
Inhibition Profiling of Retroviral Protease Inhibitors Using an HIV-2 Modular System


Abstract
:1. Introduction

{kind=link}
| Inhibitor | Abbreviation | Trade Name | Remarks |
|---|---|---|---|
| Saquinavir | SQV | Invirase | |
| Ritonavir | RTV | Norvir | Used as booster drug |
| Combination therapy | |||
| Indinavir | INV | Crixivan | |
| Nelfinavir | NFV | Viracept | |
| Amprenavir | APV | Agenerase | Discontinued |
| Lopinavir/+Ritonavir | LPV | Kaletra, Aluvia | Second-generation |
| Fixed-dose combination therapy | |||
| Atazanavir | ATV | Reyataz | Second-generation |
| Fosamprenavir | FPV | Telzir, Lexiva | Second-generation |
| Tipranavir | TPV | Aptivus | Second-generation |
| Non-peptidic inhibitor | |||
| Darunavir | DRV | Prezista | Second-generation |
| Non-peptidic inhibitor |
2. Results and Discussion
2.1. In Vitro Kinetic Assays
| Inhibitor | IC50 (nM) | Ki (nM) | IC50 (nM) | Ki (nM) | Fold Increase (Ki) |
|---|---|---|---|---|---|
| Wild-Type | Double Mutant (I54M, L90M) | ||||
| Lopinavir | 1.18 ± 0.1 | 0.03 ± 0.001 | 2.32 ± 0.1 | 0.32 ± 0.02 | 10.6 |
| Indinavir | 1.30 ± 0.5 | 0.03 ± 0.02 | 2.60 ± 1.4 | 0.36 ± 0.07 | 12 |
| Darunavir | 1.76 ± 0.1 | 0.05 ± 0.005 | 8.14 ± 1.3 | 1.11 ± 0.1 | 22.2 |
| Saquinavir | 3.42 ± 0.1 | 0.09 ± 0.001 | 14.09 ± 1.7 | 1.93 ± 0.2 | 21.4 |
| Atazanavir | 3.34 ± 1 | 0.09 ± 0.03 | 10.93 ± 0.2 | 1.50 ± 0.04 | 16.6 |
| Ritonavir | 5.24 ± 3 | 0.12 ± 0.075 | 95.35 ± 17.6 | 13.05 ± 2.4 | 108.3 |
| Nelfinavir | 38 ± 10 | 1.01 ± 0.3 | 190 ± 14.5 | 26 ± 2 | 26 |
| Tipranavir | 50 ± 2 | 1.31 ± 0.56 | 4.80 ± 1.6 | 0.66 ± 0.2 | 0.5 |
| Amprenavir | 100 ± 7 | 2.43 ± 1.9 | 152 ± 9 | 20.8 ± 1.2 | 8.5 |
2.2. Cell Culture Assays
| Inhibitor | IC50 (μM) | IC50 (μM) | Fold Increase |
|---|---|---|---|
| Wild-Type | Double Mutant (I54M, L90M) | ||
| Lopinavir | 0.1 ± 0.01 | 3.1 ± 1.1 | 20.6 |
| Darunavir | 0.4 ± 0.05 | 18.7 ± 1.6 | 44.5 |
| Saquinavir | 1.3 ± 0.2 | 3.5 ± 1.2 | 2.7 |
| Indinavir sulfate | 1.4 ± 0.3 | 16.5 ± 2.1 | 11.7 |
| Nelfinavir | 2.7 ± 0.9 | 5.1 ± 1.7 | 1.8 |
| Tipranavir | 3.7 ± 0.6 | 5.8 ± 1.7 | 1.5 |
| Atazanavir sulfate | 5.9 ± 0.5 | 28.6 ± 0.35 | 4.8 |
| Ritonavir | 7.1 ± 0.7 | 14.5 ± 0.3 | 2 |
| Amprenavir | 68.7 ± 9.2 | >100 | ‒ |
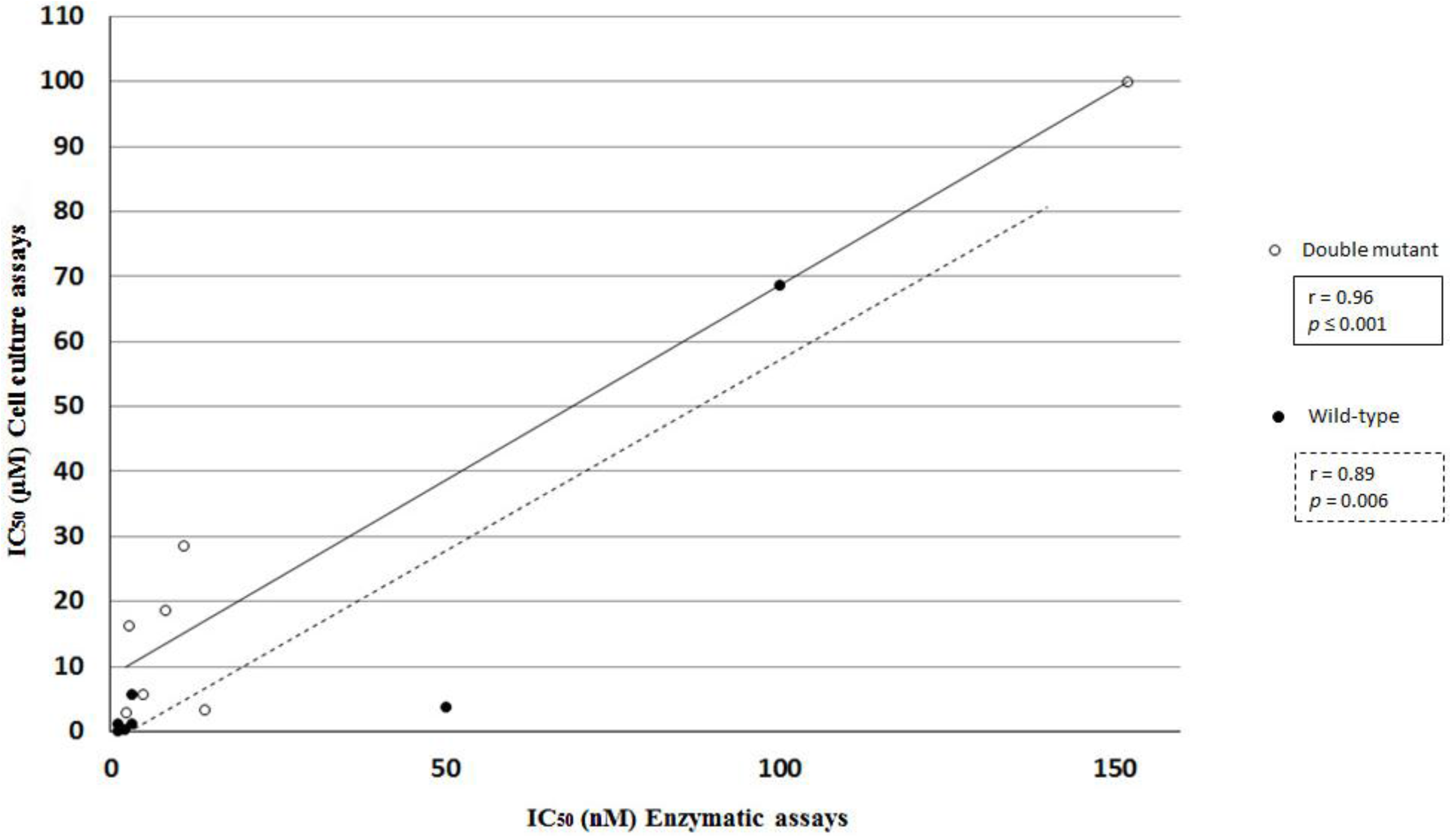
