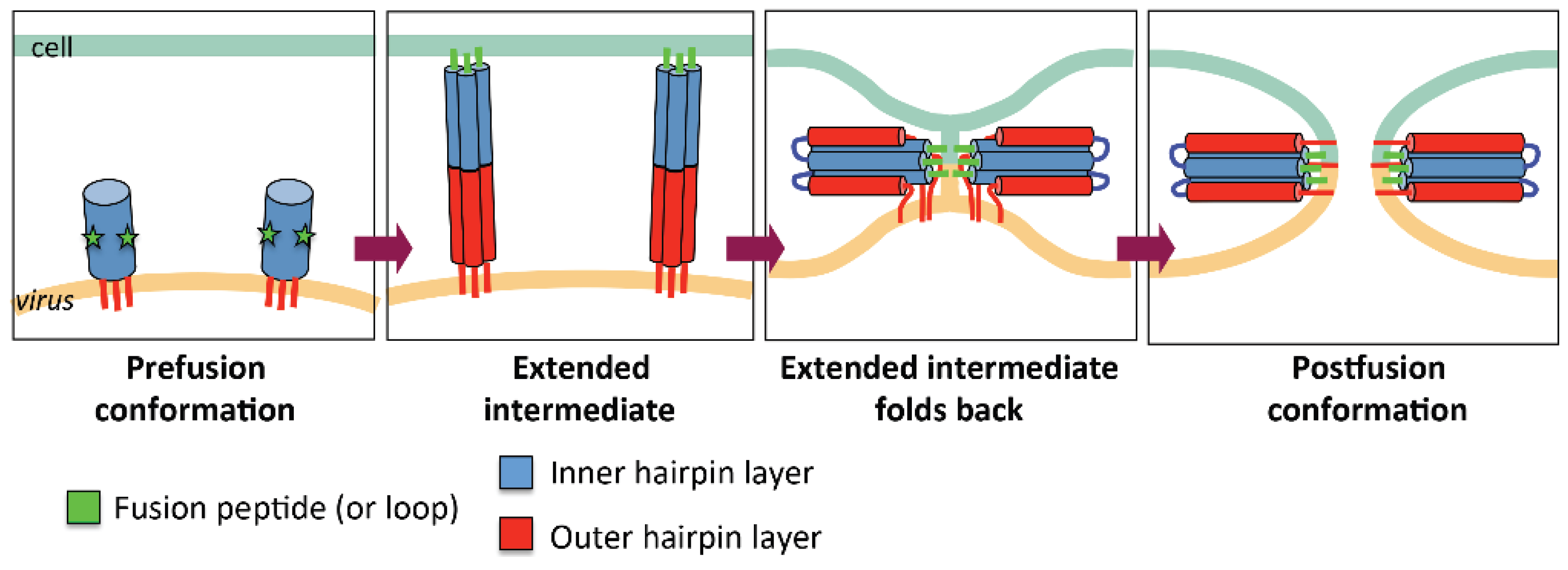
2.1. General Mechanism of Viral Fusion
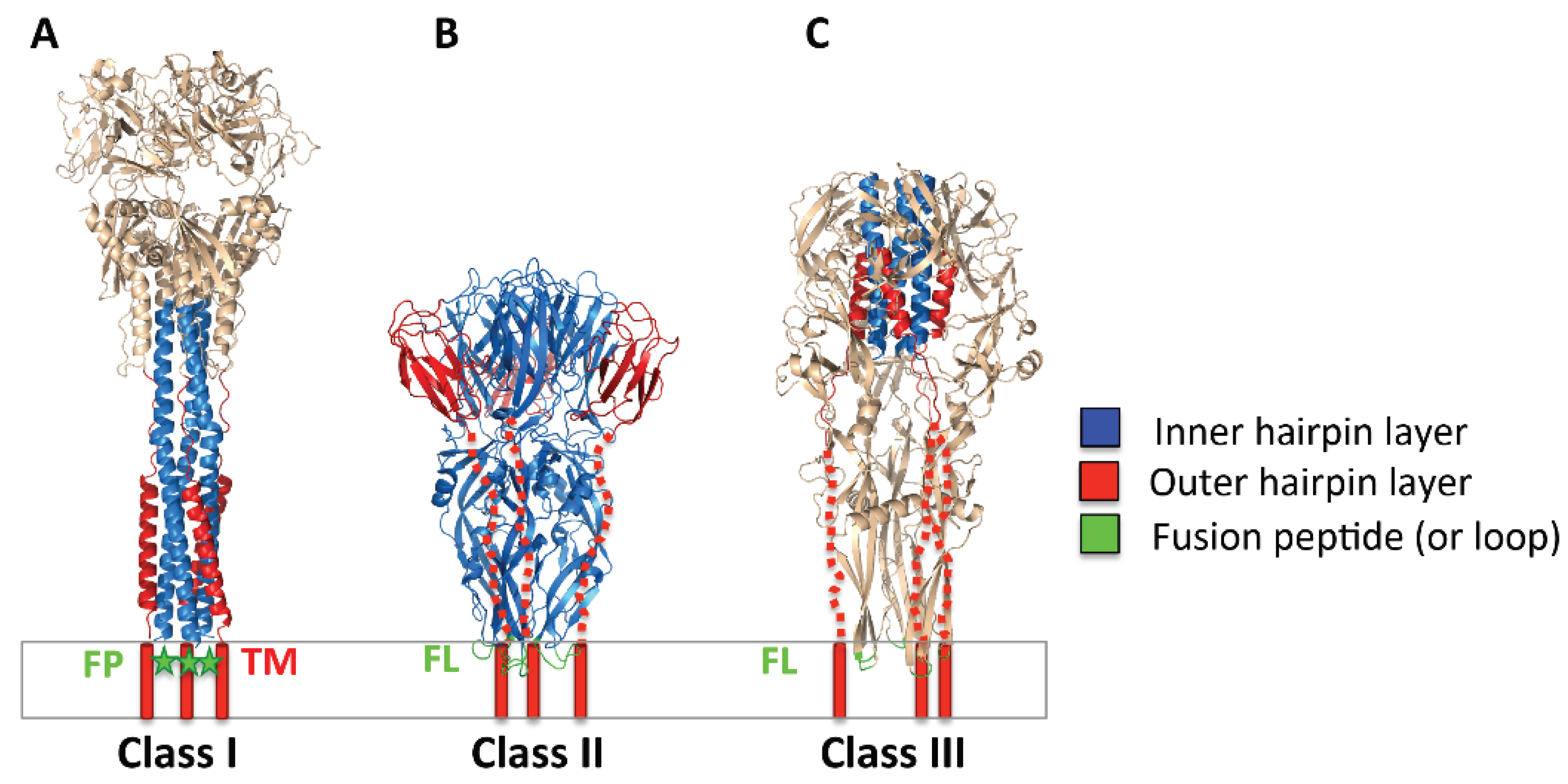
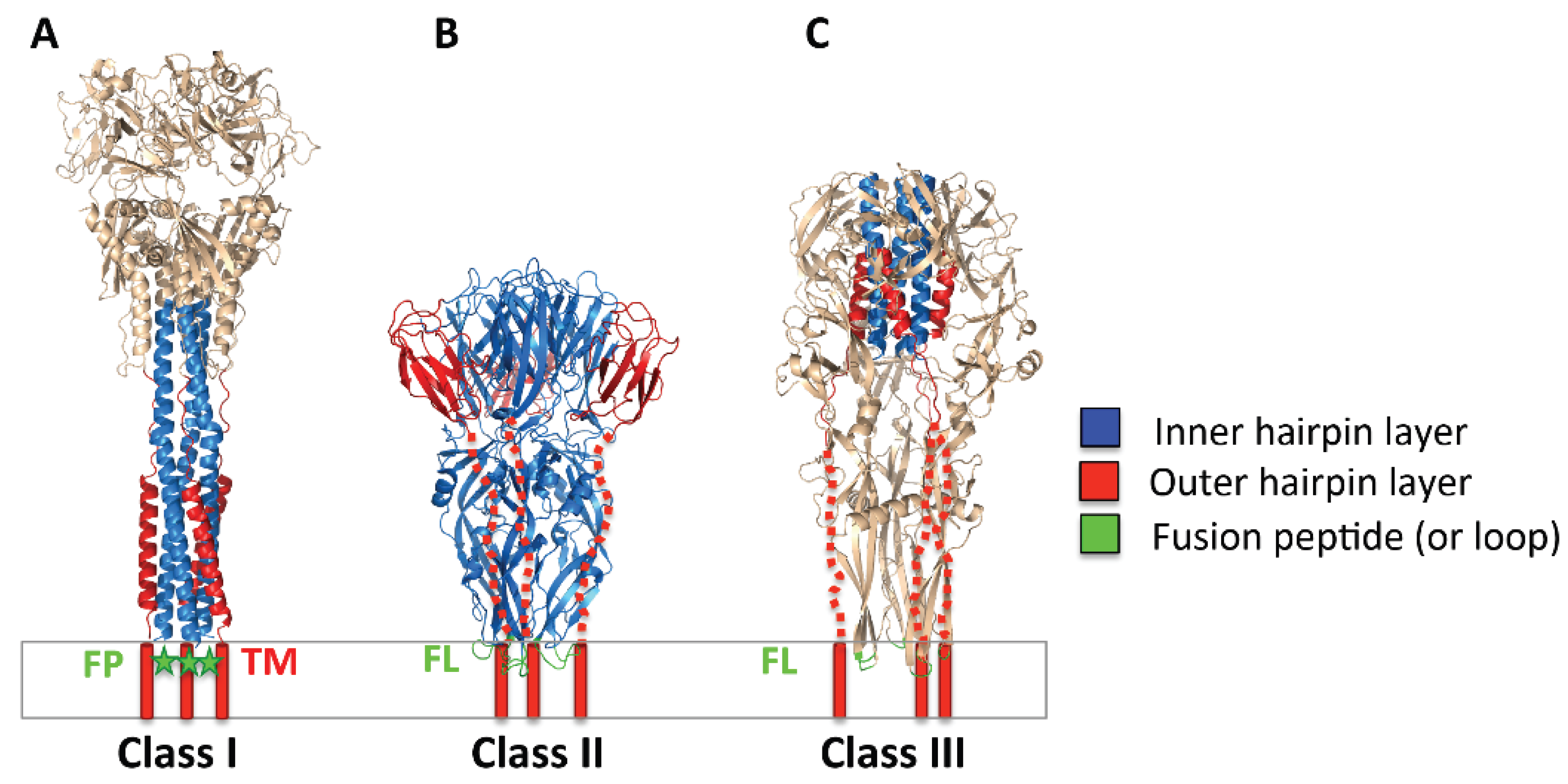
2.2. Three Classes of Viral Fusogens
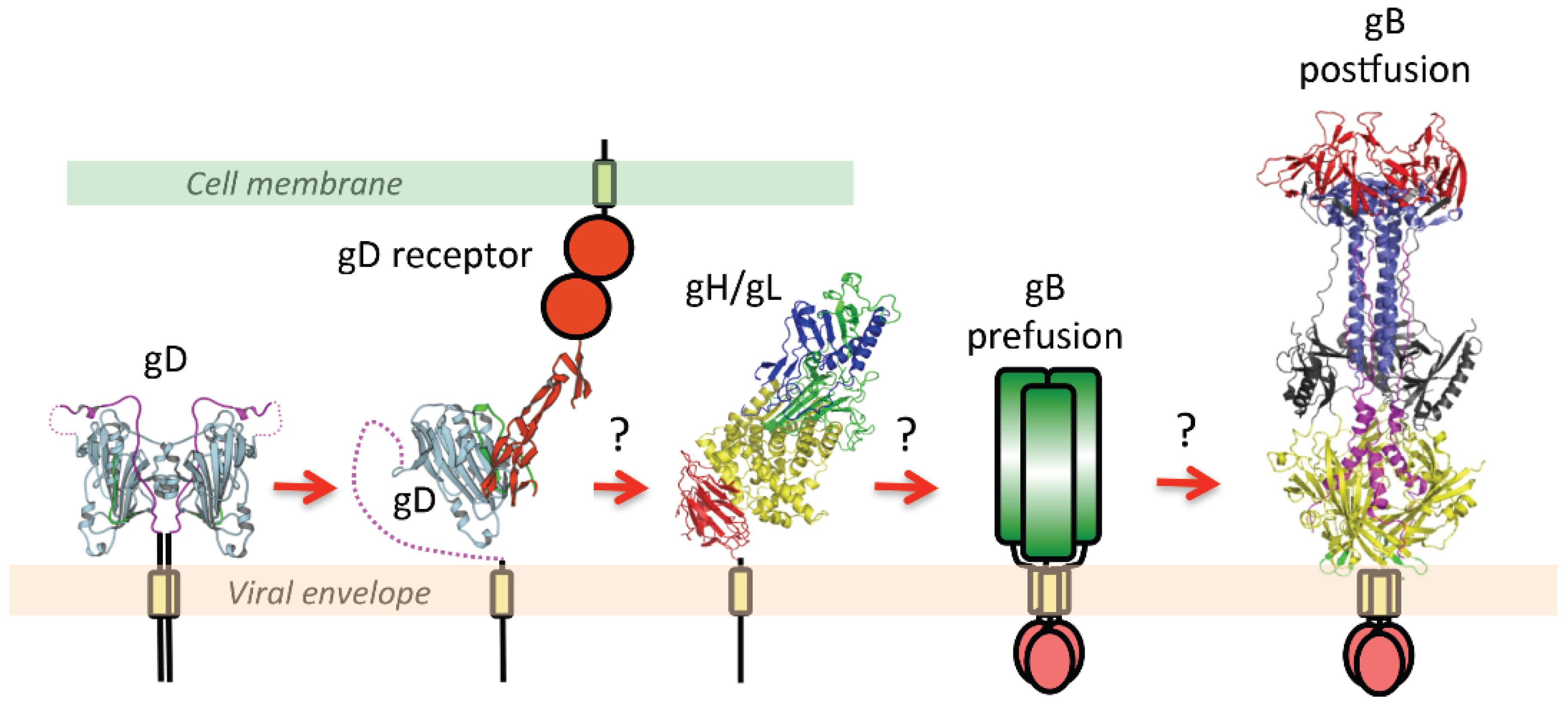
2.3. The Orchestration of Herpesvirus Membrane Fusion

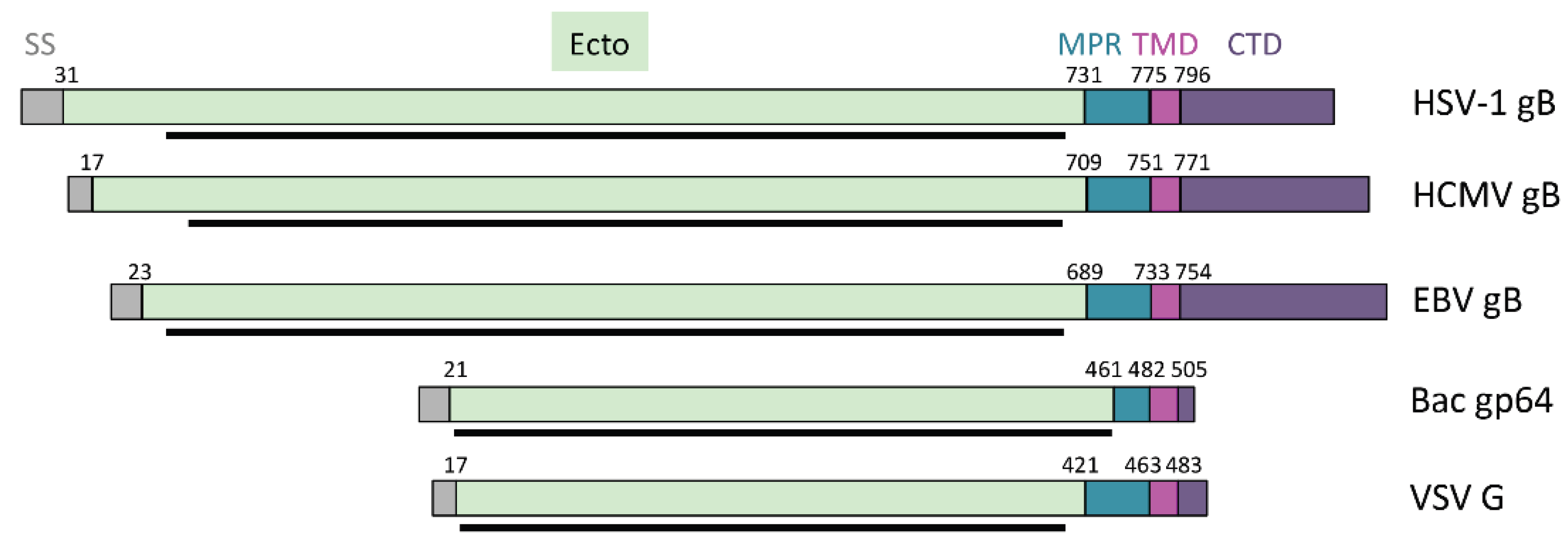
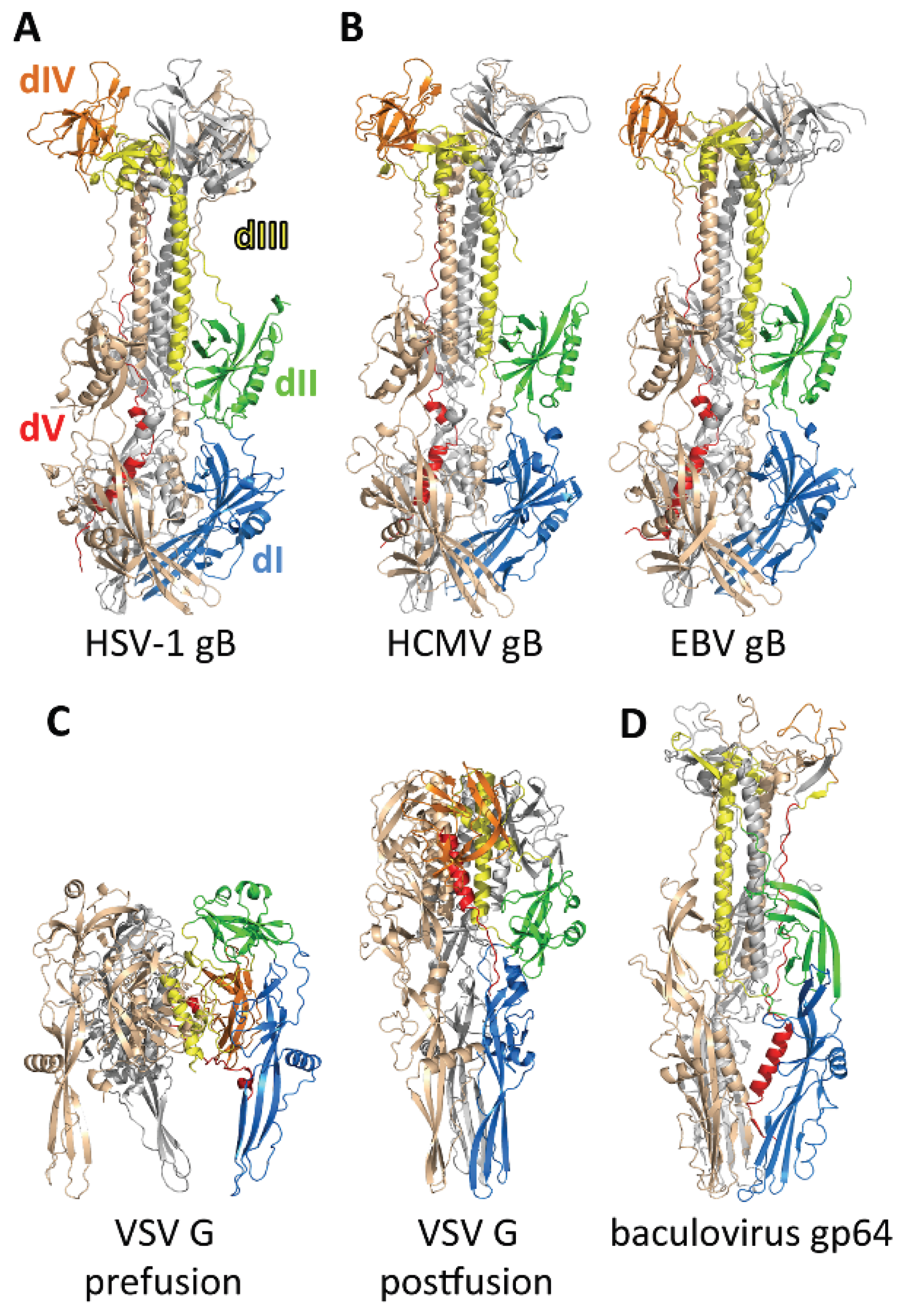
3. The gB Ectodomain
3.1. Structure of the Postfusion gB Ectodomain
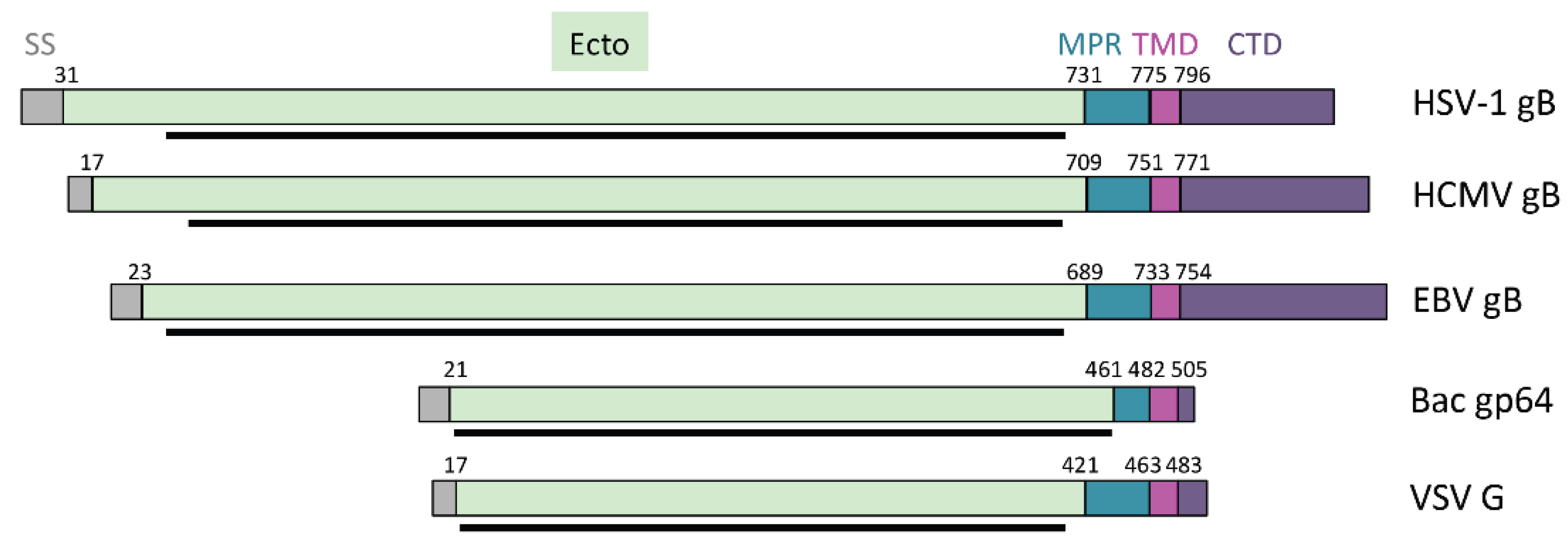
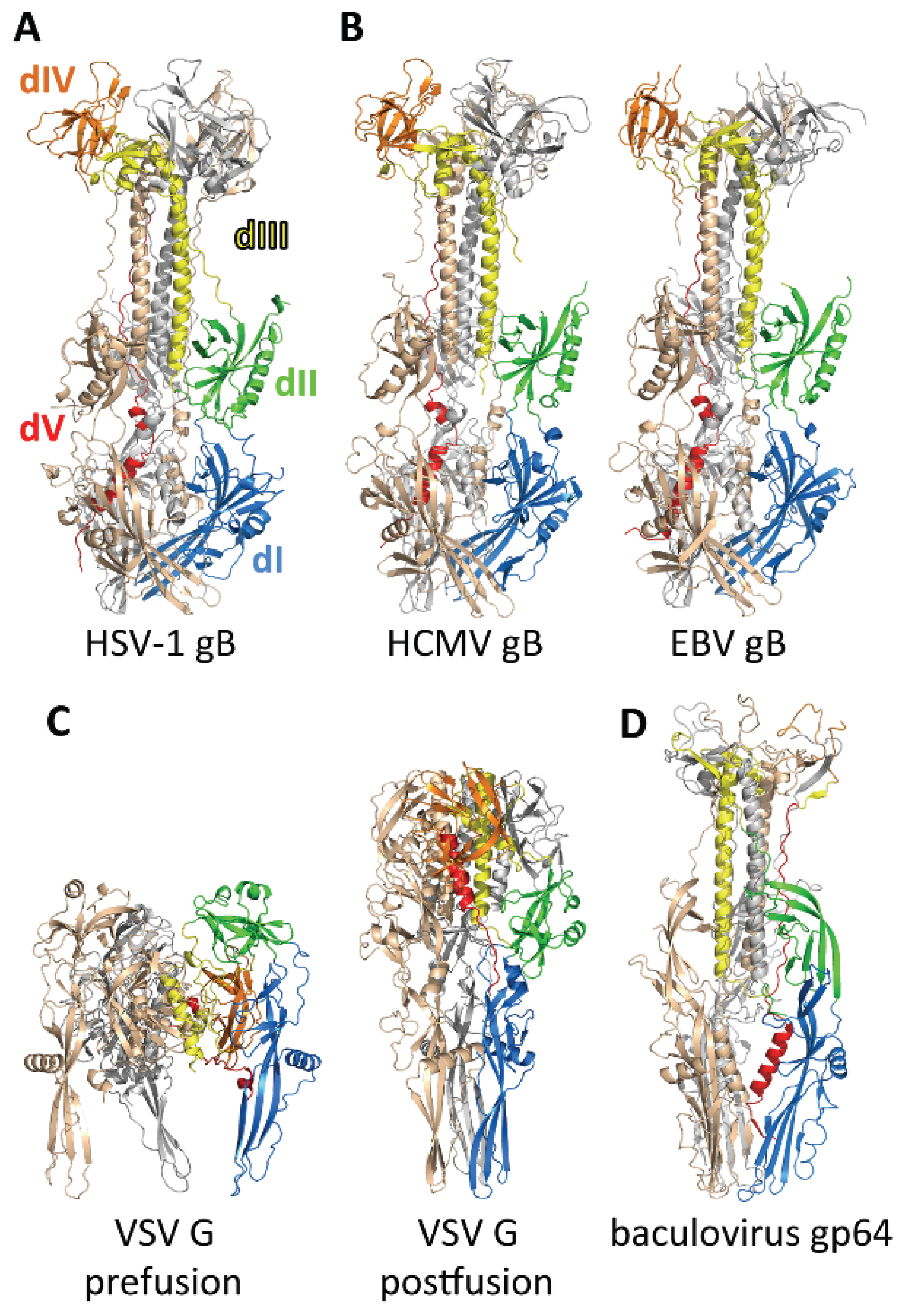
3.2. The Prefusion Form of gB
4. Regulatory Regions of gB
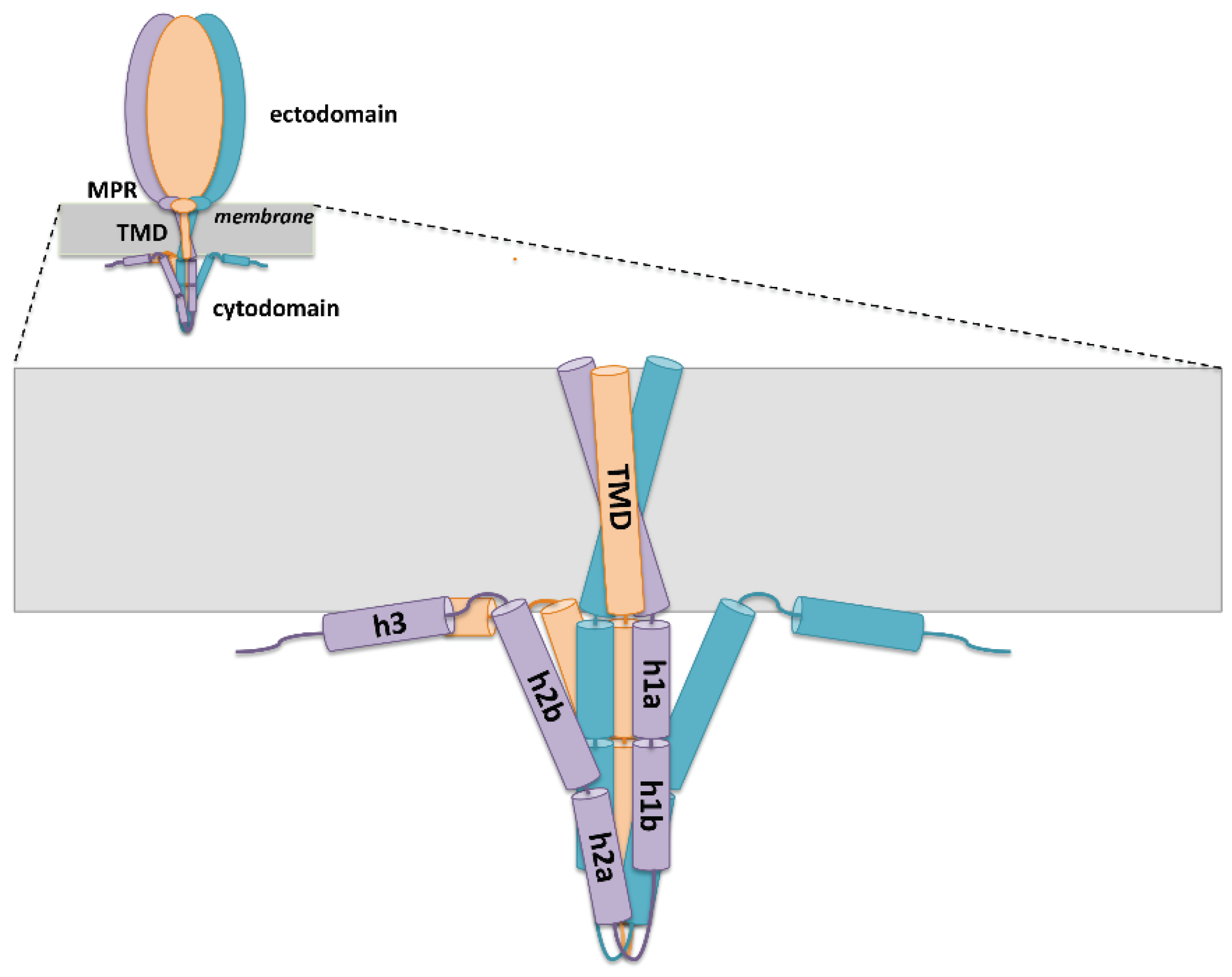
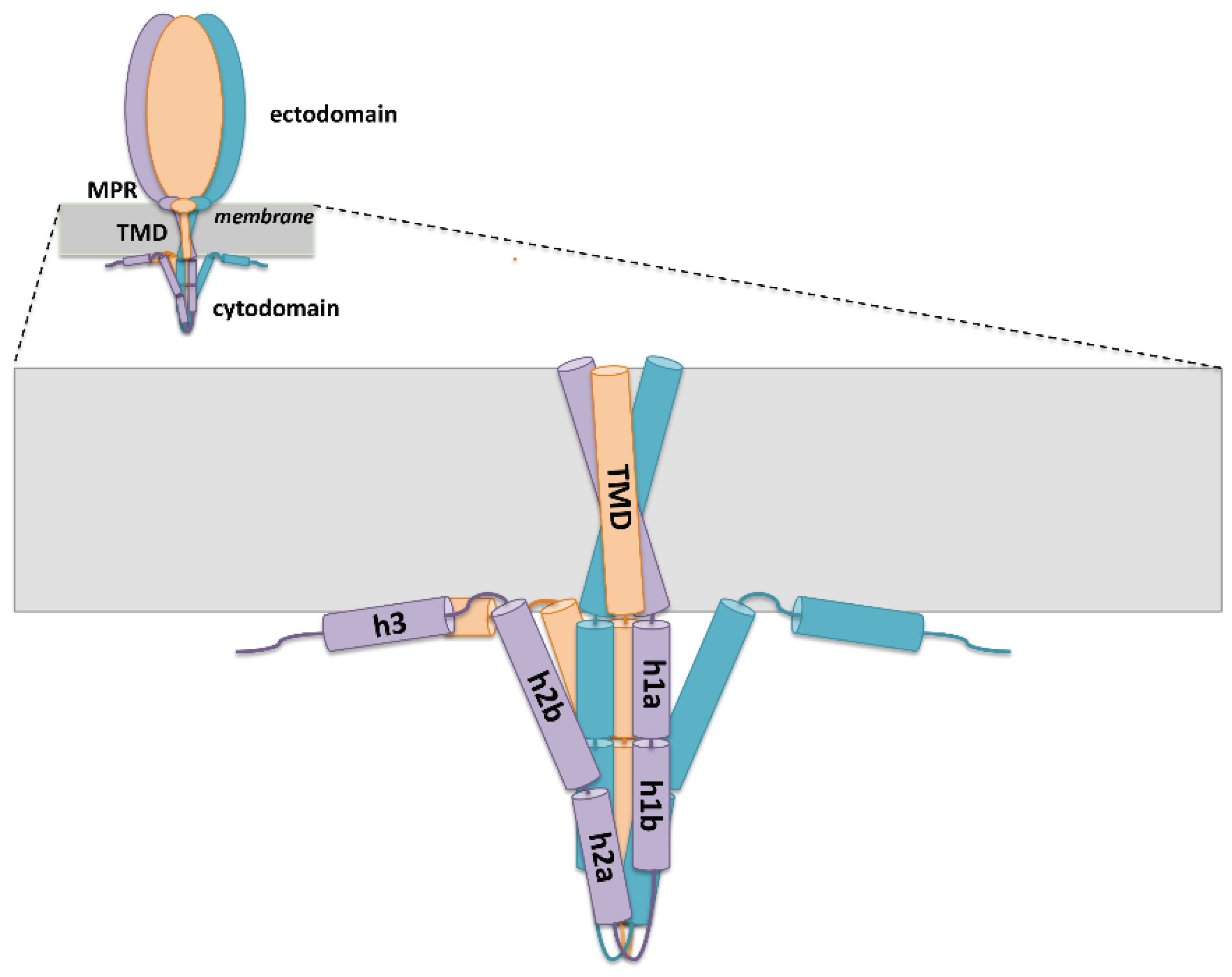
4.1. Membrane-Proximal Region
4.2. Transmembrane Domain
4.3. Cytoplasmic Domain
5. Role of gH/gL Interactions
5.1. The gH/gL Ectodomain
5.2. The gH Cytotail
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Heldwein, E.E.; Krummenacher, C. Entry of herpesviruses into mammalian cells. Cell. Mol. Life Sci. 2008, 65, 1653–1668. [Google Scholar] [CrossRef] [PubMed]
- Gianni, T.; Salvioli, S.; Chesnokova, L.S.; Hutt-Fletcher, L.M.; Campadelli-Fiume, G. Alphavbeta6- and alphavbeta8-integrins serve as interchangeable receptors for HSV gH/gL to promote endocytosis and activation of membrane fusion. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Gianni, T.; Massaro, R.; Campadelli-Fiume, G. Dissociation of HSV gL from gH by alphavbeta6- or alphavbeta8-integrin promotes gH activation and virus entry. Proc. Natl Acad Sci. USA 2015, 112, E3901–E3910. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Arii, J.; Suenaga, T.; Wang, J.; Kogure, A.; Uehori, J.; Arase, N.; Shiratori, I.; Tanaka, S.; Kawaguchi, Y.; et al. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 2008, 132, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Modis, Y. A novel membrane fusion protein family in Flaviviridae? Trends Microbiol. 2014, 22, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Jardetzky, T.S. Structural basis of viral invasion: Lessons from paramyxovirus F. Curr. Opin. Struct. Biol. 2007, 17, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Fusco, M.L.; Hessell, A.J.; Oswald, W.B.; Burton, D.R.; Saphire, E.O. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 2008, 454, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Modis, Y. Class II fusion proteins. Adv. Exp. Med. Biol. 2013, 790, 150–166. [Google Scholar] [PubMed]
- Kielian, M. Class II virus membrane fusion proteins. Virology 2006, 344, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lok, S.M.; Yu, I.M.; Zhang, Y.; Kuhn, R.J.; Chen, J.; Rossmann, M.G. The flavivirus precursor membrane-envelope protein complex: Structure and maturation. Science 2008, 319, 1830–1834. [Google Scholar] [CrossRef] [PubMed]
- Voss, J.E.; Vaney, M.C.; Duquerroy, S.; Vonrhein, C.; Girard-Blanc, C.; Crublet, E.; Thompson, A.; Bricogne, G.; Rey, F.A. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 2010, 468, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Backovic, M.; Jardetzky, T.S. Class III viral membrane fusion proteins. Curr. Opin. Struct. Biol. 2009, 19, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Roche, S.; Rey, F.A.; Gaudin, Y.; Bressanelli, S. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 2007, 315, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Roche, S.; Bressanelli, S.; Rey, F.A.; Gaudin, Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 2006, 313, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Zelus, B.D.; Schickli, J.H.; Blau, D.M.; Weiss, S.R.; Holmes, K.V. Conformational changes in the spike glycoprotein of murine coronavirus are induced at 37 degrees C either by soluble murine CEACAM1 receptors or by pH 8. J. Virol. 2003, 77, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Taguchi, F. Two-step conformational changes in a coronavirus envelope glycoprotein mediated by receptor binding and proteolysis. J. Virol. 2009, 83, 11133–11141. [Google Scholar] [CrossRef] [PubMed]
