Locally-Delivered T-Cell-Derived Cellular Vehicles Efficiently Track and Deliver Adenovirus Delta24-RGD to Infiltrating Glioma


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Cell Culture
2.2. Virus Construction and Propagation
2.3. Delta24-RGD Infection and Replication Assay
2.4. T-Cell Migration Assays
2.5. In Vivo Experiments
2.6. Immunohistochemistry
2.7. Statistics
3. Results
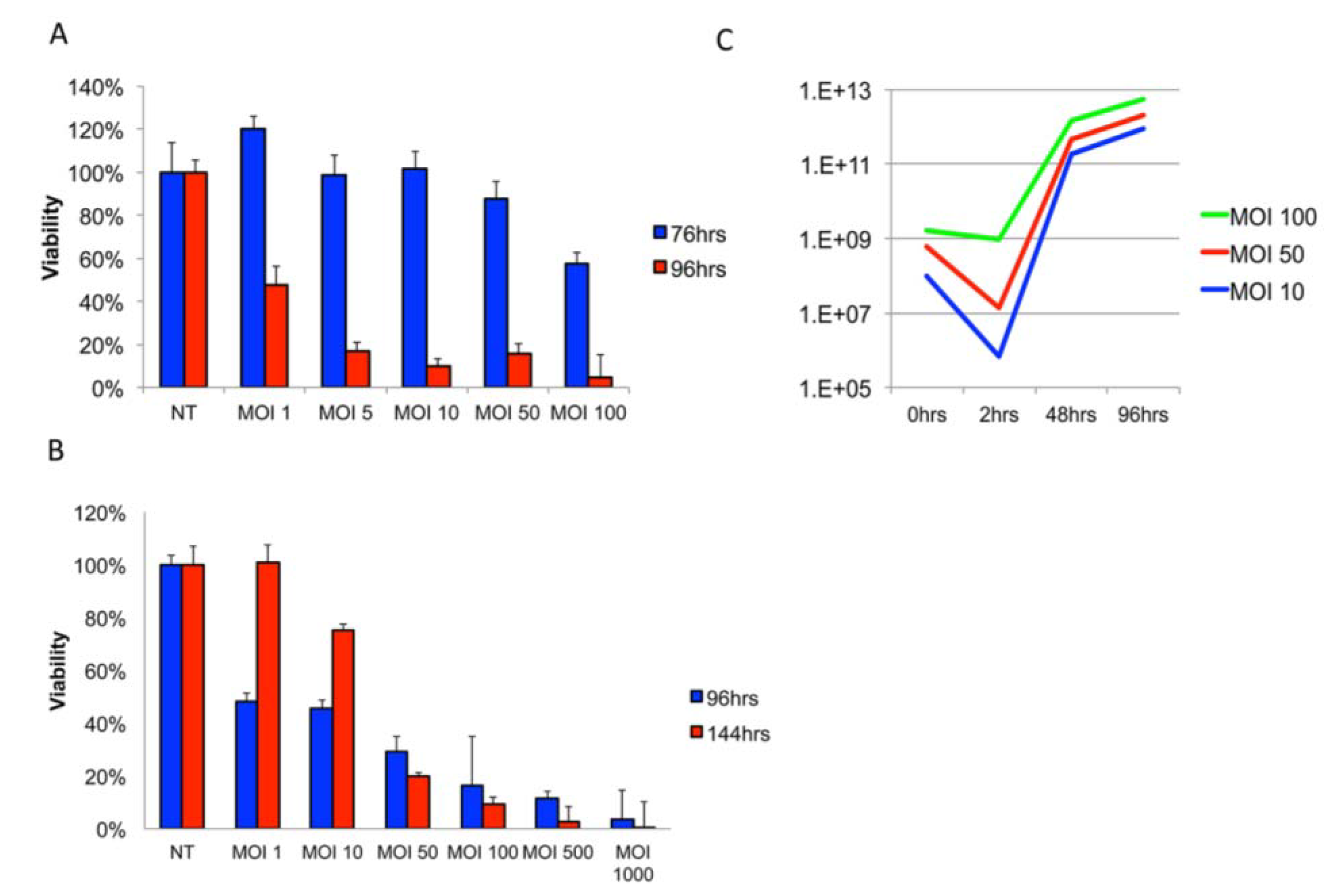
