Human Immunodeficiency Virus-1 (HIV-1)-Mediated Apoptosis: New Therapeutic Targets

Abstract
:1. Introduction
2. Mediators of Apoptosis in HIV
2.1. Viral Entry and the Cell Surface Receptors
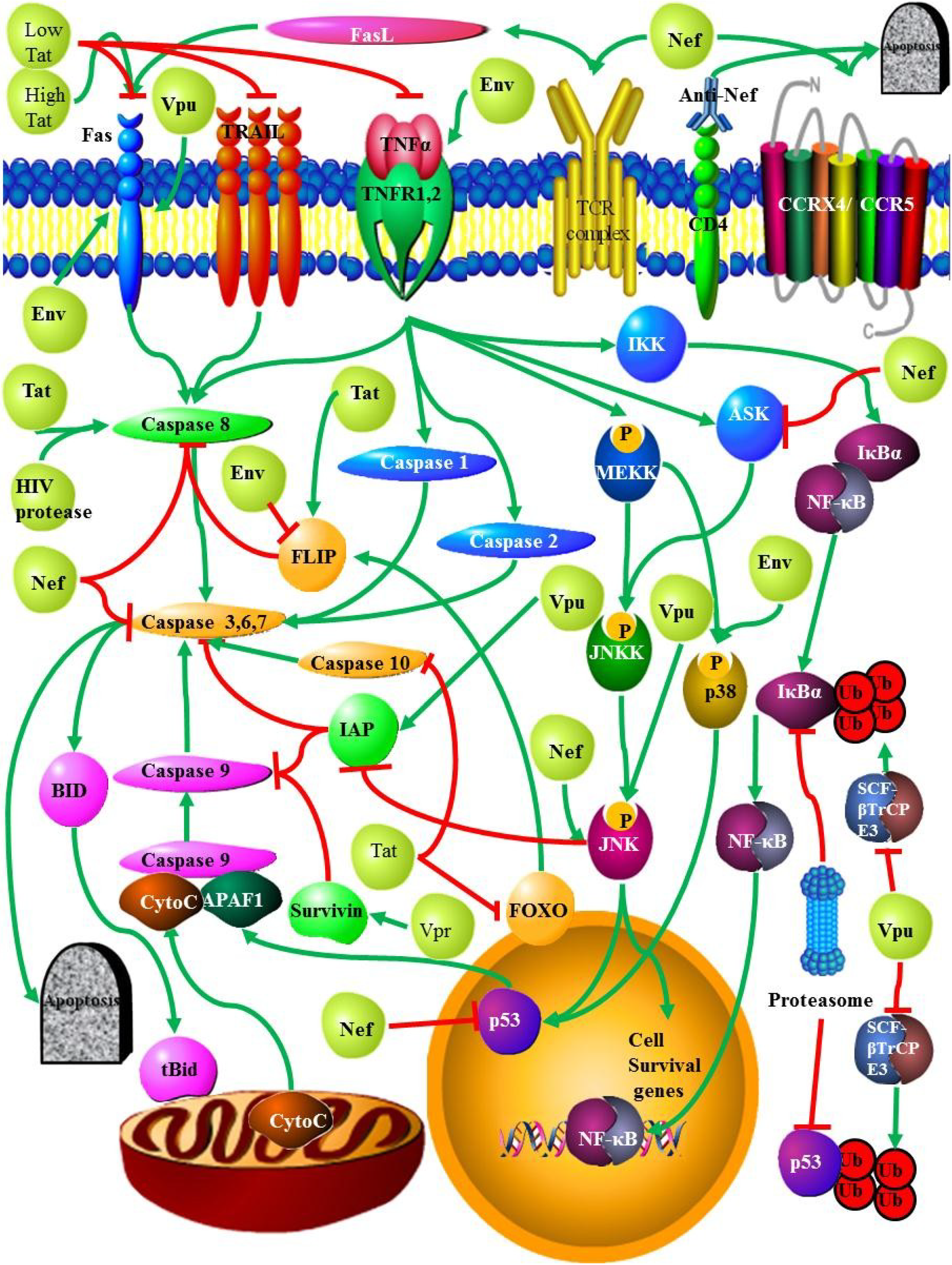
2.2. Death Receptors
2.2.1. Fas/FasL
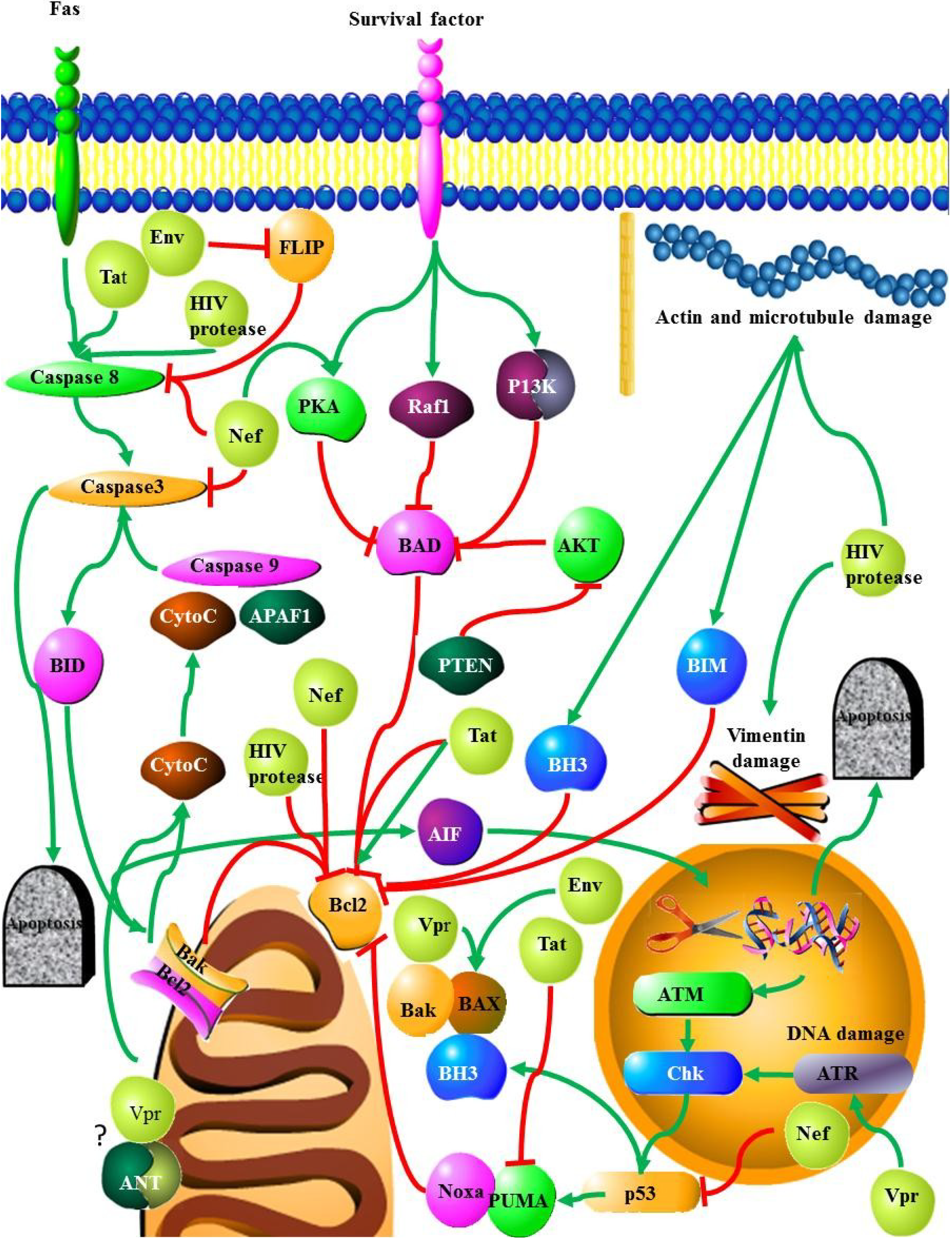
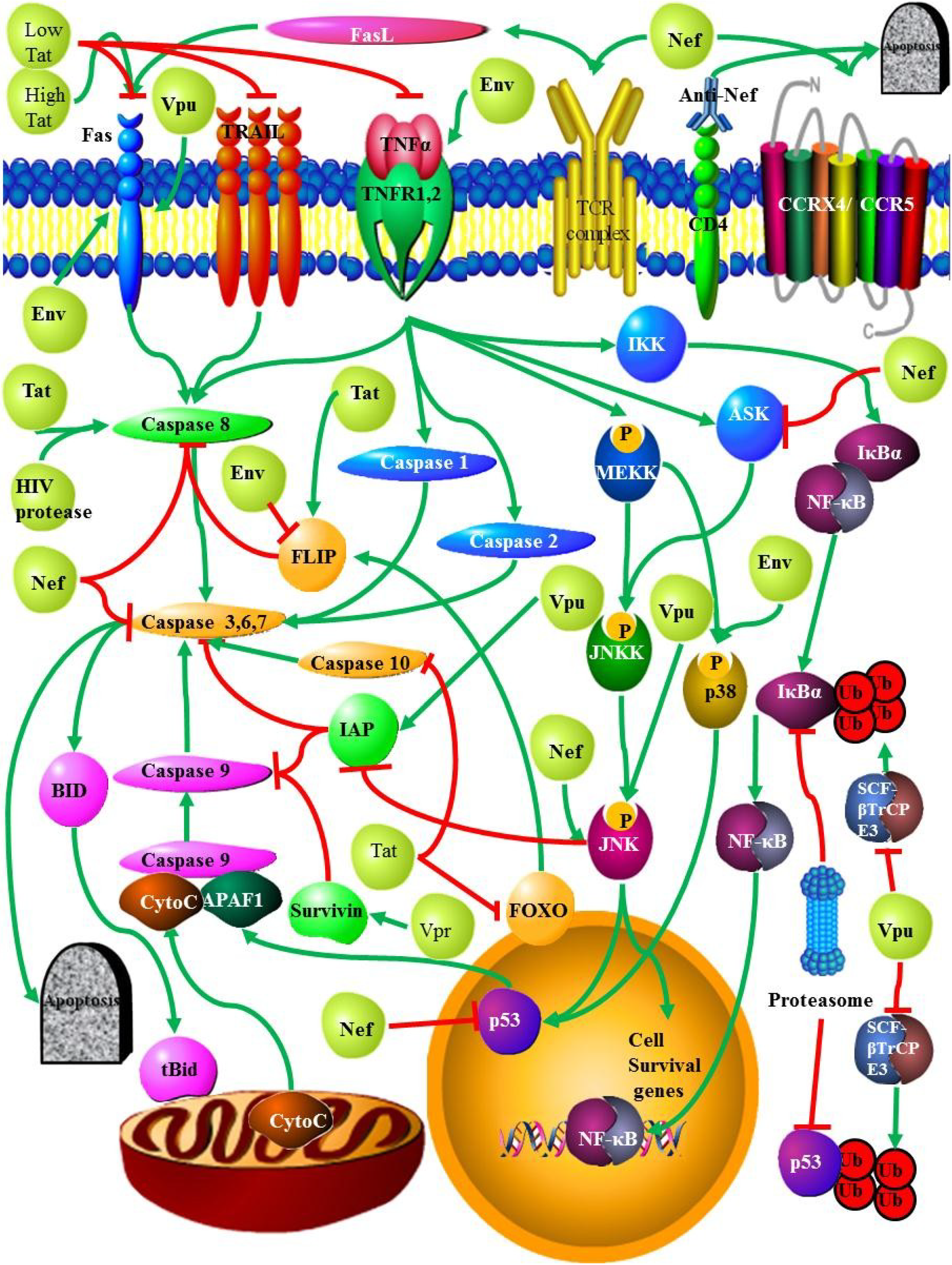
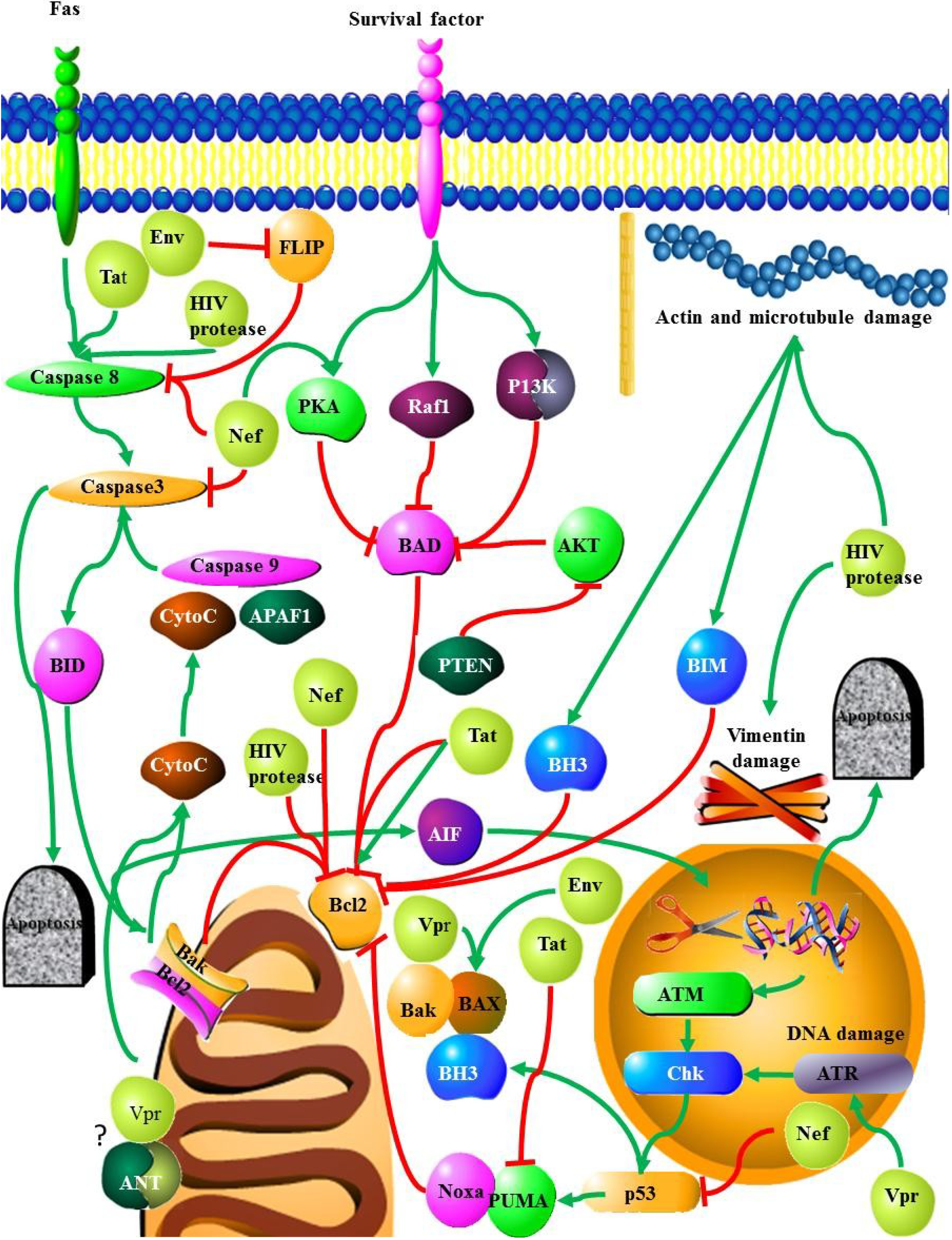
2.2.2. TNF
2.2.3. TRAIL
2.2.4. Co-Receptors CCR5/CXCR4
2.3. HIV Proteins and Apoptosis
2.3.1. HIV Protease

{kind=link}
{kind=link}
{kind=link}
| Pro/anti Apoptotic | Mechanism | Reference | |
|---|---|---|---|
| HIV protease | Pro-apoptotic | Cytoskeletal damage | [51] |
| Damage to Plasma membrane | [52] | ||
| Proteolytic cleavage of Bcl-2 | [53] | ||
| Cleavage of procaspase 8 | [54] | ||
| Anti-apoptotic | Increased NF-κβ signaling | [55,56] | |
| Tat | Pro-apoptotic | Up-regulate Caspase 3 and 8 | [57] |
| Up-regulation of FasL and RCAS | [58] | ||
| Up-regulation of Bax | [58] | ||
| Decrease in FOXO3a signaling- FLIP decrease | [59,60] | ||
| Altered microtubule stability resulting in Bcl2 inhibition | [61,62] | ||
| Increased ROS production | [63] | ||
| Anti-apoptotic | Increased resistance of cells to TNF, Fas and TRAIL | [58] | |
| Decrease in Caspase 10 | [64] | ||
| Increase in FLIP transcription | [64] | ||
| Increase in Bcl2 activity | [65] | ||
| Decrease in FOXO3 leading to a decrease in Bim and Puma transcription | [66] | ||
| Nef | Pro-apoptotic | Up-regulation of FasL | [67] |
| Increase in JNK signaling leading to increase in p53 transcription | [68] | ||
| Decrease in Bcl-2 and Bcl-xL activity | [69] | ||
| Binding to CXCR4 | [70] | ||
| Anti-apoptotic | Inhibition of caspase 3 and 8 | [71] | |
| Inhibition of Ask1 | [72] | ||
| Phosphorylation of BAD | [73,74] | ||
| Up-regulate MAPK and JNK | [75] | ||
| Bind p53 and prevent p53 mediated apoptosis | [76] | ||
| Down modulate the expression of molecules of the MHC class I | [77] | ||
| PAK activation | [78] | ||
| Inhibition of caspases 9 via increased nuclear export of TRNA via eEF1A and Exp-t | [79] | ||
| Vpr | Pro-apoptotic | ANT mitochondrial membrane permeability | [80] |
| Bax activation | [81] | ||
| Anti-apoptotic | Survivin | [82] | |
| Vpu | Pro-apoptotic | Decrease in NF-κβ signaling | [83] |
| Increase in p53 protein levels | [84] | ||
| Increases the sensitivity of cells to Fas associated apoptosis | [85] | ||
| Increase in JNK signaling | [86] | ||
| Anti-apoptotic | Unknown role but cell type dependent decrease has been observed | [87] | |
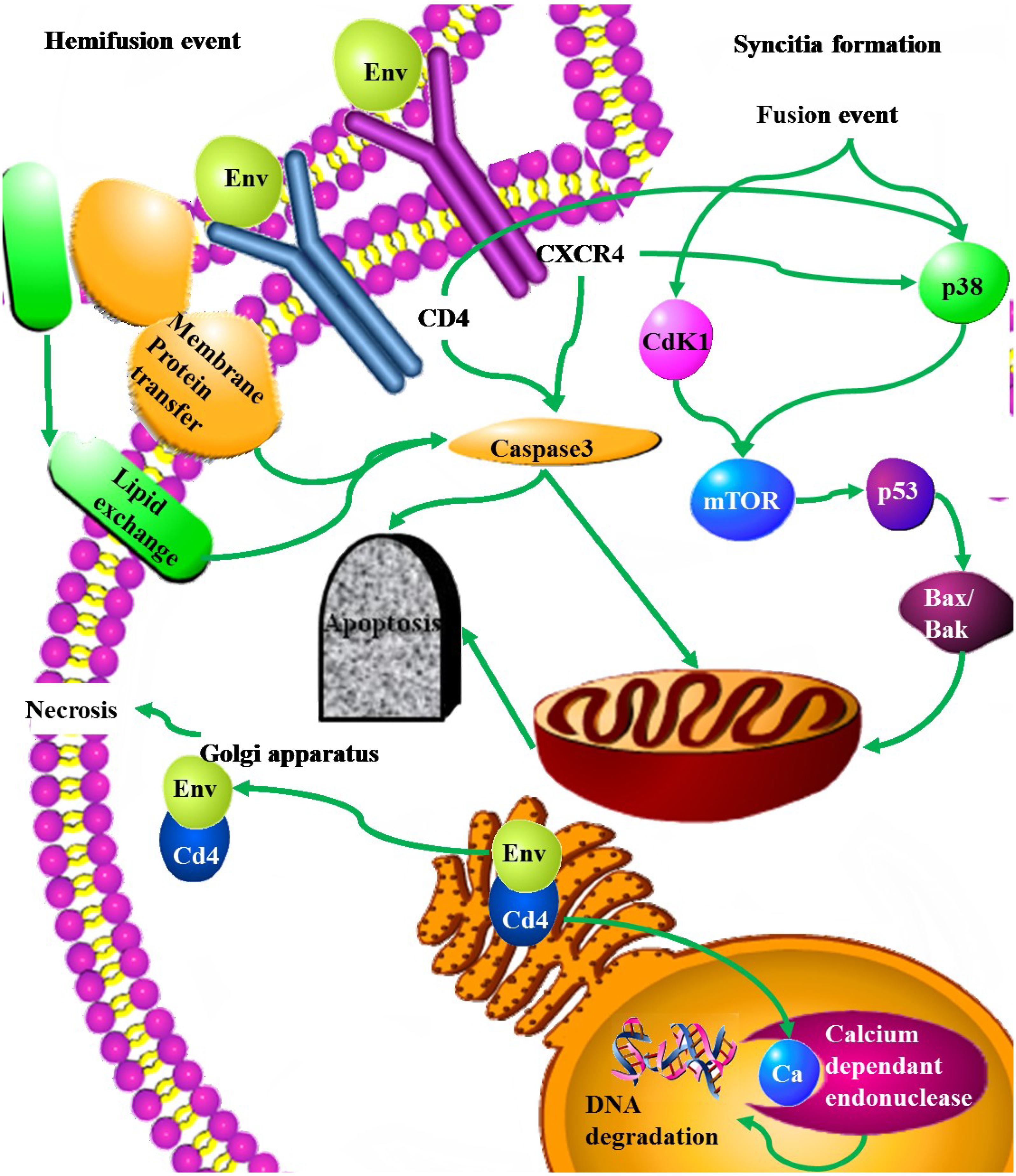
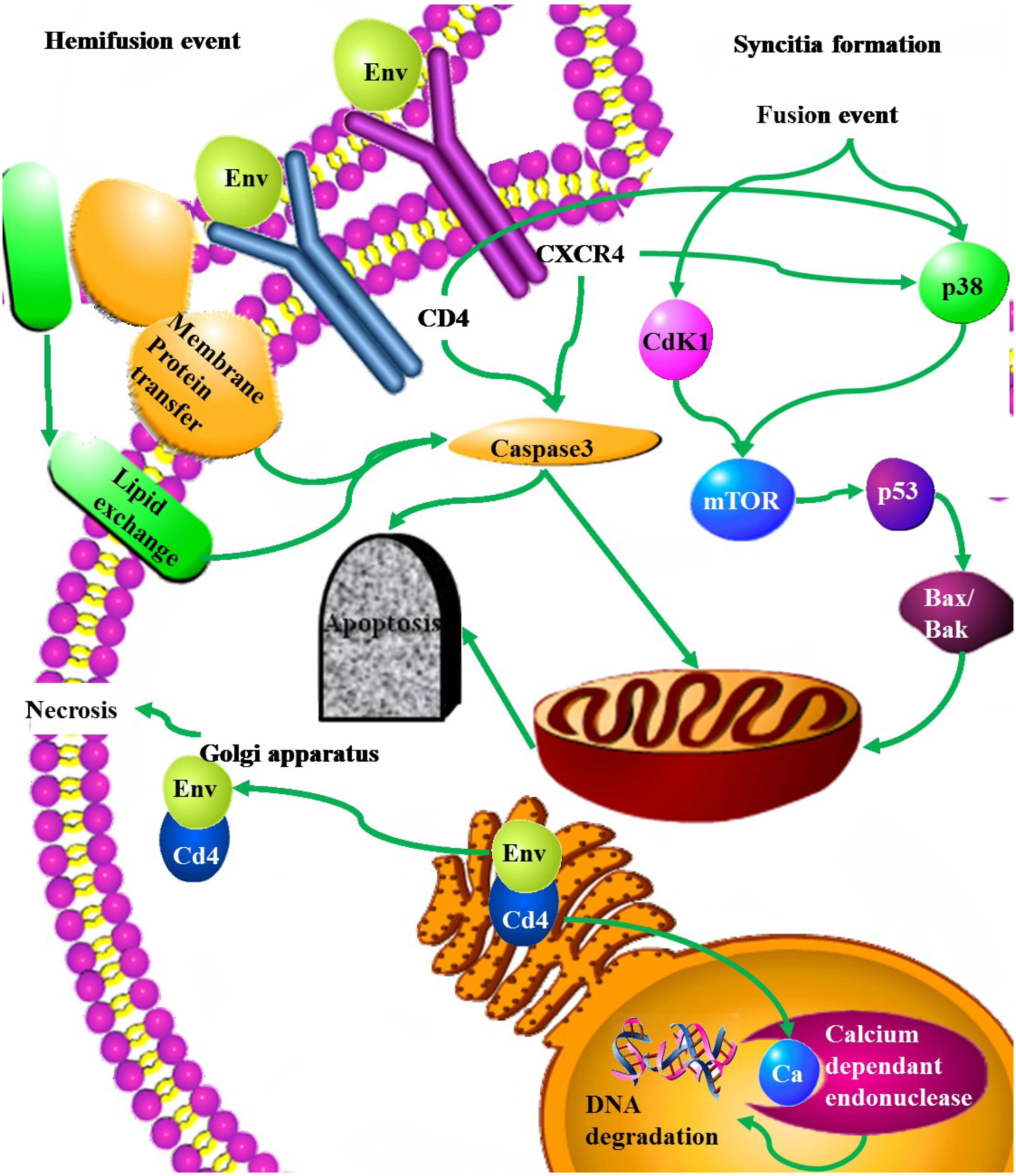
| Env | Pro-apoptotic | Up-regulation of Fas and Fas/L, and an increase in Fas mediated apoptosis | [88] |
| Decrease in the transcription of the FLICE-like inhibitory protein (FLIP) | [89] | ||
| Up-regulating Bax. Intrinsic apoptosis pathway | [9,89] | ||
| Activation of the p38 but not AKT or ERK | [90] | ||
| Induces membrane expression of TNF-α | [91] | ||
| Hemifusion cell killing associated with caspase 3 and high ROS | [92,93,94] | ||
| Syncitia formation leading fused cells to undergo apoptosis through the intrinsic pathway, involving the activation of Cdk1/cyclinB, Nk-κβ, mTOR, MAPK, p53 and PUMA | [95,96] | ||
| Molecular mimicry of Fas | [97,98] | ||
| Up-regulation of caspase 1 | [99] | ||
| gp-160 -CD4 complex blocks nuclear pores | [100] | ||
| contagious apoptosis through caspase activation and alterations in mitochondrial trans-membrane potential | [101] | ||
| Anti-apoptotic | High levels of CD4 expression lead to the retention of gp160-CD4 complexes within the Endoplasmic reticulum. | [102] |
2.3.2. Nef
2.3.3. Tat
2.3.4. Vpu
2.3.5. Vpr
2.3.6. Env
3. HIV Related Apoptotic Pathways in Immune Related Cells
3.1. CD4+ T Cells
3.2. The Contribution of HIV Proteins to Apoptosis in Macrophages
4. HIV-Associated Pathologies: The Role of Apoptosis
| HIV-Associated Neurocognitive Disorders (HAND) | ||
|---|---|---|
 | Prevalence 40%–50% of HIV-1 positive patients. | [166,167] |
| Symptoms Impaired cognitive activity, memory, learning, attention, problem solving, decision making, confusion, forgetfulness, behavioral changes, and nerve pain. | [168] | |
| Histological patterns Macrophage infiltration, activated microglia, reduced synaptic/dendritic density and selective neuronal loss. | [169] | |
Underlying causes
| [170] | |
| [171,172] | |
| [173] | |
Role of HIV proteins
| ||
| [174] [166,175] | |
| [14,166,176]. | |
| [166] [167] | |
| [177] | |
| [178] | |
| HIV-Associated Nephropathy (HIVAN) | ||
 | Prevalence Many patients with HIVAN ultimately progress to end stage renal disease (ESRD).90% of all ESRD cases attributed to HIVAN occur in African Americans | [179,180] [135] |
| Symptoms | ||
| Inflammation is the major pathology. | [181,182] | |
Histological patterns
| [183] [184,185] [186] | |
Underlying causes
| [161,162],[187] | |
| [188,189] | |
| [185] | |
Role of HIV proteins
| [190,191] [190] [192] | |
| HIV-Associated Vascular Complications | ||
 | Prevalence 1% of HIV patients. | [163] |
| Symptoms | ||
Coronary heart disease, pulmonary hypertension (PH), and atherosclerosisIncrease the risk for noninfectious pulmonary conditions, including
| [193,194,195] [196,197] [198] | |
| Underlying causes Increased production of reactive oxygen species (ROS), such as superoxide, leading to
| [164,165] | |
| Role of HIV proteins Documented, but not yet fully understood. Suspected to play a role through promoting apoptosis, growth, and proliferation of a variety of cells in vitro in vitro by interacting with molecular partners in the infected host | [160,199,200] | |
| HIV-Associated Tumors, Kaposi Sarcoma (KS) | ||
 | Prevalence For some time, Kaposi sarcoma was seen in 30%–40% of patients with AIDS, often as the presenting sign. The incidence of Kaposi sarcoma has fallen markedly in recent times, although its prevalence has not. | [201] |
| Symptoms Raised red, purple, brown, or black blotches found on the skin, mouth gastrointestinal tract and respiratory tract. | [201] | |
| Histological patterns Initially inflammatory dermatosis with signs of vasoformation. Later abnormal elongated spindle cells are present and are arranged in haphazard clusters. Dense vascularization with Hyaline globules | [202] | |
Underlying causes
| [203,204,205] [206,207,208,209] [210,211] | |
| Role of HIV proteins The de-regulation of apoptosis by HIV proteins has been shown to play a significant role in tumor development. Tat effects are cell-type dependent selectively promoting apoptosis in various cell systems. Tat increases Bcl-2 expression | [212,213,214] | |
5. Therapeutic Targets: Targeting Apoptotic Pathways
5.1. Therapies and Their Shortcomings
5.2. Drugs Targeting HIV Proteins
| Drug | Target | Reference |
|---|---|---|
| Drugs targeting Tat | ||
| Bovine Dialyzable Leukocyte Extract (bDLE) | Down regulation of Tat-protein lowers the expression of anti-apoptotic protein BCL-2 in infected cells | [81] |
| PI3K inhibitors and Akt inhibitors | Counter Tat-protein induced protection on infected cells | [82] |
| picolinic acid (PA) and fusaric acid (FA) | Target the conserved RING finger on Tat, inhibiting trans-activation | [225,226] |
| β-arrestin 2 | Reduces apoptosis | [3] |
| Drugs targeting HIV protease | ||
| Saquinavir, Ritonavir, Indinavir, Nelfinavir,Amprenavir, Lopinavir, Atazanavir, Fosamprenavir, Tipranavir, Darunavir | Inhibit HIV protease—inhibit viral maturation | [11,227] |
| Polyoxometalates | Act against HIV protease | [228] |
| Single-chain Fv (scFv) | An artificial derivative of mAb1696 | |
| P27 peptide | Peptide derivative of the C- and N-terminal domains of HIV protease which inhibit dimerisation | [11] |
| mAb1696 antibody | Uncouples the protease dimer and induces inhibition | [229] |
| 12-aminododecanoic acid (12-Ado) | Template for HIV protease dimerisation inhibition | [230] |
| C3-substituted cyclopentyltetrahydrofuranyl | Allosteric inhibitors Bind the flap region of HIV protease | [11,231] |
| GRL-02031 | Another derivative of Cp-THF | [232] |
| Drugs targeting gp120 | ||
| Sifuvirtide | Fusion Inhibitor | [233] |
| Enfuvirtide | Fusion inhibitor | [41] |
| Maraviroc | CCR5 antagonist | [41] |
| 4-phenyl-1-4-phenylbutyl piperidine (PPBP) | sigma-1 receptor agonist acting against gp120 | [167] |
| Selective serotonin reuptake inhibitors (SSRIs) | Reduces cytokine receptor expression in the nervous system, reducing gp120 binding targets | [234] |
| Other drugs | ||
| Double stranded RNA Activated Caspase Oligomerizer (DRACO), | Selects for viral infected cells only based on the length of RNA transcription helices. Increases apoptosis by caspase activation | [235] |
| NAPVSIPQ (NAP) | protects against mitochondrial release of cytochrome c. | [176] |
5.2.1. Drugs Targeting Tat
5.2.2. Drugs Targeting gp120
5.2.3. Drugs Targeting HIV Protease
5.3. Other Drugs of Interest
5.3.1. Aptamers
5.3.2. Nanoformulation of ARVs
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Pantaleo, G.; Fauci, A.S. Apoptosis in HIV infection. Nat. Med. 1995, 1, 118–120. [Google Scholar] [CrossRef]