2.3. Statistical and Correlation Analysis of the Assays
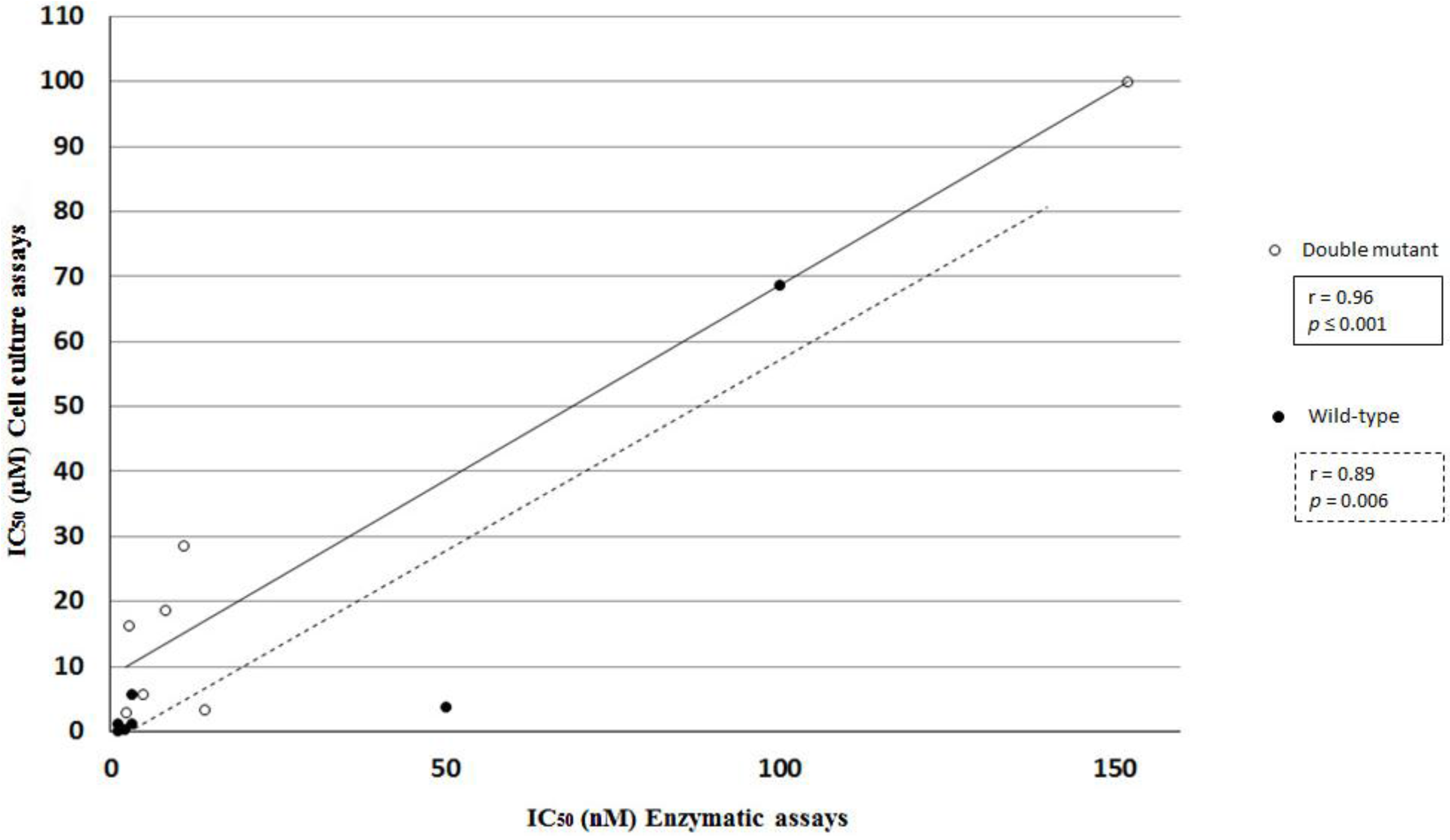
3. Materials and Methods
3.1. The Modular System
3.2. In Vitro Protease Expression and Purification
3.3. Enzymatic Assays
3.4. The Double Mutant Protease
3.5. Cell Culture Assays
3.6. Protease Inhibitors
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lemey, P.; Pybus, O.G.; Wang, B.; Saksena, N.K.; Salemi, M.; Vandamme, A.M. Tracing the origin and history of the HIV-2 epidemic. Proc. Natl. Acad. Sci. USA 2003, 100, 6588–6592. [Google Scholar] [CrossRef] [PubMed]
- Campbell-Yesufu, O.T.; Gandhi, R.T. Update on human immunodeficiency virus (HIV)-2 infection. Clin. Infect. Dis. 2011, 52, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.C.; Valadas, E.; Franca, L.; Carvalho, C.; Aleixo, M.J.; Mendez, J.; Marques, R.; Sarmento, A.; Doroana, M.; Antunes, F.; et al. Population mobility and the changing epidemics of HIV-2 in Portugal. HIV Med. 2012, 13, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Hodges-Mameletzis, I.; Silva, J.C.; Rodes, B.; Erasmus, S.; Paolucci, S.; Ruelle, J.; Pieniazek, D.; Taveira, N.; Trevino, A.; et al. Phylogeographical footprint of colonial history in the global dispersal of human immunodeficiency virus type 2 group A. J. Gen. Virol. 2012, 93, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Zagury, J.F.; Franchini, G.; Reitz, M.; Collalti, E.; Starcich, B.; Hall, L.; Fargnoli, K.; Jagodzinski, L.; Guo, H.G.; Laure, F.; et al. Genetic variability between isolates of human immunodeficiency virus (HIV) type 2 is comparable to the variability among HIV type 1. Proc. Natl. Acad. Sci. USA 1988, 85, 5941–5945. [Google Scholar] [CrossRef] [PubMed]
- Gallo, S.A.; Reeves, J.D.; Garg, H.; Foley, B.; Doms, R.W.; Blumenthal, R. Kinetic studies of HIV-1 and HIV-2 envelope glycoprotein-mediated fusion. Retrovirology 2006, 3, 90. [Google Scholar] [CrossRef] [PubMed]