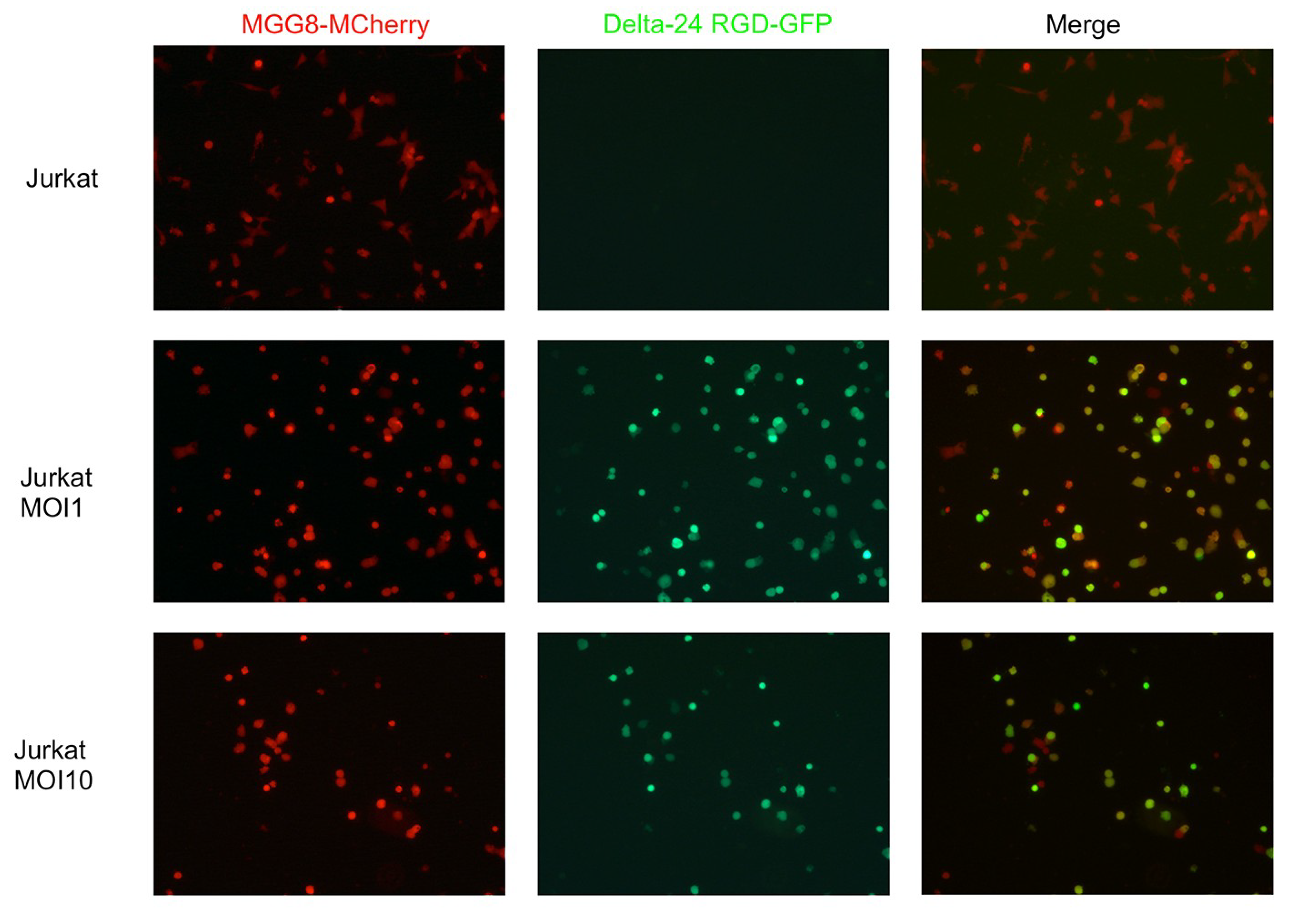
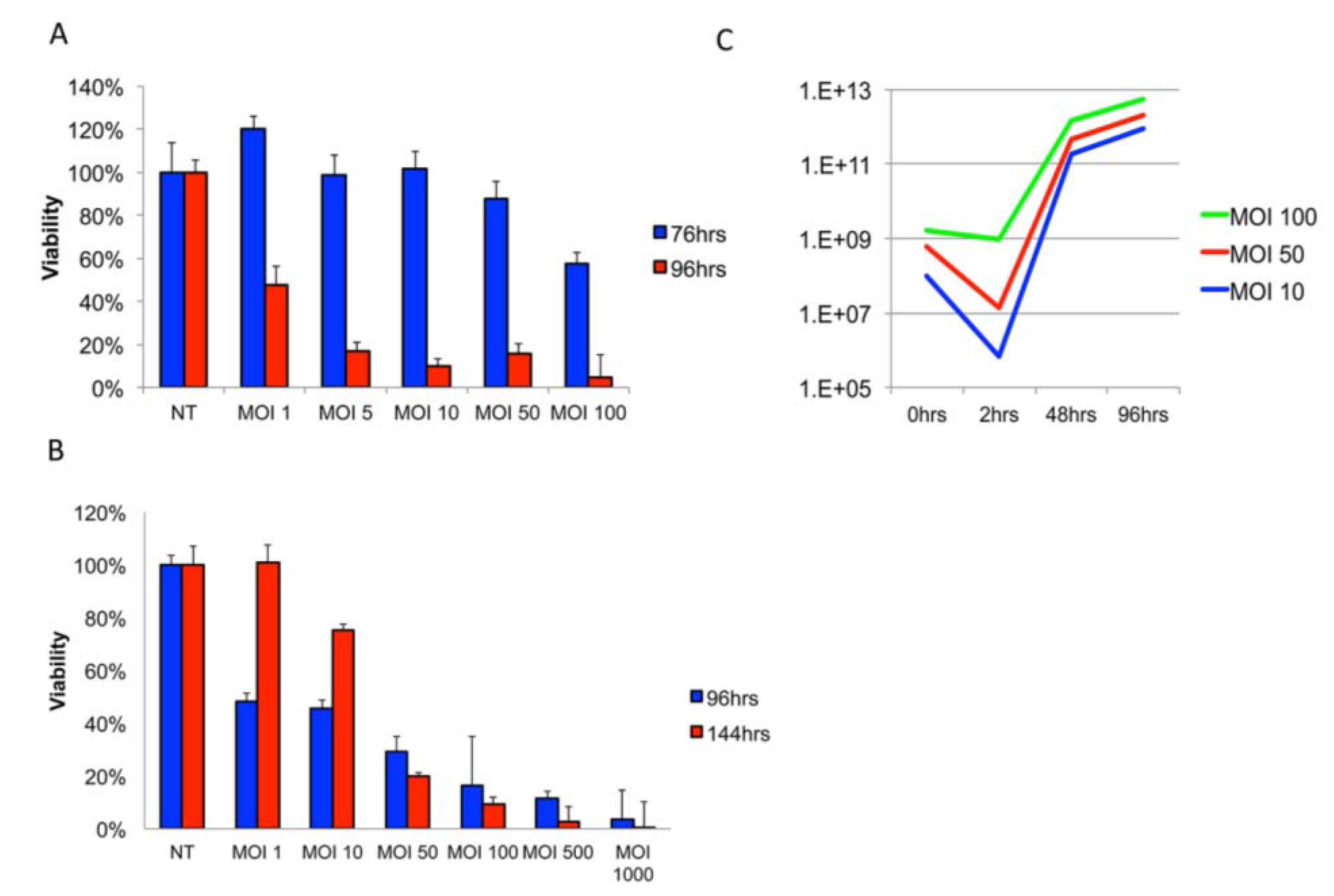
3.1. Delta24-RGD Efficiently Infects and Replicates in GSC and Jurkat T-Cells