- Meyaard, L.; Otto, S.A.; Jonker, R.R.; Mijnster, M.J.; Keet, R.P.; Miedema, F. Programmed death of T cells in HIV-1 infection. Science 1992, 257, 217–219. [Google Scholar]
- Moorman, J.; Zhang, Y.; Liu, B.; LeSage, G.; Chen, Y.; Stuart, C.; Prayther, D.; Yin, D. HIV-1 gp120 primes lymphocytes for opioid-induced, β-arrestin 2-dependent apoptosis. Biochim et Biophys Acta (BBA). Mol. Cell Res. 2009, 1793, 1366–1371. [Google Scholar]
- Izmailova, E.; Bertley, F.M.N.; Huang, Q.; Makori, N.; Miller, C.J.; Young, R.A.; Aldovini, A. HIV-1 Tat reprograms immature dendritic cells to express chemoattractants for activated T cells and macrophages. Nat. Med. 2003, 9, 191–197. [Google Scholar] [CrossRef]
- Pontillo, A.; Silva, L.T.; Oshiro, T.M.; Finazzo, C.; Crovella, S.; Duarte, A.J. HIV-1 induces NALP3-inflammasome expression and interleukin-1β secretion in dendritic cells from healthy individuals but not from HIV-positive patients. AIDS 2012, 26, 11–18. [Google Scholar] [CrossRef]
- Barreto-de-Souza, V.; Pacheco, G.J.; Silva, A.R.; Castro-Faria-Neto, H.C.; Bozza, P.T.; Saraiva, E.M.; Bou-Habib, D.C. Increased Leishmania Replication in HIV-1 Infected Macrophages Is Mediated by Tat Protein through Cyclooxygenase-2 Expression and Prostaglandin E2 Synthesis. J. Infect. Dis. 2006, 194, 846–854. [Google Scholar] [CrossRef]
- Brown, H.J.; Zack, J.A. Animal models of HIV-1 latency and persistence. Curr. Opin. HIV AIDS 2006, 1, 103–107. [Google Scholar]
- Zalar, A.; Figueroa, M.I.; Ruibal-Ares, B.; Bare, P.; Cahn, P.; de Bracco, M.M.; Belmonte, L. Macrophage HIV-1 infection in duodenal tissue of patients on long term HAART. Antivir. Res. 2010, 87, 269–271. [Google Scholar] [CrossRef]
- Roggero, R.; Robert-Hebmann, V.; Harrington, S.; Roland, J.; Vergne, L.; Jaleco, S.; Devaux, C.; Biard-Piechaczyk, M. Binding of Human Immunodeficiency Virus Type 1 gp120 to CXCR4 Induces Mitochondrial Transmembrane Depolarization and Cytochromec-Mediated Apoptosis Independently of Fas Signaling. J. Virol. 2001, 75, 7637–7650. [Google Scholar] [CrossRef]
- Gurwell, J.A.; Nath, A.; Sun, Q.; Zhang, J.; Martin, K.M.; Chen, Y.; Hauser, K. Synergistic neurotoxicity of opioids and human immunodeficiency virus-1 Tat protein in striatal neurons in vitro. Neuroscience 2001, 102, 555–563. [Google Scholar] [CrossRef]
- Yang, H.; Nkeze, J.; Zhao, R.Y. Effects of HIV-1 protease on cellular functions and their potential applications in antiretroviral therapy. Cell Biosci. 2012, 2, 32. [Google Scholar] [CrossRef]
- Nie, Z.; Bren, G.D.; Rizza, S.A.; Badley, A.D. HIV Protease Cleavage of Procaspase 8 is Necessary for Death of HIV-Infected Cells. Open Virol. J. 2008, 2, 1–7. [Google Scholar]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAF-1 Cytochrome c Multimeric Complex Is a Functional Apoptosome That Activates Procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar] [CrossRef]
- Gaardbo, J.C.; Hartling, H.J.; Gerstoft, J.; Nielsen, S.D. Incomplete Immune Recovery in HIV Infection: Mechanisms, Relevance for Clinical Care, and Possible Solutions. Clin. Dev. Immunol. 2012, 2012, 17. [Google Scholar]
- Lin, X.; Irwin, D.; Kanazawa, S.; Huang, L.; Romeo, J.; Yen, T.S.B.; Peterlin, B.M. Transcriptional Profiles of Latent Human Immunodeficiency Virus in Infected Individuals: Effects of Tat on the Host and Reservoir. J. Virol. 2003, 77, 8227–8236. [Google Scholar] [CrossRef]
- Deeks, S.G.; Walker, B.D. Human Immunodeficiency Virus Controllers: Mechanisms of Durable Virus Control in the Absence of Antiretroviral Therapy. Immunity 2007, 27, 406–416. [Google Scholar] [CrossRef]
- Lavrik, I.N. Systems biology of apoptosis signaling networks. Curr. Opin. Biotechnol. 2010, 21, 551–555. [Google Scholar] [CrossRef]
- Wood, W.G.; Igbavboa, U.; Muller, W.E.; Eckert, G.P. Statins, Bcl-2, and apoptosis: Cell death or cell protection? Mo. Neurobiol. 2013, 48, 308–314. [Google Scholar] [CrossRef]
- Wan, Z.-T.; Chen, X.-l. Mechanisms of HIV envelope-induced T lymphocyte apoptosis. Virol. Sinica. 2010, 25, 307–315. [Google Scholar] [CrossRef]
- Piguet, V.; Steinman, R.M. The interaction of HIV with dendritic cells: Outcomes and pathways. Trends Immunol. 2007, 28, 503–510. [Google Scholar] [CrossRef]
- Herbeuval, J.-P.; Grivel, J.-C.; Boasso, A.; Hardy, A.W.; Chougnet, C.; Dolan, M.J.; Yagita, H.; Lifson, J.D.; Shearer, G.M. CD4+ T-cell death induced by infectious and noninfectious HIV-1: Role of type 1 interferonâ-dependent, TRAIL/DR5-mediated apoptosis. Blood 2005, 106, 3524–3531. [Google Scholar] [CrossRef]
- Piatak, M., Jr.; Saag, M.S.; Yang, L.C.; Clark, S.J.; Kappes, J.C.; Luk, K.C.; Hahn, B.H.; Shaw, G.M.; Lifson, J.D. Determination of plasma viral load in HIV-1 infection by quantitative competitive polymerase chain reaction. AIDS 1993, 7, S65–S71. [Google Scholar] [CrossRef]
- Idriss, H.T.; Naismith, J.H. TNFα and the TNF receptor superfamily: Structure-function relationship(s). Microsc. Res. Tech. 2000, 50, 184–195. [Google Scholar] [CrossRef]
- Yonehara, S.; Ishii, A.; Yonehara, M. A cell-killing monoclonal antibody (anti-Fas) to a cell surface antigen co-downregulated with the receptor of tumor necrosis factor. J. Exp. Med. 1989, 169, 1747–1756. [Google Scholar] [CrossRef]
- Crisps, N.I. Fatal interactions: Fas-induced apoptosis of mature T cells. Immunity 1994, 1, 347–349. [Google Scholar] [CrossRef]
- Dianzani, U.; Bensi, T.; Savarino, A.; Sametti, S.; Indelicato, M.; Mesturini, R.; Chiocchetti, A. Role of FAS in HIV infection. Curr. HIV Res. 2003, 1, 405–417. [Google Scholar] [CrossRef]
- Lelievre, J.-D.; Petit, F.; Arnoult, D.; Ameisen, J.-C.; Estaquier, J. Interleukin 7 Increases Human Immunodeficiency Virus Type 1 LAI-Mediated Fas-Induced T-Cell Death. J. Virol. 2005, 79, 3195–3199. [Google Scholar] [CrossRef]
- Oyaizu, N.; Adachi, Y.; Hashimoto, F.; McCloskey, T.W.; Hosaka, N.; Kayagaki, N.; Yagita, H.; Pahwa, S. Monocytes express Fas ligand upon CD4 cross-linking and induce CD4+ T cells apoptosis: A possible mechanism of bystander cell death in HIV infection. J. Immunol. 1997, 158, 2456–2463. [Google Scholar]
- Poonia, B.; Pauza, C.D.; Salvato, M.S. Role of the Fas/FasL pathway in HIV or SIV disease. Retrovirology 2009, 6, 91. [Google Scholar] [CrossRef]
- Herbein, G.; Khan, K.A. Is HIV infection a TNF receptor signalling-driven disease? Trends Immunol. 2008, 29, 61–67. [Google Scholar] [CrossRef]
- De Oliveira Pinto, L.M.; Garcia, S.; Lecoeur, H.; Rapp, C.; Gougeon, M.-L. Increased sensitivity of T lymphocytes to tumor necrosis factor receptor 1 (TNFR1) a and TNFR2-mediated apoptosis in HIV infection: Relation to expression of Bcl-2 and active caspase-8 and caspase-3. Blood 2002, 99, 1666–1675. [Google Scholar] [CrossRef]
- Friedmann, E.; Hauben, E.; Maylandt, K.; Schleeger, S.; Vreugde, S.; Lichtenthaler, S.F.; Kuhn, P.H.; Stauffer, D.; Rovelli, G.; Martoglio, B. SPPL2a and SPPL2b promote intramembrane proteolysis of TNFα in activated dendritic cells to trigger IL-12 production. Nat. Cell. Biol. 2006, 8, 843–848. [Google Scholar]
- Lum, J.J.; Pilon, A.A.; Sanchez-Dardon, J.; Phenix, B.N.; Kim, J.E.; Mihowich, J.; Jamison, K.; Hawley-Foss, N.; Lynch, D. H.; Badley, A.D. Induction of Cell Death in Human Immunodeficiency Virus-Infected Macrophages and Resting Memory CD4 T Cells by TRAIL/Apo2L. J. Virol. 2001, 75, 11128–11136. [Google Scholar] [CrossRef]
- Miura, Y.; Misawa, N.; Maeda, N.; Inagaki, Y.; Tanaka, Y.; Ito, M.; Kayagaki, N.; Yamamoto, N.; Yagita, H.; Mizusawa, H.; Koyanagi, Y. Critical Contribution of Tumor Necrosis Factor α Related Apoptosis-Inducing Ligand (Trail) to Apoptosis of Human Cd4+T Cells in HIV-1 Infected Hu-Pbl-Nod-Scid Mice. J. Exp. Med. 2001, 193, 651–660. [Google Scholar] [CrossRef]
- Yang, Y.; Tikhonov, I.; Ruckwardt, T.J.; Djavani, M.; Zapata, J.C.; Pauza, C.D.; Salvato, M.S. Monocytes Treated with Human Immunodeficiency Virus Tat Kill Uninfected CD4+ Cells by a Tumor Necrosis Factor-Related Apoptosis-Induced Ligand-Mediated Mechanism. J. Virol. 2003, 77, 6700–6708. [Google Scholar] [CrossRef]
- Herbeuval, J.-P.; Lambert, C.; Sabido, O.; Cottier, M.L.; Fournel, P.; Dy, M.; Genin, C. Macrophages From Cancer Patients: Analysis of TRAIL, TRAIL Receptors, and Colon Tumor Cell Apoptosis. J. Nat. Cancer Inst. 2003, 95, 611–621. [Google Scholar] [CrossRef]
- Chaudhary, P.M.; Eby, M.; Jasmin, A.; Bookwalter, A.; Murray, J.; Hood, L. Death Receptor 5, a New Member of the TNFR Family, and DR4 Induce FADD-Dependent Apoptosis and Activate the NF-kB Pathway. Immunity 1997, 7, 821–830. [Google Scholar] [CrossRef]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.J. The Nature of the Principal Type 1 Interferon-Producing Cells in Human Blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef]
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. Molecular mechanisms of HIV entry. Adv. Exp. Med. Biol. 2012, 726, 223–242. [Google Scholar] [CrossRef]
- Caruz, A.; Samsom, M.; Alonso, J.M.; Alcami, J.; Baleux, F.; Virelizier, J.L.; Parmentier, M.; Arenzana-Seisdedos, F. Genomic organization and promoter characterization of human CXCR4 gene. FEBS Lett. 1998, 426, 271–278. [Google Scholar] [CrossRef]