- Bullough, P.A.; Hughson, F.M.; Skehel, J.J.; Wiley, D.C. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994, 371, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Kadlec, J.; Loureiro, S.; Abrescia, N.G.; Stuart, D.I.; Jones, I.M. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat. Struct. Mol. Biol. 2008, 15, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Mair, C.M.; Meyer, T.; Schneider, K.; Huang, Q.; Veit, M.; Herrmann, A. A histidine residue of the influenza virus hemagglutinin controls the pH dependence of the conformational change mediating membrane fusion. J. Virol. 2014, 88, 13189–13200. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, T.; Mueller, D.S.; Mark, A.E.; Young, P.R.; Kobe, B. The role of histidine residues in low-pH-mediated viral membrane fusion. Structure 2006, 14, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Roche, S.; Albertini, A.A.; Lepault, J.; Bressanelli, S.; Gaudin, Y. Structures of vesicular stomatitis virus glycoprotein: Membrane fusion revisited. Cell. Mol. Life Sci. 2008, 65, 1716–1728. [Google Scholar] [CrossRef] [PubMed]
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. Molecular mechanisms of HIV entry. Adv. Exp. Med. Biol. 2012, 726, 223–242. [Google Scholar] [PubMed]
- Mothes, W.; Boerger, A.L.; Narayan, S.; Cunningham, J.M.; Young, J.A. Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell 2000, 103, 679–689. [Google Scholar] [CrossRef]
- Chandran, K.; Sullivan, N.J.; Felbor, U.; Whelan, S.P.; Cunningham, J.M. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 2005, 308, 1643–1645. [Google Scholar] [CrossRef] [PubMed]
- Cote, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 2011, 477, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Paterson, R.G.; Jardetzky, T.S. Paramyxovirus membrane fusion: Lessons from the F and HN atomic structures. Virology 2006, 344, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, R.J.; Atanasiu, D.; Cairns, T.M.; Gallagher, J.R.; Krummenacher, C.; Cohen, G.H. Herpes virus fusion and entry: A story with many characters. Viruses 2012, 4, 800–832. [Google Scholar] [CrossRef] [PubMed]
- Spear, P.G. Herpes simplex virus: Receptors and ligands for cell entry. Cell Microbiol. 2004, 6, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; O’Donnell, C.; Copeland, R.J.; Scarlett, T.; Liu, J.; Shukla, D. Soluble 3-O-sulfated heparan sulfate can trigger herpes simplex virus type 1 entry into resistant Chinese hamster ovary (CHO-K1) cells. J. Gen. Virol. 2007, 88, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Lazear, E.; Carfi, A.; Whitbeck, J.C.; Cairns, T.M.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J. Engineered disulfide bonds in herpes simplex virus type 1 gD separate receptor binding from fusion initiation and viral entry. J. Virol. 2008, 82, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Krummenacher, C.; Supekar, V.M.; Whitbeck, J.C.; Lazear, E.; Connolly, S.A.; Eisenberg, R.J.; Cohen, G.H.; Wiley, D.C.; Carfi, A. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 2005, 24, 4144–4153. [Google Scholar] [CrossRef] [PubMed]
- Atanasiu, D.; Saw, W.T.; Cohen, G.H.; Eisenberg, R.J. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 2010, 84, 12292–12299. [Google Scholar] [CrossRef] [PubMed]