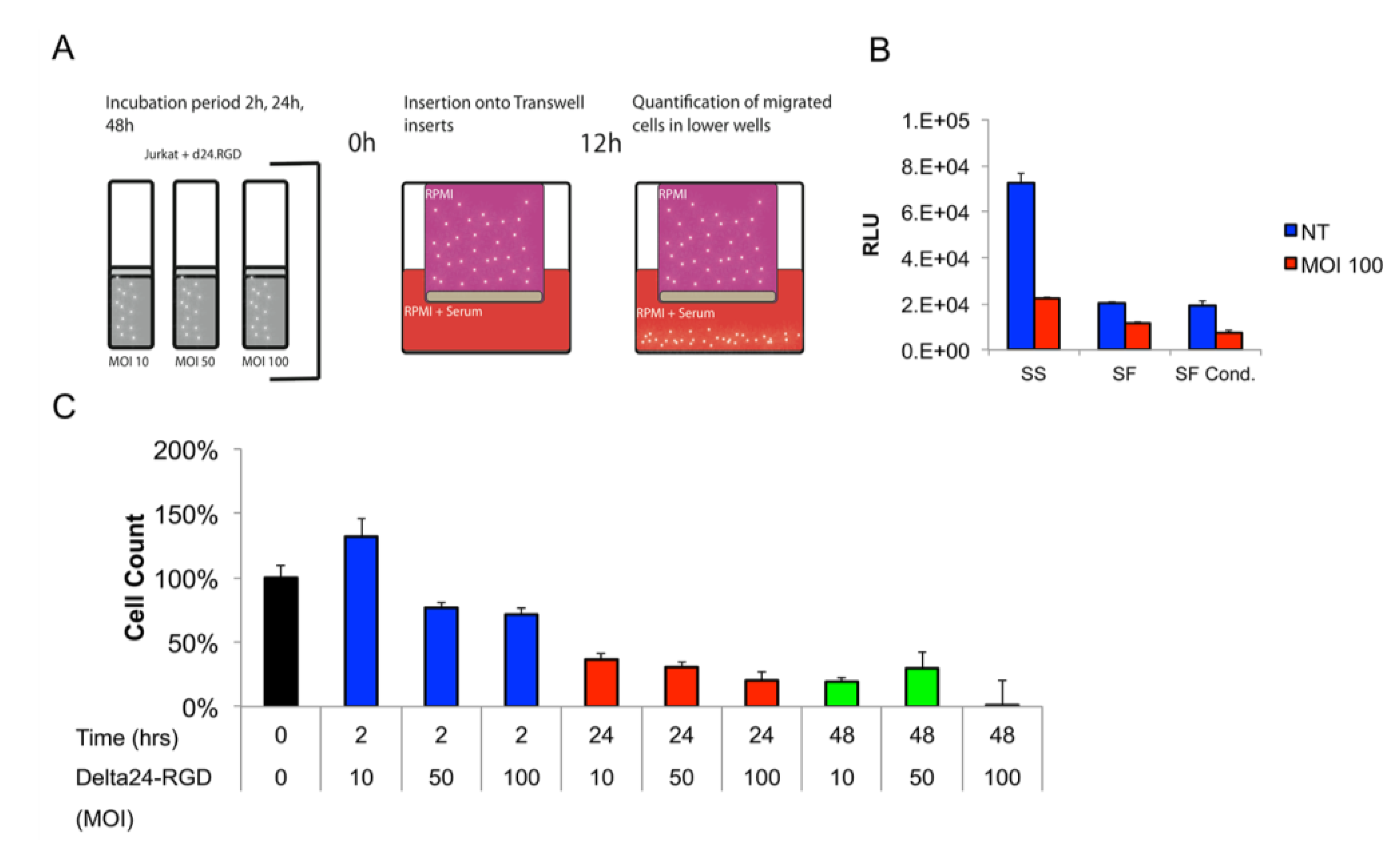
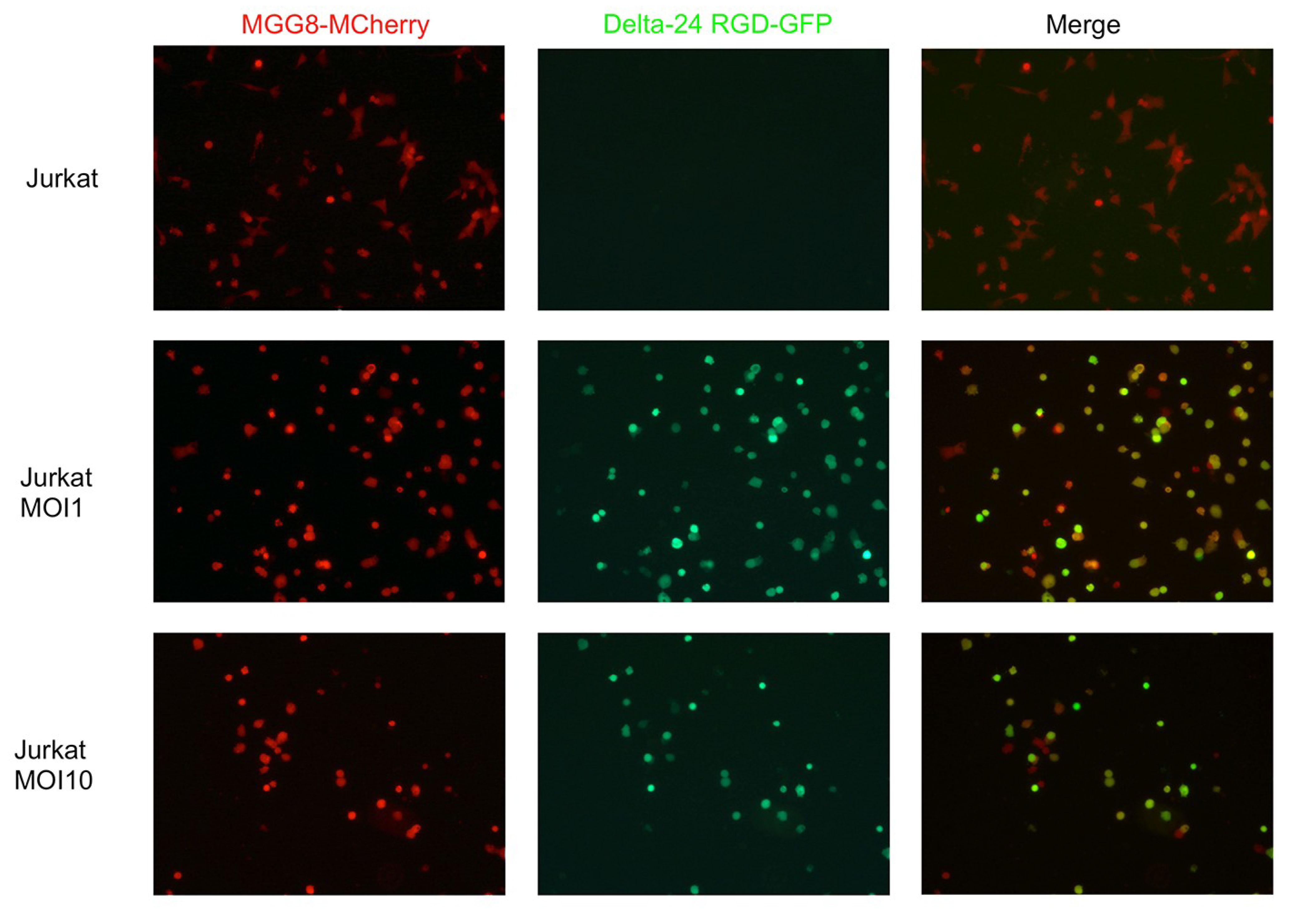
3.2. T-Cells Efficiently Deliver Delta24-RGD to GSCs In Vitro