- Yao, Q.; Compans, R.W.; Chen, C. HIV Envelope Proteins Differentially Utilize CXCR4 and CCR5 Coreceptors for Induction of Apoptosis. Virology 2001, 285, 128–137. [Google Scholar] [CrossRef]
- Garg, H.; Blumenthal, R. Role of HIV Gp41 mediated fusion/hemifusion in bystander apoptosis. Cell. Mol. Life Sci. 2008, 65, 3134–3144. [Google Scholar] [CrossRef]
- Joshi, A.; Nyakeriga, A.M.; Ravi, R.; Garg, H. HIV ENV Glycoprotein-mediated Bystander Apoptosis Depends on Expression of the CCR5 Co-receptor at the Cell Surface and ENV Fusogenic Activity. J. Biol. Chem. 2011, 286, 36404–36413. [Google Scholar] [CrossRef]
- Frankel, A.D.; Young, J.A.T. HIV-1: Fifteen Proteins and an RNA. Annu. Rev. Biochem. 1998, 67, 1–25. [Google Scholar] [CrossRef]
- Naghavi, M.H.; Goff, S.P. Retroviral proteins that interact with the host cell cytoskeleton. Curr. Opin. Immunol. 2007, 19, 402–407. [Google Scholar] [CrossRef]
- Fevrier, M.; Dorgham, K.; Rebollo, A. CD4+ T cell depletion in human immunodeficiency virus (HIV) infection: Role of apoptosis. Viruses 2011, 3, 586–612. [Google Scholar] [CrossRef]
- Cossarizza, A. Apoptosis and HIV Infection: About Molecules and Genes. Curr. Pharm. Des. 2008, 14, 237–244. [Google Scholar] [CrossRef]
- Honer, B.; Shoeman, R.L.; Traub, P. Human immunodeficiency virus type 1 protease microinjected into cultured human skin fibroblasts cleaves vimentin and affects cytoskeletal and nuclear architecture. J. Cell Sci. 1991, 100, 799–807. [Google Scholar]
- Shoeman, R.L.; Hüttermann, C.; Hartig, R.; Traub, P. Amino-terminal Polypeptides of Vimentin Are Responsible for the Changes in Nuclear Architecture Associated with Human Immunodeficiency Virus Type 1 Protease Activity in Tissue Culture Cells. Mol. Biol. Cell. 2001, 12, 143–154. [Google Scholar]
- Blanco, R.; Carrasco, L.; Ventoso, I. Cell Killing by HIV-1 Protease. J. Biol. Chem. 2003, 278, 1086–1093. [Google Scholar] [CrossRef]
- Strack, P.R.; Frey, M.W.; Rizzo, C.J.; Cordova, B.; George, H.J.; Meade, R.; Ho, S.P.; Corman, J.; Tritch, R.; Korant, B.D. Apoptosis mediated by HIV protease is preceded by cleavage of Bcl-2. Proc. Natl. Acad. Sci. USA 1996, 93, 9571–9576. [Google Scholar] [CrossRef]
- Nie, Z.; Bren, G.D.; Vlahakis, S.R.; Schimnich, A.A.; Brenchley, J.M.; Trushin, S.A.; Warren, S.; Schnepple, D.J.; Kovacs, C.M.; Loutfy, M.R.; et al. Human Immunodeficiency Virus Type 1 Protease Cleaves Procaspase 8 In Vivo. J. Virol. 2007, 81, 6947–6956. [Google Scholar] [CrossRef]
- Riviere, Y.; Blank, V.; Kourilsky, P.; Israel, A. Processing of the precursor of NF-κB by the HIV-1 protease during acute infection. Nature 1991, 350, 625–626. [Google Scholar] [CrossRef]
- Bren, G.D.; Whitman, J.; Cummins, N.; Shepard, B.; Rizza, S.A.; Trushin, S.A.; Badley, A.D. Infected Cell Killing by HIV-1 Protease Promotes NF-κB Dependent HIV-1 Replication. PLoS One 2008, 3, e2112. [Google Scholar]
- Bartz, S.R.; Emerman, M. Human Immunodeficiency Virus Type 1 Tat Induces Apoptosis and Increases Sensitivity to Apoptotic Signals by Up-Regulating FLICE/Caspase-8. J. Virol. 1999, 73, 1956–1963. [Google Scholar]
- Cummins, N.W.; Badley, A.D. Mechanisms of HIV-associated lymphocyte apoptosis: 2010. Cell Death Dis. 2010, 1, e99. [Google Scholar] [CrossRef]
- Dabrowska, A.; Kim, N.; Aldovini, A. Tat-Induced FOXO3a Is a Key Mediator of Apoptosis in HIV-1-Infected Human CD4+ T Lymphocytes. J. Immunol. 2008, 181, 8460–8477. [Google Scholar] [CrossRef]
- Skurk, C.; Maatz, H.; Kim, H.-S.; Yang, J.; Abid, M.R.; Aird, W.C.; Walsh, K. The Akt-regulated Forkhead Transcription Factor FOXO3a Controls Endothelial Cell Viability through Modulation of the Caspase-8 Inhibitor FLIP. J. Biol. Chem. 2004, 279, 1513–1525. [Google Scholar]
- Chen, D.; Wang, M.; Zhou, S.; Zhou, Q. HIV-1 Tat targets microtubules to induce apoptosis, a process promoted by the pro-apoptotic Bcl-2 relative Bim. EMBO J. 2002, 21, 6801–6810. [Google Scholar] [CrossRef]
- Huo, L.; Li, D.; Sun, L.; Liu, M.; Shi, X.; Sun, X.; Li, J.; Dong, B.; Dong, X.; Zhou, J. Tat acetylation regulates its actions on microtubule dynamics and apoptosis in T lymphocytes. J. Pathol. 2011, 223, 28–36. [Google Scholar] [CrossRef]
- Buccigrossi, V.; Laudiero, G.; Nicastro, E.; Miele, E.; Esposito, F.; Guarino, A. The HIV-1 Transactivator Factor (Tat) Induces Enterocyte Apoptosis through a Redox-Mediated Mechanism. PLoS One 2011, 6, e29436. [Google Scholar]
- Gibellini, D.; Carla Re, M.; Ponti, C.; Vitone, F.; Bon, I.; Fabbri, G.; Grazia Di Iasio, M.; Zauli, G. HIV-1 Tat protein concomitantly down-regulates apical caspase-10 and up-regulates c-FLIP in lymphoid T cells: A potential molecular mechanism to escape TRAIL cytotoxicity. J. Cell. Physiol. 2005, 203, 547–556. [Google Scholar] [CrossRef]
- Zheng, L.; Yang, Y.; Guocai, L.; Pauza, D.; Salvatob, M.S. HIV Tat Protein Increases Bcl-2 Expression in Monocytes Which Inhibits Monocyte Apoptosis Induced by Tumor Necrosis Factor-Alpha-Related Apoptosis-Induced Ligand. Intervirology 2007, 50, 224–228. [Google Scholar] [CrossRef]
- Ekoff, M.; Kaufmann, T.; Engström, M.; Motoyama, N.; Villunger, A.; Jönsson, J.-I.; Strasser, A.; Nilsson, G. The BH3-only protein Puma plays an essential role in cytokine deprivation-induced apoptosis of mast cells. Blood 2007, 110, 3209–3217. [Google Scholar] [CrossRef]
- Zauli, G.; Gibellini, D.; Secchiero, P.; Dutartre, H.L.N.; Olive, D.; Capitani, S.; Collette, Y. Human Immunodeficiency Virus Type 1 Nef Protein Sensitizes CD4+ T Lymphoid Cells to Apoptosis via Functional Upregulation of the CD95/CD95 Ligand Pathway. Blood 1999, 93, 1000–1010. [Google Scholar]
- Lee, S.B.; Park, J.; Jung, J.U.; Chung, J. Nef induces apoptosis by activating JNK signaling pathway and inhibits NF-κB-dependent immune responses in Drosophila. J. Cell. Sci. 2005, 118, 1851–1859. [Google Scholar] [CrossRef]
- Rasola, A.; Gramaglia, D.; Boccaccio, C.; Comoglio, P.M. Apoptosis Enhancement by the HIV-1 Nef Protein. J. Immunol. 2001, 166, 81–88. [Google Scholar] [CrossRef]
- James, C.O.; Huang, M.-B.; Khan, M.; Garcia-Barrio, M.; Powell, M.D.; Bond, V.C. Extracellular Nef Protein Targets CD4+ T Cells for Apoptosis by Interacting with CXCR4 Surface Receptors. J. Virol. 2004, 78, 3099–3109. [Google Scholar] [CrossRef]
- Yoon, K.; Jeong, J.G.; Kim, S. Stable expression of human immunodeficiency virus type 1 Nef confers resistance against Fas-mediated apoptosis. AIDS Res. Hum. Retrovir. 2001, 20, 99–104. [Google Scholar] [CrossRef]
- Geleziunas, R.; Xu, W.; Takeda, K.; Ichijo, H.; Greene, W.C. HIV-1 Nef inhibits ASK1-dependent death signalling providing a potential mechanism for protecting the infected host cell. Nature 2001, 410, 834–838. [Google Scholar] [CrossRef]
- Wolf, D.; Witte, V.; Laffert, B.; Blume, K.; Stromer, E.; Trapp, S.; d'Aloja, P.; Schurmann, A.; Baur, A.S. HIV-1 Nef associated PAK and PI3-Kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat. Med. 2001, 7, 1217–1224. [Google Scholar] [CrossRef]
- Olivetta, E.; Federico, M. HIV-1 Nef protects human-monocyte-derived macrophages from HIV-1-induced apoptosis. Exp. Cell. Res. 2006, 312, 890–900. [Google Scholar] [CrossRef]
- Robichaud, G.A.; Poulin, L. HIV type 1 nef gene inhibits tumor necrosis factor alpha-induced apoptosis and promotes cell proliferation through the action of MAPK and JNK in human glial cells. AIDS Res. Hum. Retrovir. 2000, 16, 1959–1965. [Google Scholar] [CrossRef]
- Greenway, A.L.; McPhee, D.A.; Allen, K.; Johnstone, R.; Holloway, G.; Mills, J.; Azad, A.; Sankovich, S.; Lambert, P. Human Immunodeficiency Virus Type 1 Nef Binds to Tumor Suppressor p53 and Protects Cells against p53-Mediated Apoptosis. J. Virol. 2002, 76, 2692–2702. [Google Scholar] [CrossRef]
- Xu, X.-N.; Laffert, B.; Screaton, G.R.; Kraft, M.; Wolf, D.; Kolanus, W.; Mongkolsapay, J.; McMichael, A.J.; Baur, A.S. Induction of Fas Ligand Expression by HIV Involves the Interaction of Nef with the T Cell Receptor ζ Chain. J. Exp. Med. 1999, 189, 1489–1496. [Google Scholar] [CrossRef]
- van den Broeke, C.; Radu, M.; Chernoff, J.; Favoreel, H.W. An emerging role for p21-activated kinases (Paks) in viral infections. Trends Cell Biol. 2011, 20, 160–169. [Google Scholar] [CrossRef]
- Abbas, W.; Khan, K.A.; Kumar, A.; Tripathy, M.K.; Dichamp, I.; Keita, M.; Mahlknecht, U.; Rohr, O.; Herbein, G. Blockade of BFA-mediated apoptosis in macrophages by the HIV-1 Nef protein. Cell Death Dis. 2014, 5, e1080. [Google Scholar] [CrossRef]
- Kwon, H.-S.; Brent, M.M.; Getachew, R.; Jayakumar, P.; Chen, L.-F.; Schnolzer, M.; McBurney, M.W.; Marmorstein, R.; Greene, W.C.; Ott, M. Human Immunodeficiency Virus Type 1 Tat Protein Inhibits the SIRT1 Deacetylase and Induces T Cell Hyperactivation. Cell Host Microbe. 2008, 3, 158–167. [Google Scholar] [CrossRef]
- Lara, H.H.; Ixtepan-Turrent, L.; Garza-Trevino, E.N.; Badillo-Almaraz, J.I.; Rodriguez-Padilla, C. Antiviral mode of action of bovine dialyzable leukocyte extract against human immunodeficiency virus type 1 infection. BMC Res. Notes 2011, 4, 474. [Google Scholar] [CrossRef]
- Ganau, M.; Prisco, L.; Pescador, D.; Ganau, L. Challenging new targets for CNS-HIV infection. Front. Neurol. 2012, 23, 3. [Google Scholar]