- Popper, S.J.; Sarr, A.D.; Travers, K.U.; Gueye-Ndiaye, A.; Mboup, S.; Essex, M.E.; Kanki, P.J. Lower human immunodeficiency virus (HIV) type 2 viral load reflects the difference in pathogenicity of HIV-1 and HIV-2. J. Infect. Dis. 1999, 180, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Bock, P.J.; Markovitz, D.M. Infection with HIV-2. AIDS 2001, 15, S35–S45. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Consolidated Guidelines on the Use of Antiretroviral Drugs for Treating and Preventing HIV Infection; Recommendations for a public health approach; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- AIDSinfo. Considerations for Antiretroviral Use in Special Patient Populations. Available online: https://aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-arv-guidelines/24/hiv-2-infection (accessed on 8 April 2015).
- Descamps, D.; Peytavin, G.; Visseaux, B.; Tubiana, R.; Damond, F.; Campa, P.; Charpentier, C.; Khuong-Josses, M.A.; Duvivier, C.; Karmochkine, M.; et al. Dolutegravir in HIV-2-infected patients with resistant virus to first-line integrase inhibitors from the French named patient program. Clin. Infect. Dis. 2015, 60, 1521–1527. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Bird, L.E.; Chamberlain, P.P.; Stewart-Jones, G.B.; Stuart, D.I.; Stammers, D.K. Structure of HIV-2 reverse transcriptase at 2.35-Å resolution and the mechanism of resistance to non-nucleoside inhibitors. Proc Natl. Acad. Sci. USA 2002, 99, 14410–14415. [Google Scholar] [CrossRef] [PubMed]
- Ekouevi, D.K.; Tchounga, B.K.; Coffie, P.A.; Tegbe, J.; Anderson, A.M.; Gottlieb, G.S.; Vitoria, M.; Dabis, F.; Eholie, S.P. Antiretroviral therapy response among HIV-2 infected patients: A systematic review. BMC Infect. Dis. 2014, 14, 461. [Google Scholar] [CrossRef] [PubMed]
- Gilleece, Y.; Chadwick, D.R.; Breuer, J.; Hawkins, D.; Smit, E.; McCrae, L.X.; Pillay, D.; Smith, N.; Anderson, J.; Subcommittee, B.G. British HIV association guidelines for antiretroviral treatment of HIV-2-positive individuals 2010. HIV Med. 2010, 11, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.; Jallow, S.; Rowland-Jones, S.L.; de Silva, T.I. Antiretroviral therapy for HIV-2 infection: Recommendations for management in low-resource settings. AIDS Res. Treat. 2011, 2011, 463704. [Google Scholar] [CrossRef] [PubMed]
- Pokorna, J.; Machala, L.; Rezacova, P.; Konvalinka, J. Current and novel inhibitors of HIV protease. Viruses 2009, 1, 1209–1239. [Google Scholar] [CrossRef] [PubMed]