- Atanasiu, D.; Cairns, T.M.; Whitbeck, J.C.; Saw, W.T.; Rao, S.; Eisenberg, R.J.; Cohen, G.H. Regulation of herpes simplex virus gB-induced cell-cell fusion by mutant forms of gH/gL in the absence of gD and cellular receptors. MBio 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Gianni, T.; Amasio, M.; Campadelli-Fiume, G. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J. Biol. Chem 2009, 284, 17370–17382. [Google Scholar] [CrossRef] [PubMed]
- Atanasiu, D.; Whitbeck, J.C.; Cairns, T.M.; Reilly, B.; Cohen, G.H.; Eisenberg, R.J. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. USA 2007, 104, 18718–18723. [Google Scholar] [CrossRef] [PubMed]
- Atanasiu, D.; Whitbeck, J.C.; de Leon, M.P.; Lou, H.; Hannah, B.P.; Cohen, G.H.; Eisenberg, R.J. Bimolecular complementation defines functional regions of herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J. Virol. 2010, 84, 3825–3834. [Google Scholar] [CrossRef] [PubMed]
- Chowdary, T.K.; Cairns, T.M.; Atanasiu, D.; Cohen, G.H.; Eisenberg, R.J.; Heldwein, E.E. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 2010, 17, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Spriggs, M.K.; Kovats, S.; Turk, S.M.; Comeau, M.R.; Nepom, B.; Hutt-Fletcher, L.M. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J. Virol. 1997, 71, 4657–4662. [Google Scholar] [PubMed]
- Mullen, M.M.; Haan, K.M.; Longnecker, R.; Jardetzky, T.S. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol. Cell 2002, 9, 375–385. [Google Scholar] [CrossRef]
- Chesnokova, L.S.; Hutt-Fletcher, L.M. Fusion of Epstein-Barr virus with epithelial cells can be triggered by αvβ5 in addition to αvβ6 and αvβ8, and integrin binding triggers a conformational change in glycoproteins gHgL. J. Virol. 2011, 85, 13214–13223. [Google Scholar] [CrossRef] [PubMed]
- Chesnokova, L.S.; Nishimura, S.L.; Hutt-Fletcher, L.M. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins αvβ6 or αvβ8. Proc. Natl. Acad. Sci. USA 2009, 106, 20464–20469. [Google Scholar] [CrossRef] [PubMed]
- Vanarsdall, A.L.; Chase, M.C.; Johnson, D.C. Human cytomegalovirus glycoprotein gO complexes with gH/gL, promoting interference with viral entry into human fibroblasts but not entry into epithelial cells. J. Virol. 2011, 85, 11638–11645. [Google Scholar] [CrossRef] [PubMed]
- Wille, P.T.; Knoche, A.J.; Nelson, J.A.; Jarvis, M.A.; Johnson, D.C. A human cytomegalovirus gO-null mutant fails to incorporate gH/gL into the virion envelope and is unable to enter fibroblasts and epithelial and endothelial cells. J. Virol. 2010, 84, 2585–2596. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 18153–18158. [Google Scholar] [CrossRef] [PubMed]
- Ryckman, B.J.; Jarvis, M.A.; Drummond, D.D.; Nelson, J.A.; Johnson, D.C. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 2006, 80, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Lanchy, J.M.; Ryckman, B.J. Human cytomegalovirus gH/gL/gO promotes the fusion step of entry into all cell types, whereas gH/gL/UL128–131 broadens virus tropism through a distinct mechanism. J. Virol. 2015, 89, 8999–9009. [Google Scholar] [CrossRef] [PubMed]
- Laquerre, S.; Argnani, R.; Anderson, D.B.; Zucchini, S.; Manservigi, R.; Glorioso, J.C. Heparan sulfate proteoglycan binding by herpes simplex virus type 1 glycoproteins B and C, which differ in their contributions to virus attachment, penetration, and cell-to-cell spread. J. Virol. 1998, 72, 6119–6130. [Google Scholar] [PubMed]
- Stampfer, S.D.; Lou, H.; Cohen, G.H.; Eisenberg, R.J.; Heldwein, E.E. Structural basis of local, pH-dependent conformational changes in glycoprotein B from herpes simplex virus type 1. J. Virol. 2010, 84, 12924–12933. [Google Scholar] [CrossRef] [PubMed]
- Heldwein, E.E.; Lou, H.; Bender, F.C.; Cohen, G.H.; Eisenberg, R.J.; Harrison, S.C. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 2006, 313, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Backovic, M.; Longnecker, R.; Jardetzky, T.S. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. USA 2009, 106, 2880–2885. [Google Scholar] [CrossRef] [PubMed]