- Akari, H.; Bour, S.; Kao, S.; Adachi, A.; Strebel, K. The Human Immunodeficiency Virus Type 1 Accessory Protein Vpu Induces Apoptosis by Suppressing the Nuclear Factor kB dependent Expression of Antiapoptotic Factors. J. Exp. Med. 2001, 194, 1299–1312. [Google Scholar] [CrossRef]
- Verma, S.; Ali, A.; Arora, S.; Banerjea, A.C. Inhibition of β-TrcP- dependent ubiquitination of p53 by HIV-1 Vpu promotes p53- mediated apoptosis in human T cells. Blood 2011, 117, 6600–6607. [Google Scholar] [CrossRef]
- Casella, C.R.; Rapaport, E.L.; Finkel, T.H. Vpu Increases Susceptibility of Human Immunodeficiency Virus Type 1-Infected Cells to Fas Killing. J. Virol. 1999, 73, 92–100. [Google Scholar]
- Marchal, C.; Vinatier, G.; Sanial, M.; Plessis, A.; Pret, A.M.; Limbourg-Bouchon, B.; Theodore, L.; Netter, S. The HIV-1 Vpu protein induces apoptosis in Drosophila via activation of JNK signaling. PLoS One 2012, 7, e34310. [Google Scholar] [CrossRef]
- Komoto, S.; Tsuji, S.; Ibrahim, M.S.; Li, Y.-G.; Warachit, J.; Taniguchi, K.; Ikuta, K. The Vpu Protein of Human Immunodeficiency Virus Type 1 Plays a Protective Role against Virus-Induced Apoptosis in Primary CD4+ T Lymphocytes. J. Virol. 2003, 77, 10304–10313. [Google Scholar] [CrossRef]
- Tateyama, M.; Oyaizu, N.; McCloskey, T.W.; Than, S.; Pahwa, S. CD4 T lymphocytes are primed to express Fas ligand by CD4 cross-linking and to contribute to CD8 T-cell apoptosis via Fas/FasL death signaling pathway. Blood 2000, 96, 195–202. [Google Scholar]
- Somma, F.; Tuosto, L.; Montani, M.S.G.; Di Somma, M.M.; Cundari, E.; Piccolella, E. Engagement of CD4 Before TCR Triggering Regulates Both Bax- and Fas (CD95)-Mediated Apoptosis. J. Immunol. 2000, 164, 5078–5087. [Google Scholar] [CrossRef]
- Trushin, S.A.; Algeciras-Schimnich, A.; Vlahakis, S.R.; Bren, G.D.; Warren, S.; Schnepple, D.J.; Badley, A.D. Glycoprotein 120 Binding to CXCR4 Causes p38-Dependent Primary T Cell Death That Is Facilitated by, but Does Not Require Cell-Associated CD4. J. Immunol. 2007, 178, 4846–4853. [Google Scholar] [CrossRef]
- Herbein, G.; Mahlknecht, U.; Batliwalla, F.; Gregersen, P.; Pappas, T.; Butler, J.; O'Brien, W.A.; Verdin, E. Apoptosis of CD8+ T cells is mediated by macrophages through interaction of HIV gp120 with chemokine receptor CXCR4. Nature 1998, 395, 189–194. [Google Scholar] [CrossRef]
- Blanco, J.; Barretina, J.; Ferri, K.F.; Jacotot, E.; Gutierrez, A.; Armand-Ugon, M.; Cabrera, C.; Kroemer, G.; Clotet, B.; Este, J. Cell-Surface-Expressed HIV-1 Envelope Induces the Death of CD4 T Cells during GP41-Mediated Hemifusion-like Events. Virology 2003, 305, 318–329. [Google Scholar] [CrossRef]
- Perfettini, J.L.; Castedo, M.; Roumier, T.; Andreau, K.; Nardacci, R.; Piacentini, M.; Kroemer, G. Mechanisms of apoptosis induction by the HIV-1 envelope. Cell Death Differ. 2005, 12, 916–923. [Google Scholar] [CrossRef]
- Garg, H.; Blumenthal, R. HIV gp41-induced apoptosis is mediated by caspase-3-dependent mitochondrial depolarization, which is inhibited by HIV protease inhibitor nelfinavir. J. Leukoc. Biol. 2006, 2, 351–362. [Google Scholar]
- Castedo, M.; Roumier, T.; Blanco, J.; Ferri, K.F.; Barretina, J.; Tintignac, L.A.; Andreau, K.; Perfettini, J.L.; Amendola, A.; Nardacci, R.; et al. Sequential involvement of Cdk1, mTOR and p53 in apoptosis induced by the HIV-1 envelope. Embo. J. 2002, 15, 4070–4080. [Google Scholar]
- Perfettini, J.-L.; Roumier, T.; Castedo, M.; Larochette, N.; Boya, P.; Raynal, B.; Lazar, V.; Ciccosanti, F.; Nardacci, R.; Penninger, J.; et al. HF-КB and p53 Are the Dominant Apoptosis-inducing Transcription Factors Elicited by the HIV-1 Envelope. J. Exp. Med. 2004, 5, 629–640. [Google Scholar]
- Szawlowski, P.W.S.; Hanke, T.; Randall, R.E. Sequence homology between HIV-1 gp120 and the apoptosis mediating protein Fas. AIDS 1993, 7, 1018. [Google Scholar] [CrossRef]
- Silvestris, F.; Nagata, S.; Cafforio, P.; Silvestris, N.; Dammacco, F. Cross-linking of Fas by antibodies to a peculiar domain of gp120 V3 loop can enhance T cell apoptosis in HIV-1-infected patients. J. Exp. Med. 1996, 6, 2287–2300. [Google Scholar]
- Michalski, C.; Li, Y.; Kang, C.Y. Induction of cytopathic effects and apoptosis in Spodoptera. frugiperda cells by the HIV-1 Env glycoprotein signal peptide. Virus Genes. 2010, 3, 341–350. [Google Scholar] [CrossRef]
- Koga, Y.; Sasaki, M.; Nakamura, K.; Kimura, G.; Nomoto, K. Intracellular distribution of the envelope glycoprotein of human immunodeficiency virus and its role in the production of cytopathic effect in CD4+ and CD4- human cell lines. J. Virol. 1990, 10, 4661–4671. [Google Scholar]
- Andreau, K.; Perfettini, J.-L.; Castedo, M.; Metivier, D.; Scott, V.; Pierron, G.; Kroemer, G. Contagious apoptosis facilitated by the HIV-1 envelope: Fusion-induced cell-to-cell transmission of a lethal signal. J. Cell. Sci. 2004, 23, 5643–5653. [Google Scholar]
- LaBonte, J.A.; Madani, N.; Sodroski, J. Cytolysis by CCR5-Using Human Immunodeficiency Virus Type 1 Envelope Glycoproteins Is Dependent on Membrane Fusion and Can Be Inhibited by High Levels of CD4 Expression. J. Virol. 2003, 12, 6645–6659. [Google Scholar] [CrossRef]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotič, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitaš, A.; Peterlin, B.M. HIV Nef is Secreted in Exosomes and Triggers Apoptosis in Bystander CD4+ T Cells. Traffic 2010, 1, 110–122. [Google Scholar]
- Fujii, Y.; Otake, K.; Tashiro, M.; Adachi, A. Human immunodeficiency virus type 1 Nef protein on the cell surface is cytocidal for human CD4+ T cells. FEBS Lett. 1996, 1, 105–108. [Google Scholar]
- Fujii, Y.; Otake, K.; Tashiro, M.; Adachi, A. Soluble Nef antigen of HIV-1 is cytotoxic for human CD4+ T cells. FEBS Lett. 1996, 1, 93–96. [Google Scholar]
- Otake, K.; Fujii, Y.; Nakaya, T.; Nishino, Y.; Zhong, Q.; Fujinaga, K.; Kameoka, M.; Ohki, K.; Ikuta, K. The carboxyl-terminal region of HIV-1 Nef protein is a cell surface domain that can interact with CD4+ T cells. J. Immunol. 1994, 12, 5826–5837. [Google Scholar]
- Okada, H.; Takei, R.; Tashiro, M. Nef protein of HIV-1 induces apoptotic cytolysis of murine lymphoid cells independently of CD95 (Fas) and its suppression by serine/threonine protein kinase inhibitors. FEBS Lett. 1997, 1, 61–64. [Google Scholar] [CrossRef]
- Kumawat, K.; Pathak, S.K.; Spetz, A.-L.; Kundu, M.; Basu, J. Exogenous Nef Is an Inhibitor of Mycobacterium tuberculosis-induced Tumor Necrosis Factor-α Production and Macrophage Apoptosis. J. Biol. Chem. 2010, 17, 12629–12637. [Google Scholar]
- Sawai, E.T.; Baur, A.; Struble, H.; Peterlin, B.M.; Levy, J.A.; Cheng-Mayer, C. Human immunodeficiency virus type 1 Nef associates with a cellular serine kinase in T lymphocytes. Proc. Natl. Acad Sci. USA 1994, 4, 1539–1543. [Google Scholar]
- Renkema, G.H.; Manninen, A.; Mann, D.A.; Harris, M.; Saksela, K. Identification of the Nef-associated kinase as p21-activated kinase 2. Curr. Biol. 1999, 23, 1407–1411. [Google Scholar]
- Arora, V.K.; Fredericksen, B.L.; Garcia, J.V. Nef: Agent of cell subversion. Microbes Infect. 2002, 2, 189–199. [Google Scholar]
- Mateyak, M.K.; Kinzy, T.G. eEF1A: Thinking outside the ribosome. J. Biol. Chem. 2010, 28, 21209–21213. [Google Scholar] [CrossRef]
- Debaisieux, S.; Rayne, F.; Yezid, H.; Beaumelle, B. The Ins and Outs of HIV-1 Tat. Traffic 2011, 3, 355–363. [Google Scholar]
- McCloskey, T.W.; Ott, M.; Tribble, E.; Khan, S.A.; Teichberg, S.; Paul, M.O.; Pahwa, S.; Verdin, E.; Chirmule, N. Dual role of HIV Tat in regulation of apoptosis in T cells. J. Immunol. 1997, 2, 1014–1019. [Google Scholar]
- Badou, A.; Bennasser, Y.; Moreau, M.; Leclerc, C.; Benkirane, M.; Bahraoui, E. Tat Protein of Human Immunodeficiency Virus Type 1 Induces Interleukin-10 in Human Peripheral Blood Monocytes: Implication of Protein Kinase C-Dependent Pathway. J. Virol. 2000, 22, 10551–10562. [Google Scholar]
- Reinhold, D.; Wrenger, S.; KÃhne, T.; Ansorge, S. HIV-1 Tat: Immunosuppression via TGF-β1 induction. Immunol. Today 1999, 8, 384. [Google Scholar]
- Westendorp, M.O.; Frank, R.; Ochsenbauer, C.; Stricker, K.; Dhein, J.; Walczak, H.; Debatin, K.M.; Krammer, P.H. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 1995, 6531, 497–500. [Google Scholar]
- Minami, R.; Yamamoto, M.; Takahama, S.; Miyamura, T.; Watanabe, H.; Suematsu, E. RCAS1 induced by HIV-Tat is involved in the apoptosis of HIV-1 infected and uninfected CD4+ T cells. Cell. Immunol. 2006, 1, 41–47. [Google Scholar]
- Terwilliger, E.F.; Cohen, E.A.; Lu, Y.C.; Sodroski, J.G.; Haseltine, W.A. Functional role of human immunodeficiency virus type 1 vpu. Proc. Natl. Acad. Sci. USA 1989, 13, 5163–5167. [Google Scholar]
- Dube, M.; Bego, M.G.; Paquay, C.; Cohen, E.A. Modulation of HIV-1-host interaction: Role of the Vpu accessory protein. Retrovirology 2010, 7, 114. [Google Scholar] [CrossRef]
- Schindler, M.; Rajan, D.; Banning, C.; Wimmer, P.; Koppensteiner, H.; Iwanski, A.; Specht, A.; Sauter, D.; Dobner, T.; Kirchhoff, F. Vpu serine 52 dependent counteraction of tetherin is required for HIV-1 replication in macrophages, but not in ex vivo ex vivo human lymphoid tissue. Retrovirology 2010, 7, 1. [Google Scholar] [CrossRef]