3.3. The Migratory Capacity of T-Cells Is Preserved Following Short Incubation Times with Delta24-RGD
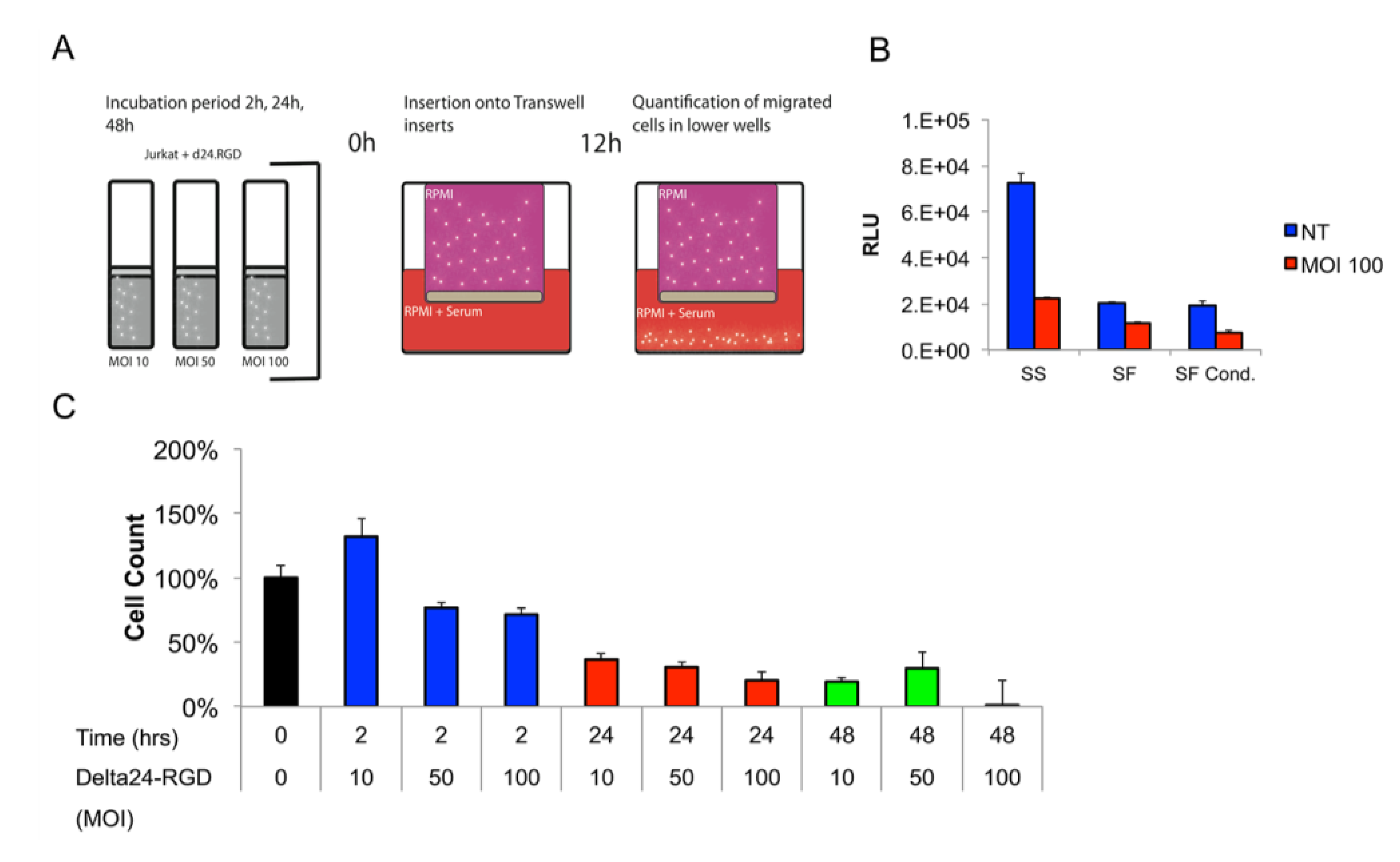