- Chandramouli, S.; Ciferri, C.; Nikitin, P.A.; Calo, S.; Gerrein, R.; Balabanis, K.; Monroe, J.; Hebner, C.; Lilja, A.E.; Settembre, E.C.; et al. Structure of HCMV glycoprotein B in the postfusion conformation bound to a neutralizing human antibody. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Burke, H.G.; Heldwein, E.E. Crystal structure of the human cytomegalovirus glycoprotein B. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Atanasiu, D.; Saw, W.T.; Gallagher, J.R.; Hannah, B.P.; Matsuda, Z.; Whitbeck, J.C.; Cohen, G.H.; Eisenberg, R.J. Dual split protein-based fusion assay reveals that mutations to herpes simplex virus (HSV) glycoprotein gB alter the kinetics of cell-cell fusion induced by HSV entry glycoproteins. J. Virol. 2013, 87, 11332–11345. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, J.R.; Atanasiu, D.; Saw, W.T.; Paradisgarten, M.J.; Whitbeck, J.C.; Eisenberg, R.J.; Cohen, G.H. Functional fluorescent protein insertions in herpes simplex virus gB report on gB conformation before and after execution of membrane fusion. PLoS Pathog. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Bender, F.C.; Samanta, M.; Heldwein, E.E.; de Leon, M.P.; Bilman, E.; Lou, H.; Whitbeck, J.C.; Eisenberg, R.J.; Cohen, G.H. Antigenic and mutational analyses of herpes simplex virus glycoprotein B reveal four functional regions. J. Virol. 2007, 81, 3827–3841. [Google Scholar] [CrossRef] [PubMed]
- Cairns, T.M.; Fontana, J.; Huang, Z.Y.; Whitbeck, J.C.; Atanasiu, D.; Rao, S.; Shelly, S.S.; Lou, H.; Ponce de Leon, M.; Steven, A.C.; et al. Mechanism of neutralization of herpes simplex virus by antibodies directed at the fusion domain of glycoprotein B. J. Virol. 2014, 88, 2677–2689. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Chen, M.; Leung, S.; Graepel, K.W.; Du, X.; Yang, Y.; Zhou, T.; Baxa, U.; Yasuda, E.; Beaumont, T.; et al. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 2013, 340, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Yang, Y.; Graham, B.S.; Kwong, P.D. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J. Virol. 2011, 85, 7788–7796. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S. Neutralizing epitopes on the respiratory syncytial virus fusion glycoprotein. Curr. Opin. Virol. 2015, 11, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Vitu, E.; Sharma, S.; Stampfer, S.D.; Heldwein, E.E. Extensive mutagenesis of the HSV-1 gB ectodomain reveals remarkable stability of its postfusion form. J. Mol. Biol. 2013, 425, 2056–2071. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.S.; Paterson, R.G.; Wen, X.; Lamb, R.A.; Jardetzky, T.S. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9288–9293. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wharton, S.A.; Weissenhorn, W.; Calder, L.J.; Hughson, F.M.; Skehel, J.J.; Wiley, D.C. A soluble domain of the membrane-anchoring chain of influenza virus hemagglutinin (HA2) folds in Escherichia coli into the low-pH-induced conformation. Proc. Natl. Acad. Sci. USA 1995, 92, 12205–12209. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.S.; Wen, X.; Paterson, R.G.; Lamb, R.A.; Jardetzky, T.S. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 2006, 439, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Sissoeff, L.; Mousli, M.; England, P.; Tuffereau, C. Stable trimerization of recombinant rabies virus glycoprotein ectodomain is required for interaction with the p75NTR receptor. J. Gen. Virol. 2005, 86, 2543–2552. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.; Corper, A.L.; Basler, C.F.; Taubenberger, J.K.; Palese, P.; Wilson, I.A. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science 2004, 303, 1866–1870. [Google Scholar] [CrossRef] [PubMed]