- Andersen, J.L.; Le Rouzic, E.; Planelles, V. HIV-1 Vpr: Mechanisms of G2 arrest and apoptosis. Exp. Mol. Path. 2008, 1, 2–10. [Google Scholar] [CrossRef]
- Roshal, M.; Kim, B.; Zhu, Y.; Nghiem, P.; Planelles, V. Activation of the ATR-mediated DNA Damage Response by the HIV-1 Viral Protein R. J. Biol. Chem. 2003, 278, 25879–25886. [Google Scholar]
- Yuan, H.; Xie, Y.-M.; Chen, I.S.Y. Depletion of Wee-1 Kinase Is Necessary for both Human Immunodeficiency Virus Type 1 Vpr- and Gamma Irradiation-Induced Apoptosis. J. Virol. 2003, 77, 2063–2070. [Google Scholar] [CrossRef]
- DeHart, J.L.; Zimmerman, E.S.; Ardon, O.; Monteiro-Filho, C.M.; Arganaraz, E.R.; Planelles, V. HIV-1 Vpr activates the G2 checkpoint through manipulation of the ubiquitin proteasome system. Virol. J. 2007, 4, 57. [Google Scholar] [CrossRef]
- Snyder, A.; Alsauskas, Z.C.; Leventhal, J.S.; Rosenstiel, P.E.; Gong, P.; Chan, J.J.; Barley, K.; He, J.C.; Klotman, M.E.; Ross, M.J.; et al. HIV-1 viral protein r induces ERK and caspase-8-dependent apoptosis in renal tubular epithelial cells. AIDS 2010, 24, 1107–1119. [Google Scholar] [CrossRef]
- Cagnol, S.; Van Obberghen-Schilling, E.; Chambard, J.C. Prolonged activation of ERK1,2 induces FADD-independent caspase 8 activation and cell death. Apoptosis 2006, 11, 337–346. [Google Scholar] [CrossRef]
- Muthumani, K.; Hwang, D.S.; Desai, B.M.; Zhang, D.; Dayes, N.; Green, D.R.; Weiner, D.B. HIV-1 Vpr Induces Apoptosis through Caspase 9 in T Cells and Peripheral Blood Mononuclear Cells. J. Biol. Chem. 2002, 277, 37820–37831. [Google Scholar]
- Muthumani, K.; Zhang, D.; Hwang, D.S.; Kudchodkar, S.; Dayes, N.S.; Desai, B.M.; Malik, A.S.; Yang, J.S.; Chattergoon, M.A.; Maguire, H.C., Jr.; et al. Adenovirus encoding HIV-1 Vpr activates caspase 9 and induces apoptotic cell death in both p53 positive and negative human tumor cell lines. Oncogene 2002, 21, 4613–4625. [Google Scholar] [CrossRef]
- Zhu, Y.; Gelbard, H.A.; Roshal, M.; Pursell, S.; Jamieson, B.D.; Planelles, V. Comparison of Cell Cycle Arrest, Transactivation, and Apoptosis Induced by the Simian Immunodeficiency Virus SIVagm and Human Immunodeficiency Virus Type 1 vpr Genes. J. Virol. 2001, 75, 3791–3801. [Google Scholar] [CrossRef]
- Jacotot, E.; Ferri, K.F.; El Hamel, C.; Brenner, C.; Druillennec, S.; Hoebeke, J.; Rustin, P.; Metivier, D.; Lenoir, C.; Geuskens, M.; et al. Control of Mitochondrial Membrane Permeabilization by Adenine Nucleotide Translocator Interacting with HIV-1 Viral Protein R and Bcl-2. J. Exp. Med. 2001, 193, 509–520. [Google Scholar] [CrossRef]
- Roumier, T.; Vieira, H.L.; Castedo, M.; Ferri, K.F.; Boya, P.; Andreau, K.; Druillennec, S.; Joza, N.; Penninger, J.M.; Roques, B.; et al. The C-terminal moiety of HIV-1 Vpr induces cell death via a caspase-independent mitochondrial pathway. Cell Death Differ. 2002, 9, 1212–1219. [Google Scholar] [CrossRef]
- Andersen, J.L.; DeHart, J.L.; Zimmerman, E.S.; Ardon, O.; Kim, B.; Jacquot, G.; Benichou, S.; Planelles, V. HIV-1 Vpr-Induced Apoptosis Is Cell Cycle Dependent and Requires Bax but Not ANT. PLoS Pathog. 2006, 2, e127. [Google Scholar] [CrossRef]
- Zhu, Y.; Roshal, M.; Li, F.; Blackett, J.; Planelles, V. Upregulation of survivin by HIV-1 Vpr. Apoptosis 2003, 8, 71–79. [Google Scholar] [CrossRef]
- Conti, L.; Matarrese, P.; Varano, B.; Gauzzi, M.C.; Sato, A.; Malorni, W.; Belardelli, F.; Gessani, S. Dual Role of the HIV-1 Vpr Protein in the Modulation of the Apoptotic Response of T Cells. J. Immunol. 2000, 165, 3293–3300. [Google Scholar] [CrossRef]
- Guha, D.; Nagilla, P.; Redinger, C.; Srinivasan, A.; Schatten, G.P.; Ayyavoo, V. Neuronal apoptosis by HIV-1 Vpr: Contribution of proinflammatory molecular networks from infected target cells. J. Neuroinflamm. 2012, 9, 138. [Google Scholar] [CrossRef]
- Kim, K.; Heo, K.; Choi, J.; Jackson, S.; Kim, H.; Xiong, Y.; Ari, W. Vpr-Binding Protein Antagonizes p53-Mediated Transcription via Direct Interaction with H3 Tail. Mol. Cell Biol. 2012, 32, 783–796. [Google Scholar] [CrossRef]
- Chirmule, N.; Pahwa, S. Envelope glycoproteins of human immunodeficiency virus type 1: Profound influences on immune functions. Microbiol. Rev. 1996, 60, 386–406. [Google Scholar]
- Garg, H.; Joshi, A.; Ye, C.; Shankar, P.; Manjunath, N. Single amino acid change in gp41 region of HIV-1 alters bystander apoptosis and CD4 decline in humanized mice. Virol. J. 2011, 8, 34. [Google Scholar] [CrossRef]
- Ferri, K.F.; Jacotot, E.; Blanco, J.; Este, J.A.; Zamzami, N.; Susin, S.A.; Xie, Z.; Brothers, G.; Reed, J.C.; Penninger, J.M.; et al. Apoptosis Control in Syncytia Induced by the HIV Type-1 Envelope Glycoprotein Complex. J. Exp. Med. 2000, 192, 1081–1092. [Google Scholar] [CrossRef]
- Melki, M.T.; Saidi, H.; Dufour, A.; Olivo-Marin, J.C.; Gougeon, M.L. Escape of HIV-1-infected dendritic cells from TRAIL-mediated NK cell cytotoxicity during NK-DC cross-talk—A pivotal role of HMGB1. PLoS Pathog. 2010, 6, e1000862. [Google Scholar] [CrossRef]
- Donaghy, H.; Pozniak, A.; Gazzard, B.; Qazi, N.; Gilmour, J.; Gotch, F.; Patterson, S. Loss of blood CD11c(+) myeloid and CD11c(–) plasmacytoid dendritic cells in patients with HIV-1 infection correlates with HIV-1 RNA virus load. Blood 2001, 98, 2574–2576. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.H.; Kwon, D.S.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.F.; Middel, J.; Cornelissen, I.L.M.H.A.; Nottet, H.S.L.M.; KewalRamani, V.N.; Littman, D.R.; et al. DC-SIGN, a Dendritic Cell-Specific HIV-1-Binding Protein that Enhances trans-Infection of T Cells. Cell 2000, 100, 587–597. [Google Scholar] [CrossRef]
- Favaloro, B.; Allocati, N.; Graziano, V.; di Ilio, C.; de Laurenzi, V. Role of apoptosis in disease. Aging 2012, 4, 330–349. [Google Scholar]
- Kumar, A.; Herbein, G. The macrophage: A therapeutic target in HIV-1 infection. Mol. Cell Ther. 2014, 2, 10. [Google Scholar] [CrossRef]
- Cummins, N.W.; Jiang, W.; McGinty, J.; Bren, G.D.; Bosch, R.J.; Landay, A.; Deeks, S.G.; Martin, J.N.; Douek, D.; Lederman, M.M.; et al. Intracellular Casp8p41 Content Is Inversely Associated with CD4 T Cell Count. J. Infect. Dis. 2010, 202, 386–391. [Google Scholar] [CrossRef]
- Natesampillai, S.; Nie, Z.; Cummins, N.W.; Jochmans, D.; Bren, G.D.; Angel, J.B.; Badley, A.D. Patients with Discordant Responses to Antiretroviral Therapy Have Impaired Killing of HIV-Infected T Cells. PLoS Pathog. 2010, 6, e1001213. [Google Scholar] [CrossRef]
- Sainski, A.M.; Natesampillai, S.; Cummins, N.W.; Bren, G.D.; Taylor, J.; Saenz, D.T.; Poeschla, E.M.; Badley, A.D. The HIV-1-Specific Protein Casp8p41 Induces Death of Infected Cells through Bax/Bak. J. Virol. 2011, 85, 7965–7975. [Google Scholar] [CrossRef]
- Stary, G.; Klein, I.; Kohlhofer, S.; Koszik, F.; Scherzer, T.; Müllauer, L.; Quendler, H.; Kohrgruber, N.; Stingl, G. Plasmacytoid dendritic cells express TRAIL and induce CD4+ T-cell apoptosis in HIV-1 viremic patients. Blood 2009, 114, 3854–3863. [Google Scholar] [CrossRef]
- Kawamura, T.; Gatanaga, H.; Borris, D.L.; Connors, M.; Mitsuya, H.; Blauvelt, A. Decreased Stimulation of CD4+ T Cell Proliferation and IL-2 Production by Highly Enriched Populations of HIV-Infected Dendritic Cells. J. Immunol. 2003, 170, 4260–4266. [Google Scholar] [CrossRef]
- Doitsh, G.; Cavrois, M.; Lassen, K.G.; Zepeda, O.; Yang, Z.; Santiago, M.L.; Hebbeler, A.M.; Greene, W.C. Abortive HIV Infection Mediates CD4 T Cell Depletion and Inflammation in Human Lymphoid Tissue. Cell 2010, 143, 789–801. [Google Scholar] [CrossRef]
- Monroe, K.M.; Yang, Z.; Johnson, J.R.; Geng, X.; Doitsh, G.; Krogan, N.J.; Greene, W.C. IFI16 DNA Sensor Is Required for Death of Lymphoid CD4 T Cells Abortively Infected with HIV. Science 2014, 343, 428–432. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description of Dead and Dying Eukaryotic Cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef]
- Cooper, A.; Garcia, M.; Petrovas, C.; Yamamoto, T.; Koup, R.A.; Nabel, G.J. HIV-1 causes CD4 cell death through DNA-dependent protein kinase during viral integration. Nature 2013, 498, 376–379. [Google Scholar] [CrossRef]
- Sakurai, Y.; Komatsu, K.; Agematsu, K.; Matsuoka, M. DNA double strand break repair enzymes function at multiple steps in retroviral infection. Retrovirology 2009, 6, 114. [Google Scholar] [CrossRef]
- Verani, A.; Gras, G.; Pancino, G. Macrophages and HIV-1: Dangerous liaisons. Mol Immunol. 2005, 42, 195–212. [Google Scholar] [CrossRef]
- Wang, X.; Ye, L.; Hou, W.; Zhou, Y.; Wang, Y.-J.; Metzger, D.S.; Ho, W.Z. Cellular microRNA expression correlates with susceptibility of monocytes/macrophages to HIV-1 infection. Blood 2009, 113, 671–674. [Google Scholar] [CrossRef]
- Benaroch, P.; Billard, E.; Gaudin, R.; Schindler, M.; Jouve, M. HIV-1 assembly in macrophages. Retrovirology 2010, 7, 29. [Google Scholar] [CrossRef]
- Groot, F.; Welsch, S.; Sattentau, Q.J. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 2008, 111, 4660–4663. [Google Scholar] [CrossRef]
- Porter, K.M.; Sutliff, R.L. HIV-1, reactive oxygen species, and vascular complications. Free Radic Biol. Med. 2012, 53, 143–159. [Google Scholar] [CrossRef]