- Tie, Y.; Wang, Y.F.; Boross, P.I.; Chiu, T.Y.; Ghosh, A.K.; Tozser, J.; Louis, J.M.; Harrison, R.W.; Weber, I.T. Critical differences in HIV-1 and HIV-2 protease specificity for clinical inhibitors. Protein Sci. 2012, 21, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Ntemgwa, M.; Brenner, B.G.; Oliveira, M.; Moisi, D.; Wainberg, M.A. Natural polymorphisms in the human immunodeficiency virus type 2 protease can accelerate time to development of resistance to protease inhibitors. Antimicrob. Agents Chemother. 2007, 51, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Desbois, D.; Roquebert, B.; Peytavin, G.; Damond, F.; Collin, G.; Benard, A.; Campa, P.; Matheron, S.; Chene, G.; Brun-Vezinet, F.; et al. In vitro phenotypic susceptibility of human immunodeficiency virus type 2 clinical isolates to protease inhibitors. Antimicrob. Agents Chemother. 2008, 52, 1545–1548. [Google Scholar] [CrossRef] [PubMed]
- Rodes, B.; Sheldon, J.; Toro, C.; Jimenez, V.; Alvarez, M.A.; Soriano, V. Susceptibility to protease inhibitors in HIV-2 primary isolates from patients failing antiretroviral therapy. J. Antimicrob. Chemother. 2006, 57, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Brower, E.T.; Bacha, U.M.; Kawasaki, Y.; Freire, E. Inhibition of HIV-2 protease by HIV-1 protease inhibitors in clinical use. Chem. Biol. Drug Des. 2008, 71, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Witvrouw, M.; Pannecouque, C.; Switzer, W.M.; Folks, T.M.; De Clercq, E.; Heneine, W. Susceptibility of HIV-2, SIV and SHIV to various anti-HIV-1 compounds: Implications for treatment and postexposure prophylaxis. Antivir. Ther. 2004, 9, 57–65. [Google Scholar] [PubMed]
- Raugi, D.N.; Smith, R.A.; Ba, S.; Toure, M.; Traore, F.; Sall, F.; Pan, C.; Blankenship, L.; Montano, A.; Olson, J.; et al. Complex patterns of protease inhibitor resistance among antiretroviral treatment-experienced HIV-2 patients from Senegal: Implications for second-line therapy. Antimicrob. Agents Chemother. 2013, 57, 2751–2760. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, M.; Matuz, K.; Toth, F.; Tozser, J. A modular system to evaluate the efficacy of protease inhibitors against HIV-2. PLoS ONE 2014, 9, e113221. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A.; Miller, M.; Jaskolski, M.; Sathyanarayana, B.K.; Baldwin, E.; Weber, I.T.; Selk, L.M.; Clawson, L.; Schneider, J.; Kent, S.B. Conserved folding in retroviral proteases: Crystal structure of a synthetic hiv-1 protease. Science 1989, 245, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Arias, L.; Tozser, J. HIV-1 protease inhibitors: Effects on HIV-2 replication and resistance. Trends Pharmacol. Sci. 2008, 29, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Gustchina, A.; Weber, I.T. Comparative analysis of the sequences and structures of HIV-1 and HIV-2 proteases. Proteins 1991, 10, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Nijhuis, M.; Schuurman, R.; de Jong, D.; Erickson, J.; Gustchina, E.; Albert, J.; Schipper, P.; Gulnik, S.; Boucher, C.A.B. Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. Aids 1999, 13, 2349–2359. [Google Scholar] [CrossRef] [PubMed]
- Romano, L.; Venturi, G.; Giomi, S.; Pippi, L.; Valensin, P.E.; Zazzi, M. Development and significance of resistance to protease inhibitors in HIV-1-infected adults under triple-drug therapy in clinical practice. J. Med. Virol. 2002, 66, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Ridky, T.; Leis, J. Development of drug resistance to HIV-1 protease inhibitors. J. Biol. Chem. 1995, 270, 29621–29623. [Google Scholar] [CrossRef] [PubMed]
- Adje-Toure, C.A.; Cheingsong, R.; Garcia-Lerma, J.G.; Eholie, S.; Borget, M.Y.; Bouchez, J.M.; Otten, R.A.; Maurice, C.; Sassan-Morokro, M.; Ekpini, R.E.; et al. Antiretroviral therapy in HIV-2-infected patients: Changes in plasma viral load, CD4+ cell counts, and drug resistance profiles of patients treated in Abidjan, Cote d’Ivoire. AIDS 2003, 17, S49–S54. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; Henry, M.; Tourres, C.; Lozachmeur, D.; Gallais, H.; Gastaut, J.A.; Moreau, J.; Tamalet, C. Polymorphism and drug-selected mutations in the protease gene of human immunodeficiency virus type 2 from patients living in Southern France. J. Clin. Microbiol. 2004, 42, 570–577. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shen, C.H.; Wang, Y.F.; Kovalevsky, A.Y.; Harrison, R.W.; Weber, I.T. Amprenavir complexes with HIV-1 protease and its drug-resistant mutants altering hydrophobic clusters. FEBS J. 2010, 277, 3699–3714. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.D.; Marousek, G.I.; Chou, S. HIV protease mutations associated with amprenavir resistance during salvage therapy: Importance of I54M. J. Clin. Virol. 2004, 30, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, C.; Camacho, R.; Ruelle, J.; Kaiser, R.; Eberle, J.; Gurtler, L.; Pironti, A.; Sturmer, M.; Brun-Vezinet, F.; Descamps, D.; et al. HIV-2EU: Supporting standardized HIV-2 drug resistance interpretation in Europe. Clin. Infect. Dis. 2013, 56, 1654–1658. [Google Scholar] [CrossRef] [PubMed]
- Ode, H.; Neya, S.; Hata, M.; Sugiura, W.; Hoshino, T. Computational simulations of HIV-1 proteases—Multi-drug resistance due to nonactive site mutation L90M. J. Am. Chem. Soc. 2006, 128, 7887–7895. [Google Scholar] [CrossRef] [PubMed]
- Ode, H.; Ota, M.; Neya, S.; Hata, M.; Sugiura, W.; Hoshino, T. Resistant mechanism against nelfinavir of human immunodeficiency virus type 1 proteases. J. Phys. Chem. B 2005, 109, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Zhang, X.C.; Hartsuck, J.A.; Tang, J. Crystal structure of an in vivo HIV-1 protease mutant in complex with saquinavir: Insights into the mechanisms of drug resistance. Protein Sci. 2000, 9, 1898–1904. [Google Scholar] [CrossRef] [PubMed]
- Wood, R. Atazanavir: Its role in HIV treatment. Exp. Rev. Anti. Infect. Ther. 2008, 6, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Poppe, S.M.; Slade, D.E.; Chong, K.T.; Hinshaw, R.R.; Pagano, P.J.; Markowitz, M.; Ho, D.D.; Mo, H.; Gorman, R.R., 3rd; Dueweke, T.J.; et al. Antiviral activity of the dihydropyrone PNU-140690, a new nonpeptidic human immunodeficiency virus protease inhibitor. Antimicrob. Agents Chemother. 1997, 41, 1058–1063. [Google Scholar] [PubMed]
- Luna, B.; Townsend, M.U. Tipranavir: The first nonpeptidic protease inhibitor for the treatment of protease resistance. Clin. Ther. 2007, 29, 2309–2318. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Z.; Brunzelle, J.S.; Kovari, I.A.; Dewdney, T.G.; Reiter, S.J.; Kovari, L.C. The higher barrier of darunavir and tipranavir resistance for HIV-1 protease. Biochem. Biophys. Res. Commun. 2011, 412, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Stanford. HIV drug resistance databse. Available online: http://hivdb.stanford.edu/DR/PIResiNote.html (accessed on 2 March 2014).