- Wanas, E.; Efler, S.; Ghosh, K.; Ghosh, H.P. Mutations in the conserved carboxy-terminal hydrophobic region of glycoprotein gB affect infectivity of herpes simplex virus. J. Gen. Virol. 1999, 80, 3189–3198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ge, P.; Yu, X.; Brannan, J.M.; Bi, G.; Zhang, Q.; Schein, S.; Zhou, Z.H. Cryo-EM structure of the mature dengue virus at 3.5-A resolution. Nat. Struct. Mol. Biol. 2013, 20, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. Structure of the dengue virus envelope protein after membrane fusion. Nature 2004, 427, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Maurer, U.E.; Zeev-Ben-Mordehai, T.; Pandurangan, A.P.; Cairns, T.M.; Hannah, B.P.; Whitbeck, J.C.; Eisenberg, R.J.; Cohen, G.H.; Topf, M.; Huiskonen, J.T.; et al. The structure of herpesvirus fusion glycoprotein B-bilayer complex reveals the protein-membrane and lateral protein-protein interaction. Structure 2013, 21, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- Shelly, S.S.; Cairns, T.M.; Whitbeck, J.C.; Lou, H.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J. The membrane-proximal region (MPR) of herpes simplex virus gB regulates association of the fusion loops with lipid membranes. MBio 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Hannah, B.P.; Cairns, T.M.; Bender, F.C.; Whitbeck, J.C.; Lou, H.; Eisenberg, R.J.; Cohen, G.H. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J. Virol. 2009, 83, 6825–6836. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Blissard, G.W. The pre-transmembrane domain of the Autographa californica multicapsid nucleopolyhedrovirus GP64 protein is critical for membrane fusion and virus infectivity. J. Virol. 2009, 83, 10993–11004. [Google Scholar] [CrossRef] [PubMed]
- Jeetendra, E.; Ghosh, K.; Odell, D.; Li, J.; Ghosh, H.P.; Whitt, M.A. The membrane-proximal region of vesicular stomatitis virus glycoprotein G ectodomain is critical for fusion and virus infectivity. J. Virol. 2003, 77, 12807–12818. [Google Scholar] [CrossRef] [PubMed]
- Jeetendra, E.; Robison, C.S.; Albritton, L.M.; Whitt, M.A. The membrane-proximal domain of vesicular stomatitis virus G protein functions as a membrane fusion potentiator and can induce hemifusion. J. Virol. 2002, 76, 12300–12311. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Maidji, E.; Tugizov, S.; Pereira, L. Mutations in the carboxyl-terminal hydrophobic sequence of human cytomegalovirus glycoprotein B alter transport and protein chaperone binding. J. Virol. 1996, 70, 8029–8040. [Google Scholar] [PubMed]
- Rasile, L.; Ghosh, K.; Raviprakash, K.; Ghosh, H.P. Effects of deletions in the carboxy-terminal hydrophobic region of herpes simplex virus glycoprotein gB on intracellular transport and membrane anchoring. J. Virol. 1993, 67, 4856–4866. [Google Scholar] [PubMed]
- Lin, E.; Spear, P.G. Random linker-insertion mutagenesis to identify functional domains of herpes simplex virus type 1 glycoprotein B. Proc. Natl. Acad. Sci. USA 2007, 104, 13140–13145. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.; Ghosh, K.; Rasile, L.; Ghosh, H.P. Membrane anchoring domain of herpes simplex virus glycoprotein gB is sufficient for nuclear envelope localization. J. Virol. 1994, 68, 2272–2285. [Google Scholar] [PubMed]
- Arkin, I.T.; Brunger, A.T. Statistical analysis of predicted transmembrane α-helices. Biochim. Biophys. Acta 1998, 1429, 113–128. [Google Scholar] [CrossRef]