- Wyatt, C.M.; Meliambro, K.; Klotman, P.E. Recent Progress in HIV-Associated Nephropathy. Ann. Rev. Med. 2012, 63, 147–159. [Google Scholar] [CrossRef]
- Winston, J.A.; Bruggeman, L.A.; Ross, M.D.; Jacobson, J.; Ross, L.; D’Agati, V.D.; Klotman, P.E.; Klotman, M.E. Nephropathy and Establishment of a Renal Reservoir of HIV Type 1 during Primary Infection. N. Eng. J. Med. 2001, 344, 1979–1984. [Google Scholar] [CrossRef]
- Kline, E.R.; Kleinhenz, D.J.; Liang, B.; Dikalov, S.; Guidot, D.M.; Hart, C.M.; Jones, D.P.; Sutliff, R.L. Vascular oxidative stress and nitric oxide depletion in HIV-1 transgenic rats are reversed by glutathione restoration. Am. J. Physiol. 2008, 294, H2792–H2804. [Google Scholar]
- Zafari, A.M.; Ushio-Fukai, M.; Akers, M.; Yin, Q.; Shah, A.; Harrison, D.G.; Taylor, W.R.; Griendling, K.K. Role of NADH/NADPH Oxidase-Derived H2O2 in Angiotensin II-Induced Vascular Hypertrophy. Hypertension 1998, 32, 488–495. [Google Scholar] [CrossRef]
- Shankar, S.S.; Dube, M.P. Clinical aspects of endothelial dysfunction associated with human immunodeficiency virus infection and antiretroviral agents. Cardiovasc. Toxicol. 2004, 4, 261–269. [Google Scholar] [CrossRef]
- Irish, B.P.; Khan, Z.K.; Jain, P.; Nonnemacher, M.R.; Pirrone, V.; Rahman, S.; Rajagopalan, N.; Suchitra, J.B.; Mostoller, K.; Wigdahl, B. Molecular Mechanisms of Neurodegenerative Diseases Induced by Human Retroviruses: A Review. Am. J. Infect. Dis. 2009, 5, 231–258. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, Y.; Qiao, L.; Sun, Y.; Ding, W.; Zhang, H.; Li, N.; Chen, D. Sigma-1 receptor agonists provide neuroprotection against gp120 via a change in bcl-2 expression in mouse neuronal cultures. Brain Res. 2011, 1431, 13–22. [Google Scholar]
- The Lancet Infectious Diseases. The challenge of HIV associated neurocognitive disorder. Lancet Infect. Dis. 2013, 13, 907. [Google Scholar] [CrossRef]
- Glass, J.D.; Fedor, H.; Wesselingh, S.L.; McArthur, J.C. Immunocytochemical quantitation of human immunodeficiency virus in the brain: Correlations with dementia. Ann. Neurol. 1995, 38, 755–762. [Google Scholar] [CrossRef]
- Raivich, G.; Jones, L.L.; Kloss, C.U.A.; Werner, A.; Neumann, H.; Kreutzberg, G.W. Immune Surveillance in the Injured Nervous System: T-Lymphocytes Invade the Axotomized Mouse Facial Motor Nucleus and Aggregate around Sites of Neuronal Degeneration. J. Neurosci. 1998, 18, 5804–5816. [Google Scholar]
- Lipton, S.A. Similarity of neuronal cell injury and death in AIDS dementia and focal cerebral ischemia: Potential treatment with NMDA open-channel blockers and nitric oxide-related species. Brain Pathol. 1996, 6, 507–517. [Google Scholar] [CrossRef]
- Sui, Z.; Fan, S.; Sniderhan, L.; Reisinger, E.; Litzburg, A.; Schifitto, G.; Gelbard, H.A.; Dewhurst, S.; Maggirwar, S.B. Inhibition of Mixed Lineage Kinase 3 Prevents HIV-1 Tat-Mediated Neurotoxicity and Monocyte Activation. J. Immunol. 2006, 177, 702–711. [Google Scholar] [CrossRef]
- Cinque, P.; Vago, L.; Mengozzi, M.; Torri, V.; Ceresa, D.; Vicenzi, E.; Transidico, P.; Vagani, A.; Sozzani, S.; Mantovani, A.; et al. Elevated cerebrospinal fluid levels of monocyte chemotactic protein-1 correlate with HIV-1 encephalitis and local viral replication. AIDS 1998, 12, 1327–1332. [Google Scholar] [CrossRef]
- Nath, A. Human Immunodeficiency Virus (HIV) Proteins in Neuropathogenesis of HIV Dementia. J. Infect. Dis. 2002, 186, S193–S198. [Google Scholar] [CrossRef]
- Pocernich, C.B.; Sultana, R.; Mohmmad-Abdul, H.; Nath, A.; Butterfield, D.A. HIV-dementia, Tat-induced oxidative stress, and antioxidant therapeutic considerations. Brain Res. Rev. 2005, 50, 14–26. [Google Scholar] [CrossRef]
- Zemlyak, I.; Sapolsky, R.; Gozes, I. NAP protects against cytochrome c release: Inhibition of the initiation of apoptosis. Eur. J. Pharmacol. 2009, 618, 9–14. [Google Scholar] [CrossRef]
- Jones, G.J.; Barsby, N.L.; Cohen, Ã.r.A.; Holden, J.; Harris, K.; Dickie, P.; Jhamandas, J.; Power, C. HIV-1 Vpr Causes Neuronal Apoptosis and In Vivo Neurodegeneration. J. Neurosci. 2007, 27, 3703–3711. [Google Scholar] [CrossRef]
- Zink, W.E.; Zheng, J.; Persidsky, Y.; Poluektova, L.; Gendelman, H.E. The neuropathogenesis of HIV-1 infection. FEMS Immunol. Med. Micro. 1999, 26, 233–241. [Google Scholar] [CrossRef]
- Lucas, G.M.; Eustace, J.A.; Sozio, S.; Mentari, E.K.; Appiah, K.A.; Moore, R.D. Highly active antiretroviral therapy and the incidence of HIV-1-associated nephropathy: A 12-year cohort study. AIDS 2004, 18, 541–546. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, R.; Zhong, Y.; Plotnikov, A.N.; Zhang, W.; Zeng, L.; Rusinova, E.; Gerona-Nevarro, G.; Moshkina, N.; Joshua, J.; et al. Down-regulation of NF-κβ Transcriptional Activity in HIV-associated Kidney Disease by BRD4 Inhibition. J. Biol. Chem. 2012, 287, 28840–28851. [Google Scholar] [CrossRef]
- Bruggeman, L.A.; Ross, M.D.; Tanji, N.; Cara, A.; Dikman, S.; Gordon, R.E.; Burns, G.C.; D'Agati, V.D.; Winston, J.A.; Klotman, M.E.; et al. Renal Epithelium Is a Previously Unrecognized Site of HIV-1 Infection. J. Am. Soc. Nephrol. 2000, 11, 2079–2087. [Google Scholar]
- Mikula, M.; Fuchs, E.; Huber, H.; Beug, H.; Schulte-Hermann, R.; Mikulits, W. Immortalized p19ARF null hepatocytes restore liver injury and generate hepatic progenitors after transplantation. Hepatology 2004, 39, 628–634. [Google Scholar] [CrossRef]
- Agati, V.D.D.; Fogo, A.B.; Bruijn, J.A.; Jennette, J.C. Pathologic classification of focal segmental glomerulosclerosis: A working proposal. Am. J. Kidney Dis. 2004, 43, 368–382. [Google Scholar] [CrossRef]
- Barisoni, L.; Kriz, W.; Mundel, P.; D’Agati, V. The Dysregulated Podocyte Phenotype. J. Am. Soc. Nephrol. 1999, 10, 51–61. [Google Scholar]
- Bruggeman, L.A.; Nelson, P.J. Controversies in the pathogenesis of HIV-associated renal diseases. Nat. Rev. Nephrol. 2009, 5, 574–581. [Google Scholar] [CrossRef]
- Gharavi, A.G.; Ahmad, T.; Wong, R.D.; Hooshyar, R.; Vaughn, J.; Oller, S.; Frankel, R.Z.; Bruggeman, L.A.; D'Agati, V.D.; Klotman, P.E.; et al. Mapping a locus for susceptibility to HIV-1-associated nephropathy to mouse chromosome 3. Proc. Natl. Acad. Sci. USA 2004, 101, 2488–2493. [Google Scholar] [CrossRef]
- Papeta, N.; Sterken, R.; Kiryluk, K.; Kalyesubula, R.; Gharavi, A. The molecular pathogenesis of HIV-1 associated nephropathy: Recent advances. J. Mol. Med. 2011, 89, 429–436. [Google Scholar] [CrossRef]
- Ross, M.J.; Martinka, S.; D’Agati, V.D.; Bruggeman, L.A. NF-κB Regulates Fas-Mediated Apoptosis in HIV-Associated Nephropathy. J. Am. Soc. Nephrol. 2005, 16, 2403–2411. [Google Scholar] [CrossRef]
- Albaqumi, M.; Soos, T.J.; Barisoni, L.; Nelson, P.J. Collapsing Glomerulopathy. J. Am. Soc. Nephrol. 2006, 17, 2854–2863. [Google Scholar] [CrossRef]
- Zuo, Y.; Matsusaka, T.; Zhong, J.; Ma, J.; Ma, L.-J.; Hanna, Z.; Jolicoeur, P.; Fogo, A.B.; Ichikawa, I. HIV-1 Genes vpr and nef Synergistically Damage Podocytes, Leading to Glomerulosclerosis. J. Am. Soc. Nephrol. 2006, 17, 2832–2843. [Google Scholar] [CrossRef]
- Rosenstiel, P.E.; Gruosso, T.; Letourneau, A.M.; Chan, J.J.; LeBlanc, A.; Husain, M.; Najfeld, V.; Planelles, V.; D'Agati, V.D.; Klotman, M.E.; et al. HIV-1 Vpr inhibits cytokinesis in human proximal tubule cells. Kidney Int. 2008, 74, 1049–1058. [Google Scholar] [CrossRef]
- Ross, M.J.; Wosnitzer, M.S.; Ross, M.D.; Granelli, B.; Gusella, G.L.; Husain, M.; Kaufman, L.; Vasievich, M.; D'Agati, V.D.; Wilson, P.D.; et al. Role of Ubiquitin-Like Protein FAT10 in Epithelial Apoptosis in Renal Disease. J. Am. Soc. Nephrol. 2006, 17, 996–1004. [Google Scholar] [CrossRef]
- Barbaro, G. Cardiovascular Manifestations of HIV Infection. Circulation 2002, 106, 1420–1425. [Google Scholar] [CrossRef]
- Krishnaswamy, G.; Chi, D.S.; Kelley, J.L.; Sarubbi, F.; Smith, J.K.; Peiris, A. The cardiovascular and metabolic complications of HIV infection. Cardiol. Rev. 2000, 8, 260–268. [Google Scholar] [CrossRef]
- Lo, J.; Plutzky, J. The Biology of Atherosclerosis: General Paradigms and Distinct Pathogenic Mechanisms Among HIV-Infected Patients. J. Infect. Dis. 2012, 205, S368–S374. [Google Scholar] [CrossRef]
- Diaz, P.T.; King, M.A.; Pacht, E.R.; Wewers, M.D.; Gadek, J.E.; Nagaraja, H.N.; Drake, J.; Clanton, T.L. Increased Susceptibility to Pulmonary Emphysema among HIV-Seropositive Smokers. Ann. Int. Med. 2000, 132, 369–372. [Google Scholar]
- Crothers, K.; Butt, A.A.; Gibert, C.L.; Rodriguez-Barradas, M.C.; Crystal, S.; Justice, A.C. Increased COPD Among HIV-Positive Compared to HIV-Negative Veterans. CHEST J. 2006, 130, 1326–1333. [Google Scholar] [CrossRef]
- Kirk, G.D.; Merlo, C.; O’Driscoll, P.; Mehta, S.H.; Galai, N.; Vlahov, D.; Samet, J.; Engels, E.A. HIV Infection Is Associated with an Increased Risk for Lung Cancer, Independent of Smoking. Clin. Infect. Dis. 2007, 45, 103–110. [Google Scholar] [CrossRef]
- Rusnati, M.; Presta, M. HIV-1 Tat protein and endothelium: From protein/cell interaction to AIDS-associated pathologies. Angiogenesis 2002, 5, 141–151. [Google Scholar] [CrossRef]
- Kanmogne, G.D.; Primeaux, C.; Grammas, P. Induction of apoptosis and endothelin-1 secretion in primary human lung endothelial cells by HIV-1 gp120 proteins. Biochem. Biophys. Res. Comm. 2005, 333, 1107–1115. [Google Scholar] [CrossRef]