- Hsu, A.; Granneman, G.R.; Bertz, R.J. Ritonavir. Clinical pharmacokinetics and interactions with other anti-HIV agents. Clin. Pharmacokinet. 1998, 35, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.E.; Wu, E.; Patick, A.K.; Kerr, B.; Zorbas, M.; Lankford, A.; Kobayashi, T.; Maeda, Y.; Shetty, B.; Webber, S. Circulating metabolites of the human immunodeficiency virus protease inhibitor nelfinavir in humans: Structural identification, levels in plasma, and antiviral activities. Antimicrob. Agents Chemother. 2001, 45, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Hirani, V.N.; Raucy, J.L.; Lasker, J.M. Conversion of the HIV protease inhibitor nelfinavir to a bioactive metabolite by human liver CYP2C19. Drug Metab. Dispos. 2004, 32, 1462–1467. [Google Scholar] [CrossRef] [PubMed]
- Denissen, J.F.; Grabowski, B.A.; Johnson, M.K.; Buko, A.M.; Kempf, D.J.; Thomas, S.B.; Surber, B.W. Metabolism and disposition of the HIV-1 protease inhibitor ritonavir (ABT-538) in rats, dogs, and humans. Drug Metab. Dispos. 1997, 25, 489–501. [Google Scholar] [PubMed]
- Wu, C.; Orozco, C.; Boyer, J.; Leglise, M.; Goodale, J.; Batalov, S.; Hodge, C.L.; Haase, J.; Janes, J.; Huss, J.W., 3rd; et al. Biogps: An extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol 2009, 10, R130. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Lee, H.L.; Pacchia, A.L.; Ron, Y.; Dougherty, J.P. A HIV-2-based self-inactivating vector for enhanced gene transduction. J. Biotechnol. 2007, 127, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Louis, J.M.; Ishima, R.; Aniana, A.; Sayer, J.M. Revealing the dimer dissociation and existence of a folded monomer of the mature HIV-2 protease. Protein Sci. 2009, 18, 2442–2453. [Google Scholar] [CrossRef] [PubMed]
- Miklossy, G.; Tozser, J.; Kadas, J.; Ishima, R.; Louis, J.M.; Bagossi, P. Novel macromolecular inhibitors of human immunodeficiency virus-1 protease. Protein Eng. Des. Sel. 2008, 21, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Sokal, R.R.; Rohlf, F.J. Introduction to Biostatistics, 2nd ed.; W.H. Freeman and Company: Mineola, NY, USA, 1969. [Google Scholar]
- Day, R.W.; Quinn, G.P. Comparisons of treatments after an analysis of variance in ecology. Ecol. Monogr. 1989, 59, 433–463. [Google Scholar] [CrossRef]
- Cohen, J. Statistical power analysis. Curr. Dir. Psychol. Sci. 1992, 1, 98–101. [Google Scholar] [CrossRef]
- Field, A. Discovering Statistics; SAGE Publications: London, UK, 2009. [Google Scholar]
- Hammer, Ř.; Harper, D.A.T.; Ryan, P.D. Past: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahdi, M.; Szojka, Z.; Mótyán, J.A.; Tőzsér, J. Inhibition Profiling of Retroviral Protease Inhibitors Using an HIV-2 Modular System. Viruses 2015, 7, 6152-6162. https://doi.org/10.3390/v7122931
Mahdi M, Szojka Z, Mótyán JA, Tőzsér J. Inhibition Profiling of Retroviral Protease Inhibitors Using an HIV-2 Modular System. Viruses. 2015; 7(12):6152-6162. https://doi.org/10.3390/v7122931
Chicago/Turabian StyleMahdi, Mohamed, Zsófia Szojka, János András Mótyán, and József Tőzsér. 2015. "Inhibition Profiling of Retroviral Protease Inhibitors Using an HIV-2 Modular System" Viruses 7, no. 12: 6152-6162. https://doi.org/10.3390/v7122931
APA StyleMahdi, M., Szojka, Z., Mótyán, J. A., & Tőzsér, J. (2015). Inhibition Profiling of Retroviral Protease Inhibitors Using an HIV-2 Modular System. Viruses, 7(12), 6152-6162. https://doi.org/10.3390/v7122931