- Engelman, D.M.; Steitz, T.A.; Goldman, A. Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu. Rev. Biophys. Biophys. Chem. 1986, 15, 321–353. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.A.; Geraghty, R.J. Fusion activity of lipid-anchored envelope glycoproteins of herpes simplex virus type 1. Virology 2004, 324, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Kemble, G.W.; Danieli, T.; White, J.M. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell 1994, 76, 383–391. [Google Scholar] [CrossRef]
- Odell, D.; Wanas, E.; Yan, J.; Ghosh, H.P. Influence of membrane anchoring and cytoplasmic domains on the fusogenic activity of vesicular stomatitis virus glycoprotein G. J. Virol. 1997, 71, 7996–8000. [Google Scholar] [PubMed]
- Markosyan, R.M.; Cohen, F.S.; Melikyan, G.B. The lipid-anchored ectodomain of influenza virus hemagglutinin (GPI-HA) is capable of inducing nonenlarging fusion pores. Mol. Biol. Cell 2000, 11, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, M.L.; Donald, J.E.; DeGrado, W.F.; Jardetzky, T.S.; Lamb, R.A. Functional analysis of the transmembrane domain in paramyxovirus F protein-mediated membrane fusion. J. Mol. Biol. 2009, 386, 14–36. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Blissard, G.W. The Autographa californica multicapsid nucleopolyhedrovirus GP64 protein: Analysis of transmembrane domain length and sequence requirements. J. Virol. 2009, 83, 4447–4461. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, K.; Komano, J.; Yokomaku, Y.; Sugiura, W.; Yamamoto, N.; Matsuda, Z. Role of the specific amino acid sequence of the membrane-spanning domain of human immunodeficiency virus type 1 in membrane fusion. J. Virol. 2005, 79, 4720–4729. [Google Scholar] [CrossRef] [PubMed]
- Cleverley, D.Z.; Lenard, J. The transmembrane domain in viral fusion: Essential role for a conserved glycine residue in vesicular stomatitis virus G protein. Proc. Natl. Acad. Sci. USA 1998, 95, 3425–3430. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.G. Lipid-protein interactions in biological membranes: A structural perspective. Biochim. Biophys. Acta 2003, 1612, 1–40. [Google Scholar] [CrossRef]
- Ulmschneider, M.B.; Sansom, M.S. Amino acid distributions in integral membrane protein structures. Biochim. Biophys. Acta 2001, 1512, 1–14. [Google Scholar] [CrossRef]
- Armstrong, R.T.; Kushnir, A.S.; White, J.M. The transmembrane domain of influenza hemagglutinin exhibits a stringent length requirement to support the hemifusion to fusion transition. J. Cell Biol. 2000, 151, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Scholtz, J.M. A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 1998, 75, 422–427. [Google Scholar] [CrossRef]
- Yi, M.; Cross, T.A.; Zhou, H.X. Conformational heterogeneity of the M2 proton channel and a structural model for channel activation. Proc. Natl. Acad. Sci. USA 2009, 106, 13311–13316. [Google Scholar] [CrossRef] [PubMed]
- Quint, S.; Widmaier, S.; Minde, D.; Hornburg, D.; Langosch, D.; Scharnagl, C. Residue-specific side-chain packing determines the backbone dynamics of transmembrane model helices. Biophys. J. 2010, 99, 2541–2549. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Kielian, M. The conserved glycine residues in the transmembrane domain of the Semliki Forest virus fusion protein are not required for assembly and fusion. Virology 2005, 332, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Cymer, F.; Veerappan, A.; Schneider, D. Transmembrane helix-helix interactions are modulated by the sequence context and by lipid bilayer properties. Biochim. Biophys. Acta 2012, 1818, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Senes, A.; Engel, D.E.; DeGrado, W.F. Folding of helical membrane proteins: The role of polar, GxxxG-like and proline motifs. Curr. Opin. Struct. Biol. 2004, 14, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, W.; Langosch, D. Sequence-dependent backbone