- Dezube, B.J. Clinical presentation and natural history of AIDS-related Kaposi’s sarcoma. Hematol. Oncol. Clin. N. Am. 1996, 10, 1023–1029. [Google Scholar] [CrossRef]
- Grayson, W.; Pantanowitz, L. Histological variants of cutaneous Kaposi sarcoma. Diagn. Pathol. 2008, 3, 31. [Google Scholar] [CrossRef]
- Suster, S.; Fisher, C.; Moran, C.A. Expression of bcl-2 oncoprotein in benign and malignant spindle cell tumors of soft tissue, skin, serosal surfaces, and gastrointestinal tract. Am. J. Surg. Pathol. 1998, 22, 863–872. [Google Scholar] [CrossRef]
- Pillay, P.; Chetty, R.; Re, R. Bcl-2 and p53 Immunoprofile in Kaposi’s Sarcoma. Pathol. Oncol. Res. 1999, 5, 17–20. [Google Scholar] [CrossRef]
- Kaaya, E.; Castanos-Velez, E.; Heiden, T.; Ekman, M.; Catrina, A.I.; Kitinya, J.; Andersson, L.; Biberfeld, P. Proliferation and apoptosis in the evolution of endemic and acquired immunodeficiency syndrome-related Kaposi’s sarcoma. Med. Oncol. 2000, 17, 325–332. [Google Scholar] [CrossRef]
- Mori, S.; Murakami-Mori, K.; Nakamura, S.; Ashkenazi, A.; Bonavida, B. Sensitization of AIDS-Kaposi’s Sarcoma Cells to Apo-2 Ligand-Induced Apoptosis by Actinomycin D. J. Immunol. 1999, 162, 5616–5623. [Google Scholar]
- Sgadari, C.; Barillari, G.; Palladino, C.; Bellino, S.; Taddeo, B.; Toschi, E.; Ensoli, B. Fibroblast Growth Factor-2 and the HIV-1 Tat Protein Synergize in Promoting Bcl-2 Expression and Preventing Endothelial Cell Apoptosis: Implications for the Pathogenesis of AIDS-Associated Kaposi’s Sarcoma. Int. J. Vasc. Med. 2011, 2011. [Google Scholar] [CrossRef]
- Nor, J.E.; Christensen, J.; Liu, J.; Peters, M.; Mooney, D.J.; Strieter, R.M.; Polverini, P.J. Up-Regulation of Bcl-2 in Microvascular Endothelial Cells Enhances Intratumoral Angiogenesis and Accelerates Tumor Growth. Cancer Res. 2001, 61, 2183–2188. [Google Scholar]
- Sakai, Y.; Goodison, S.; Kusmartsev, S.; Fletcher, B.; Eruslanov, E.; Cao, W.; Porvasnik, S.; Namiki, K.; Anai, S.; Rosser, C.J. Bcl-2 mediated modulation of vascularization in prostate cancer xenografts. Prostate 2009, 69, 459–470. [Google Scholar] [CrossRef]
- Michel, J.-B. Anoikis in the Cardiovascular System. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 2146–2154. [Google Scholar] [CrossRef]
- Eccles, S.A. Parallels in invasion and angiogenesis provide pivotal points for therapeutic intervention. Int. J. Dev. Biol. 2004, 48, 583–598. [Google Scholar]
- Almodovar, S.; Knight, R.; Allshouse, A.A.; Roemer, S.; Lozupone, C.; McDonald, D.; Widmann, J.; Voelkel, N.F.; Shelton, R.J.; Suarez, E.B.; et al. Human Immunodeficiency Virus Nef signature sequences are associated with pulmonary hypertension. AIDS Res. Hum. Retrovir. 2012, 28, 607–618. [Google Scholar] [CrossRef]
- Li, C.J.; Friedman, D.J.; Wang, C.; Metelev, V.; Pardee, A.B. Induction of apoptosis in uninfected lymphocytes by HIV-1 Tat protein. Science 1995, 268, 429–431. [Google Scholar]
- Jajoo, S.; Mukherjea, D.; Brewer, G.J.; Ramkumar, V. Pertussis toxin B-oligomer suppresses human immunodeficiency virus-1 Tat-induced neuronal apoptosis through feedback inhibition of phospholipase C-β by protein kinase C. Neuroscience 2008, 151, 525–532. [Google Scholar]
- Liu, H.; Yu, S.; Zhang, H.; Xu, J. Angiogenesis Impairment in Diabetes: Role of Methylglyoxal-Induced Receptor for Advanced Glycation Endproducts, Autophagy and Vascular Endothelial Growth Factor Receptor 2. PLoS One 2012, 7, e46720. [Google Scholar]
- Rizza, S.A.; Badley, A.D. HIV protease inhibitors impact on apoptosis. Med. Chem. 2008, 4, 75–79. [Google Scholar] [CrossRef]
- Vocero-Akbani, A.M.; Heyden, N.V.; Lissy, N.A.; Ratner, L.; Dowdy, S.F. Killing HIV-infected cells by transduction with an HIV protease-activated caspase-3 protein. Nat. Med. 1999, 5, 29–33. [Google Scholar] [CrossRef]
- Miyauchi, K.; Urano, E.; Takizawa, M.; Ichikawa, R.; Komano, J. Therapeutic potential of HIV protease-activable CASP3. Sci. Rep. 2012, 2, 359. [Google Scholar]
- Rehman, S.; Husain, M.; Yadav, A.; Kasinath, B.S.; Malhotra, A.; Singhal, P.C. HIV-1 Promotes Renal Tubular Epithelial Cell Protein Synthesis: Role of mTOR Pathway. PLoS One 2012, 7, e30071. [Google Scholar]
- Passaes, C.P.; Sáez-Ciriòn, A. HIV cure research: Advances and prospects. Virology 2014, 454–455, 340–352. [Google Scholar] [CrossRef]
- Cummins, N.; Badley, A. Anti-apoptotic mechanisms of HIV: Lessons and novel approaches to curing HIV. Cell. Mol. Life Sci. 2013, 70, 3355–3363. [Google Scholar] [CrossRef]
- Badley, A.D.; Sainski, A.; Wightman, F.; Lewin, S.R. Altering cell death pathways as an approach to cure HIV infection. Cell Death Dis. 2013, 4, e718. [Google Scholar]
- Garg, H.; Joshi, A.; Blumenthal, R. Altered bystander apoptosis induction and pathogenesis of enfuvirtide-resistant HIV type 1 Env mutants. AIDS Res. Hum. Retrovir. 2009, 25, 811–817. [Google Scholar] [CrossRef]
- Chinsembu, K.C.; Hedimbi, M. A Survey of Plants with Anti-HIV Active Compounds and their Modes of Action. Med. J. Zambia 2009, 36, 178–186. [Google Scholar]
- Frankel, A.D.; Bredt, D.S.; Pabo, C.O. Tat protein from human immunodeficiency virus forms a metal-linked dimer. Science 1988, 240, 70–73. [Google Scholar]
- Ramautar, A.; Mabandla, M.; Blackburn, J.; Daniels, W.M.U. Inhibition of HIV-1 Tat-induced transactivation and apoptosis by the divalent metal chelators, fusaric acid and picolinic acid Implications for HIV-1 dementia. Neurosci. Res. 2012, 74, 59–63. [Google Scholar] [CrossRef]
- De Clercq, E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int. J. Antimicrob. Agents 2009, 33, 307–320. [Google Scholar] [CrossRef]
- Judd, D.A.; Nettles, J.H.; Nevins, N.; Snyder, J.P.; Liotta, D.C.; Tang, J.; Ermolieff, J.; Schinazi, R.F.; Hill, C.L. Polyoxometalate HIV-1 Protease Inhibitors. A New Mode of Protease Inhibition. J. Am. Chem. Soc. 2001, 123, 886–897. [Google Scholar] [CrossRef]
- Sayer, J.M.; Aniana, A.; Louis, J.M. Mechanism of Dissociative Inhibition of HIV Protease and Its Autoprocessing from a Precursor. J. Mol. Biol. 2012, 422, 230–244. [Google Scholar] [CrossRef]
- Hwang, Y.S.; Chmielewski, J. A unidirectional crosslinking strategy for HIV-1 protease dimerization inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 4297–4300. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Chapsal, B.D.; Steffey, M.; Agniswamy, J.; Wang, Y.-F.; Amano, M.; Weber, I.T.; Mitsuya, H. Substituent effects on P2-cyclopentyltetrahydrofuranyl urethanes: Design, synthesis, and X-ray studies of potent HIV-1 protease inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 2308–2311. [Google Scholar] [CrossRef]
- Koh, Y.; Das, D.; Leschenko, S.; Nakata, H.; Ogata-Aoki, H.; Amano, M.; Nakayama, M.; Ghosh, A.K.; Mitsuya, H. GRL-02031, a novel nonpeptidic protease inhibitor (PI) containing a stereochemically defined fused cyclopentanyltetrahydrofuran potent against multi-PI-resistant human immunodeficiency virus type 1 in vitro. Antimicrob. Agents Chemother. 2009, 53, 997–1006. [Google Scholar] [CrossRef]
- He, Y.; Xiao, Y.; Song, H.; Liang, Q.; Ju, D.; Chen, X.; Lu, H.; Jing, W.; Jiang, S.; Zhang, L. Design and Evaluation of Sifuvirtide, a Novel HIV-1 Fusion Inhibitor. J. Biol. Chem. 2008, 283, 11126–11134. [Google Scholar]
- Janda, E.; Visalli, V.; Colica, C.; Aprigliano, S.; Musolino, V.; Vadalà, N.; Muscoli, C.; Sacco, I.; Iannone, M.; Rotiroti, D. The protective effect of tianeptine on Gp120-induced apoptosis in astroglial cells: Role of GS and NOS, and NF-κB suppression. Br. J. Pharmacol. 2011, 164, 1590–1599. [Google Scholar]
- Rider, T.H.; Zook, C.E.; Boettcher, T.L.; Wick, S.T.; Pancoast, J.S.; Zusman, B.D. Broad-spectrum antiviral therapeutics. PLoS One 2011, 6, e22572. [Google Scholar]
- Thakur, B.K.; Chandra, A.; Dittrich, T.; Welte, K.; Chandra, P. Inhibition of SIRT1 by HIV-1 viral protein Tat results in activation of p53 pathway. Biochem. Biophys. Res. Comm. 2012, 424, 245–250. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.-i.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative Control of p53 by SirT1 Promotes Cell Survival under Stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Van Leeuwen, I.; Lain, S. Sirtuins and p53. In Advances in Cancer Research; Vande Woude, G.F., Klein, G., Eds.; Academic Press: New York, NY, USA, 2009; Volume 102, pp. 171–195. [Google Scholar]
- Vaziri, H.; Dessain, S.K.; Eaton, E.N.; Imai, S.-I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2SIRT1 Functions as an NAD-Dependent p53 Deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- Peck, B.; Chen, C.Y.; Ho, K.K.; Di Fruscia, P.; Myatt, S.S.; Coombes, R.C.; Fuchter, M.J.; Hsiao, C.D.; Lam, E.W. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol. Cancer Ther. 2010, 9, 844–855. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; Li, L.; Wang, Z.; Ho, Y.; McDonald, T.; Holyoake, T.L.; Chen, W.Y.; Bhatia, R. Activation of p53 by SIRT1 Inhibition Enhances Elimination of CML Leukemia Stem Cells in Combination with Imatinib. Cancer Cell. 2012, 21, 266–281. [Google Scholar] [CrossRef]