dynamics of a viral fusogen transmembrane helix. Protein Sci. 2012, 21, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Russ, W.P.; Engelman, D.M. The GxxxG motif: A framework for transmembrane helix-helix association. J. Mol. Biol. 2000, 296, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Unterreitmeier, S.; Fuchs, A.; Schaffler, T.; Heym, R.G.; Frishman, D.; Langosch, D. Phenylalanine promotes interaction of transmembrane domains via GxxxG motifs. J. Mol. Biol. 2007, 374, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Wang, L.; Gu, C.; Herschhorn, A.; Desormeaux, A.; Finzi, A.; Xiang, S.H.; Sodroski, J.G. Molecular architecture of the uncleaved HIV-1 envelope glycoprotein trimer. Proc. Natl. Acad. Sci. USA 2013, 110, 12438–12443. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.L.; Greene, N.G.; King, D.S.; Heldwein, E.E. Membrane requirement for folding of the herpes simplex virus 1 gB cytodomain suggests a unique mechanism of fusion regulation. J. Virol. 2012, 86, 8171–8184. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.H.; Gu, B.; Person, S. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J. Virol. 1988, 62, 2596–2604. [Google Scholar] [PubMed]
- Baghian, A.; Huang, L.; Newman, S.; Jayachandra, S.; Kousoulas, K.G. Truncation of the carboxy-terminal 28 amino acids of glycoprotein B specified by herpes simplex virus type 1 mutant amb1511-7 causes extensive cell fusion. J. Virol. 1993, 67, 2396–2401. [Google Scholar] [PubMed]
- Diakidi-Kosta, A.; Michailidou, G.; Kontogounis, G.; Sivropoulou, A.; Arsenakis, M. A single amino acid substitution in the cytoplasmic tail of the glycoprotein B of herpes simplex virus 1 affects both syncytium formation and binding to intracellular heparan sulfate. Virus Res. 2003, 93, 99–108. [Google Scholar] [CrossRef]
- Gage, P.J.; Levine, M.; Glorioso, J.C. Syncytium-inducing mutations localize to two discrete regions within the cytoplasmic domain of herpes simplex virus type 1 glycoprotein B. J. Virol. 1993, 67, 2191–2201. [Google Scholar] [PubMed]
- Nixdorf, R.; Klupp, B.G.; Karger, A.; Mettenleiter, T.C. Effects of truncation of the carboxy terminus of pseudorabies virus glycoprotein B on infectivity. J. Virol. 2000, 74, 7137–7145. [Google Scholar] [CrossRef] [PubMed]
- Garcia, N.J.; Chen, J.; Longnecker, R. Modulation of Epstein-Barr virus glycoprotein B (gB) fusion activity by the gB cytoplasmic tail domain. MBio 2013, 4, e00571–e00612. [Google Scholar] [CrossRef] [PubMed]
- Heineman, T.C.; Hall, S.L. Role of the varicella-zoster virus gB cytoplasmic domain in gB transport and viral egress. J. Virol. 2002, 76, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Chowdary, T.K.; Heldwein, E.E. Syncytial phenotype of C-terminally truncated herpes simplex virus type 1 gB is associated with diminished membrane interactions. J. Virol. 2010, 84, 4923–4935. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, J.; Zhang, X.; Jardetzky, T.S.; Longnecker, R. The Epstein-Barr virus (EBV) glycoprotein B cytoplasmic C-terminal tail domain regulates the energy requirement for EBV-induced membrane fusion. J. Virol. 2014, 88, 11686–11695. [Google Scholar] [CrossRef] [PubMed]
- Rogalin, H.B.; Heldwein, E.E. The interplay between the HSV-1 gB cytodomain and the gH cytotail during cell-cell fusion. J. Virol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Waning, D.L.; Russell, C.J.; Jardetzky, T.S.; Lamb, R.A. Activation of a paramyxovirus fusion protein is modulated by inside-out signaling from the cytoplasmic tail. Proc. Natl. Acad. Sci. USA 2004, 101, 9217–9222. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, H.C.; Anderson, W.F.; Cannon, P.M. Cytoplasmic tail of Moloney murine leukemia virus envelope protein influences the conformation of the extracellular domain: Implications for mechanism of action of the R Peptide. J. Virol. 2003, 77, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Kirchmeier, M.; Fluckiger, A.C.; Soare, C.; Bozic, J.; Ontsouka, B.; Ahmed, T.; Diress, A.; Pereira, L.; Schodel, F.; Plotkin, S.; Dalba, C.; Klatzmann, D.; Anderson, D.E. Enveloped virus-like particle expression of human cytomegalovirus glycoprotein B antigen induces antibodies with potent and broad neutralizing activity. Clin. Vaccine Immunol. 2014, 21, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Loving, R.; Wu, S.R.; Sjoberg, M.; Lindqvist, B.; Garoff, H. Maturation cleavage of the murine leukemia virus Env precursor separates the transmembrane subunits to prime it for receptor triggering. Proc. Natl. Acad. Sci. USA 2012, 109, 7735–7740. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kovacs, J.M.; Peng, H.; Rits-Volloch, S.; Lu, J.; Park, D.; Zablowsky, E.; Seaman, M.S.; Chen, B. Effect of the cytoplasmic domain on antigenic characteristics of HIV-1 envelope glycoprotein. Science 2015, 349, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Longnecker, R.; Connolly, S.A. A functional interaction between herpes simplex virus 1 glycoprotein gH/gL domains I and II and gD is defined by using alphaherpesvirus gH and gL chimeras. J. Virol. 2015, 89, 7159–7169. [Google Scholar] [CrossRef] [PubMed]
- Harman, A.; Browne, H.; Minson, T. The transmembrane domain and cytoplasmic tail of herpes simplex virus type 1 glycoprotein H play a role in membrane fusion. J. Virol. 2002, 76, 10708–10716. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.W.; Davis-Poynter, N.; Minson, A.C. Mutations in the cytoplasmic tail of herpes simplex virus glycoprotein H suppress cell fusion by a syncytial strain. J. Virol. 1994, 68, 6985–6993. [Google Scholar] [PubMed]
- Browne, H.M.; Bruun, B.C.; Minson, A.C. Characterization of herpes simplex virus type 1 recombinants with mutations in the cytoplasmic tail of glycoprotein H. J. Gen. Virol. 1996, 77, 2569–2573. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.L.; Heldwein, E.E. Mutations in the cytoplasmic tail of herpes simplex virus 1 gH reduce the fusogenicity of gB in transfected cells. J. Virol. 2013, 87, 10139–10147. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Arvin, A.M.; Oliver, S.L. The cytoplasmic domain of varicella-zoster virus glycoprotein H regulates syncytia formation and skin pathogenesis. PLoS Pathog. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Kamen, D.E.; Gross, S.T.; Girvin, M.E.; Wilson, D.W. Structural basis for the physiological temperature dependence of the association of VP16 with the cytoplasmic tail of herpes simplex virus glycoprotein H. J. Virol. 2005, 79, 6134–6141. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.O.; Lin, E.; Spear, P.G.; Longnecker, R. Insertion mutations in herpes simplex virus 1 glycoprotein H reduce cell surface expression, slow the rate of cell fusion, or abrogate functions in cell fusion and viral entry. J. Virol. 2010, 84, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cooper, R.S.; Heldwein, E.E. Herpesvirus gB: A Finely Tuned Fusion Machine. Viruses 2015, 7, 6552-6569. https://doi.org/10.3390/v7122957
Cooper RS, Heldwein EE. Herpesvirus gB: A Finely Tuned Fusion Machine. Viruses. 2015; 7(12):6552-6569. https://doi.org/10.3390/v7122957
Chicago/Turabian StyleCooper, Rebecca S., and Ekaterina E. Heldwein. 2015. "Herpesvirus gB: A Finely Tuned Fusion Machine" Viruses 7, no. 12: 6552-6569. https://doi.org/10.3390/v7122957
APA StyleCooper, R. S., & Heldwein, E. E. (2015). Herpesvirus gB: A Finely Tuned Fusion Machine. Viruses, 7(12), 6552-6569. https://doi.org/10.3390/v7122957