- Audrito, V.; Vaisitti, T.; Rossi, D.; Gottardi, D.; D’Arena, G.; Laurenti, L.; Gaidano, G.; Malavasi, F.; Deaglio, S. Nicotinamide Blocks Proliferation and Induces Apoptosis of Chronic Lymphocytic Leukemia Cells through Activation of the p53/miR-34a/SIRT1 Tumor Suppressor Network. Cancer Res. 2011, 71, 4473–4483. [Google Scholar] [CrossRef]
- Phenix, B.N.; Angel, J.B.; Mandy, F.; Kravcik, S.; Parato, K.; Chambers, K.A.; Gallicano, K.; Hawley-Foss, N.; Cassol, S.; Cameron, D.W.; et al. Decreased HIV-associated T cell apoptosis by HIV protease inhibitors. AIDS Res. Hum. Retrovir. 2000, 16, 559–567. [Google Scholar] [CrossRef]
- Pope, M.T.; Müller, A. Polyoxometalates: From Platonic Solids to Anti-Retroviral Activity (Topics in Molecular Organization and Engineering); Kluwer Academic Publishers: Dordrecht, The Netherlands, 1994. [Google Scholar]
- Moskovitz, B.L. Clinical trial of tolerance of HPA-23 in patients with acquired immune deficiency syndrome. Antimicrob. Agents Chemo. 1988, 32, 1300–1303. [Google Scholar] [CrossRef]
- Flutsch, A.; Schroeder, T.; Grütter, M.G.; Patzke, G.R. HIV-1 protease inhibition potential of functionalized polyoxometalates. Bioorg. Med. Chem. Lett. 2011, 21, 1162–1166. [Google Scholar]
- Inouye, Y.; Tokutake, Y.; Kunihara, J.; Yoshida, T.; Yamase, T.; Nakata, A.; Nakamura, S. Suppressive effect of polyoxometalates on the cytopathogenicity of human immunodeficiency virus type 1 (HIV-1) in vitro and their inhibitory activity against HIV-1 reverse transcriptase. Chem. Pharm. Bull. (Tokyo) 1992, 40, 805–807. [Google Scholar] [CrossRef]
- Gustchina, A.; Weber, I.T. Comparative analysis of the sequences and structures of HIV-1 and HIV-2 proteases. Proteins 1991, 10, 325–339. [Google Scholar] [CrossRef]
- Tang, C.; Louis, J.M.; Aniana, A.; Suh, J.-Y.; Clore, G.M. Visualizing transient events in amino-terminal autoprocessing of HIV-1 protease. Nature 2008, 455, 693–696. [Google Scholar] [CrossRef]
- Babé, L.M.; Rosé, J.; Craik, C.S. Synthetic “interface” peptides alter dimeric assembly of the HIV 1 and 2 proteases. Protein Sci. 1992, 1, 1244–1253. [Google Scholar] [CrossRef]
- Todd, M.J.; Semo, N.; Freire, E. The structural stability of the HIV-1 protease. J. Mol. Biol. 1998, 283, 475–488. [Google Scholar] [CrossRef]
- Louis, J.M.; Ishima, R.; Torchia, D.A.; Weber, I.T. HIV-1 Protease: Structure, Dynamics, and Inhibition. Adv. Pharmacol. 2007, 55, 261–298. [Google Scholar]
- Davis, D.A.; Brown, C.A.; Singer, K.E.; Wang, V.; Kaufman, J.; Stahl, S.J.; Wingfield, P.; Maeda, K.; Harada, S.; Yoshimura, K.; et al. Inhibition of HIV-1 replication by a peptide dimerization inhibitor of HIV-1 protease. Antivir. Res. 2006, 72, 89–99. [Google Scholar] [CrossRef]
- Wei, Y.; Ma, C.-M.; Hattori, M. Synthesis and evaluation of A-seco type triterpenoids for anti-HIV-1protease activity. Eur. J. Med. Chem. 2009, 44, 4112–4120. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Chapsal, B.D.; Baldridge, A.; Ide, K.; Koh, Y.; Mitsuya, H. Design and Synthesis of Stereochemically Defined Novel Spirocyclic P2-Ligands for HIV-1 Protease Inhibitors. Org. Lett. 2008, 10, 5135–5138. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Leshchenko-Yashchuk, S.; Anderson, D.D.; Baldridge, A.; Noetzel, M.; Miller, H.B.; Tie, Y.; Wang, Y.-F.; Koh, Y.; Weber, I.T.; et al. Design of HIV-1 Protease Inhibitors with Pyrrolidinones and Oxazolidinones as Novel P1 Ligands To Enhance Backbone-Binding Interactions with Protease: Synthesis, Biological Evaluation, and Protein Ligand X-ray Studies. J. Med. Chem. 2009, 52, 3902–3914. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Takayama, J. Enantioselective Synthesis of Cyclopentyltetrahydrofuran (Cp-THF), an Important High-Affinity P2-Ligand for HIV-1 Protease Inhibitors. Tetrahedron Lett. 2008, 49, 3409–3412. [Google Scholar]
- Mosing, R.K.; Mendonsa, S.D.; Bowser, M.T. Capillary Electrophoresis-SELEX Selection of Aptamers with Affinity for HIV-1 Reverse Transcriptase. Anal. Chem. 2005, 77, 6107–6112. [Google Scholar] [CrossRef]
- Baron, M.; Kinsley, N.; Sewell, B.; Jaffer, M.; Capovilla, A.; James, W.; Khati, M. Molecular Interaction of gp120 and B40 Aptamer: A Potential New HIV-1 Entry Inhibitor Drug. In Proceedings of the 2nd CSIR Biennial Conference: CSIR International Convention Centre Pretoria, South Africa, November 2008; pp. 1–12.
- Berges, B.K.; Akkina, S.R.; Remling, L.; Akkina, R. Humanized Rag2−/−γc−/− (RAG-hu) mice can sustain long-term chronic HIV-1 infection lasting more than a year. Virology 2010, 397, 100–103. [Google Scholar] [CrossRef]
- Dey, A.K.; Griffiths, C.; Lea, S.M.; James, W. Structural characterization of an anti-gp120 RNA aptamer that neutralizes R5 strains of HIV-1. RNA 2005, 11, 873–884. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, Z.; Wei, H.; Hu, Q.; Deng, J.; Guo, D.; Cui, Z.; Zhang, X.-E. Aptamer beacons for visualization of endogenous protein HIV-1 reverse transcriptase in living cells. Biosens. Bioelectron. 2011, 28, 270–276. [Google Scholar] [CrossRef]
- Neff, C.P.; Zhou, J.; Remling, L.; Kuruvilla, J.; Zhang, J.; Li, H.; Smith, D.D.; Swiderski, P.; Rossi, J.J.; Akkina, R. An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4(+) T cell decline in humanized mice. Sci. Transl. Med. 2011, 3, 66ra6. [Google Scholar]
- Rahim Ruslinda, A.; Tanabe, K.; Ibori, S.; Wang, X.; Kawarada, H. Effects of diamond-FET-based RNA aptamer sensing for detection of real sample of HIV-1 Tat protein. Biosens. Bioelectron. 2013, 40, 277–282. [Google Scholar] [CrossRef]
- Tombelli, S.; Minunni, M.; Luzi, E.; Mascini, M. Aptamer-based biosensors for the detection of HIV-1 Tat protein. Bioelectrochemistry 2005, 67, 135–141. [Google Scholar] [CrossRef]
- Wheeler, L.A.; Trifonova, R.; Vrbanac, V.; Basar, E.; McKernan, S.; Xu, Z.; Seung, E.; Deruaz, M.; Dudek, T.; Einarsson, J.I.; et al. Inhibition of HIV transmission in human cervicovaginal explants and humanized mice using CD4aptamer-siRNA chimeras. J. Clin. Invest. 2011, 121, 2401–2412. [Google Scholar] [CrossRef]
- Zhou, J.; Shu, Y.; Guo, P.; Smith, D.D.; Rossi, J.J. Dual functional RNA nanoparticles containing phi29 motor pRNA and anti-gp120 aptamer for cell-type specific delivery and HIV-1 Inhibition. Methods 2011, 54, 284–294. [Google Scholar] [CrossRef]
- Zhu, Q.; Shibata, T.; Kabashima, T.; Kai, M. Inhibition of HIV-1 protease expression in T cells owing to DNA aptamer-mediated specific delivery of siRNA. Eur. J. Med. Chem. 2012, 56, 396–399. [Google Scholar] [CrossRef]
- Castaldello, A.; Brocca-Cofano, E.; Voltan, R.; Triulzi, C.; Altavilla, G.; Laus, M.; Sparnacci, K.; Ballestri, M.; Tondelli, L.; Fortini, C.; et al. DNA prime and protein boost immunization with innovative polymeric cationic core-shell nanoparticles elicits broad immune responses and strongly enhance cellular responses of HIV-1 Tat DNA vaccination. Vaccine 2006, 24, 5655–5669. [Google Scholar]
- Chattopadhyay, N.; Zastre, J.; Wong, H.L.; Wu, X.Y.; Bendayan, R. Solid lipid nanoparticles enhance the delivery of the HIV protease inhibitor, atazanavir, by a human brain endothelial cell line. Pharm. Res. 2008, 25, 2262–2271. [Google Scholar] [CrossRef]
- Weyermann, J.; Lochmann, D.; Zimmer, A. Comparison of antisense oligonucleotide drug delivery systems. J. Controll. Release 2004, 100, 411–423. [Google Scholar] [CrossRef]
- Stanberry, L.R.; Strugnell, R. Vaccines of the future. Perspect. Vaccinol. 2011, 1, 151–199. [Google Scholar] [CrossRef]
- Christie, R.J.; Grainger, D.W. Design strategies to improve soluble macromolecular delivery constructs. Adv. Drug Del. Rev. 2003, 55, 421–437. [Google Scholar] [CrossRef]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Contr. Release 2008, 132, 171–183. [Google Scholar] [CrossRef]
- das Neves, J.; Amiji, M.M.; Bahia, M.F.; Sarmento, B. Nanotechnology-based systems for the treatment and prevention of HIV/AIDS. Adv. Drug Del. Rev. 2010, 62, 458–477. [Google Scholar] [CrossRef]
- Panyam, J.; Labhasetwar, V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Del. Rev. 2003, 55, 329–347. [Google Scholar] [CrossRef]
- Lobenberg, R.; Kreuter, J. Macrophage targeting of azidothymidine: A promising strategy for AIDS therapy. AIDS Res. Hum. Retrovir. 1996, 12, 1709–1715. [Google Scholar] [CrossRef]
- Kim, S.; Scheerer, S.; Geyer, M.A.; Howell, S.B. Direct Cerebrospinal Fluid Delivery of an Antiretroviral Agent Using Multivesicular Liposomes. J. Infect. Dis. 1990, 162, 750–752. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mbita, Z.; Hull, R.; Dlamini, Z. Human Immunodeficiency Virus-1 (HIV-1)-Mediated Apoptosis: New Therapeutic Targets. Viruses 2014, 6, 3181-3227. https://doi.org/10.3390/v6083181
Mbita Z, Hull R, Dlamini Z. Human Immunodeficiency Virus-1 (HIV-1)-Mediated Apoptosis: New Therapeutic Targets. Viruses. 2014; 6(8):3181-3227. https://doi.org/10.3390/v6083181
Chicago/Turabian StyleMbita, Zukile, Rodney Hull, and Zodwa Dlamini. 2014. "Human Immunodeficiency Virus-1 (HIV-1)-Mediated Apoptosis: New Therapeutic Targets" Viruses 6, no. 8: 3181-3227. https://doi.org/10.3390/v6083181
APA StyleMbita, Z., Hull, R., & Dlamini, Z. (2014). Human Immunodeficiency Virus-1 (HIV-1)-Mediated Apoptosis: New Therapeutic Targets. Viruses, 6(8), 3181-3227. https://doi.org/10.3390/v6083181