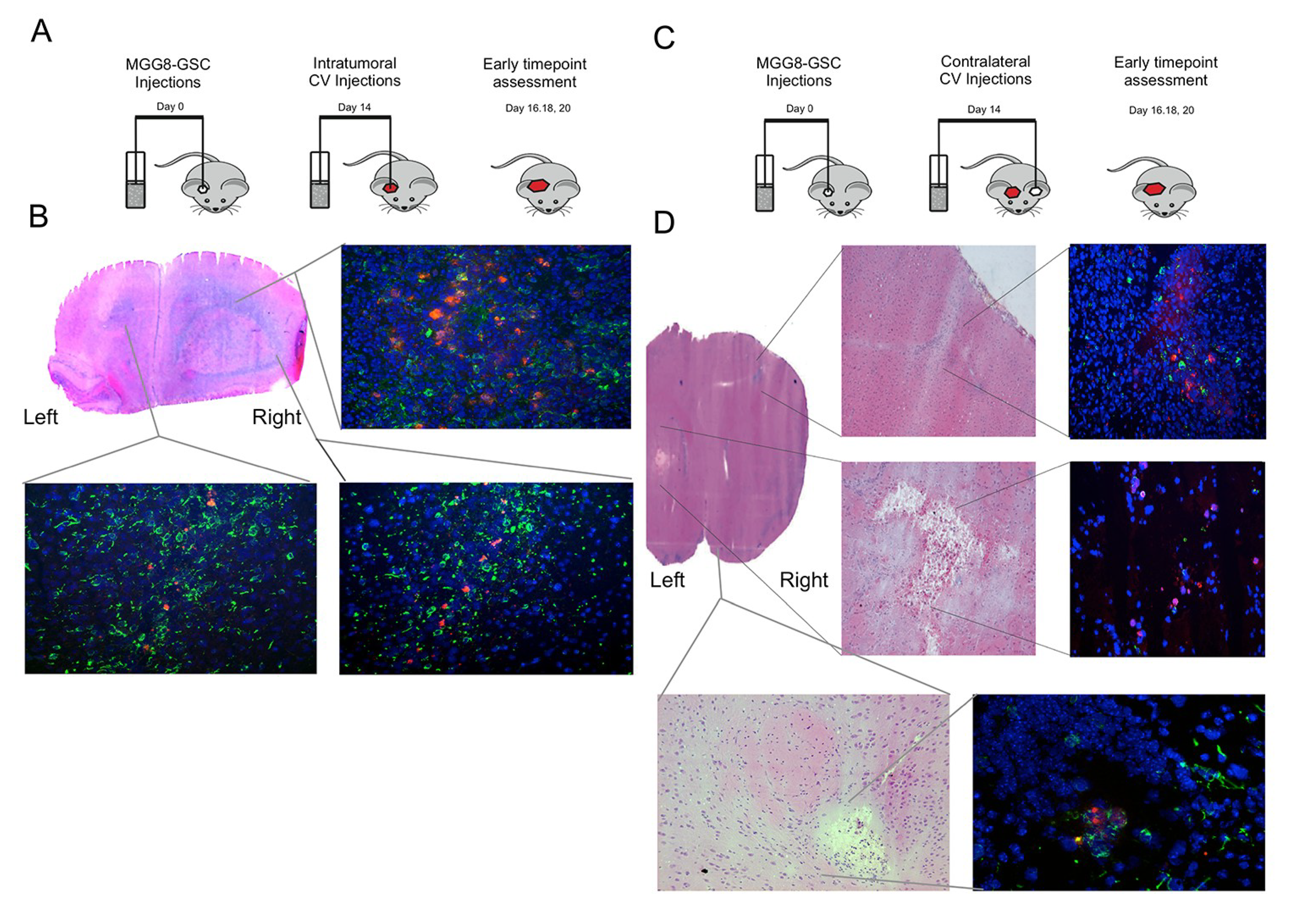
3.4. Delta24-RGD-Loaded T-Cells Demonstrate Intraparenchymal Tumor Tropism and In Vivo Viral Transfer
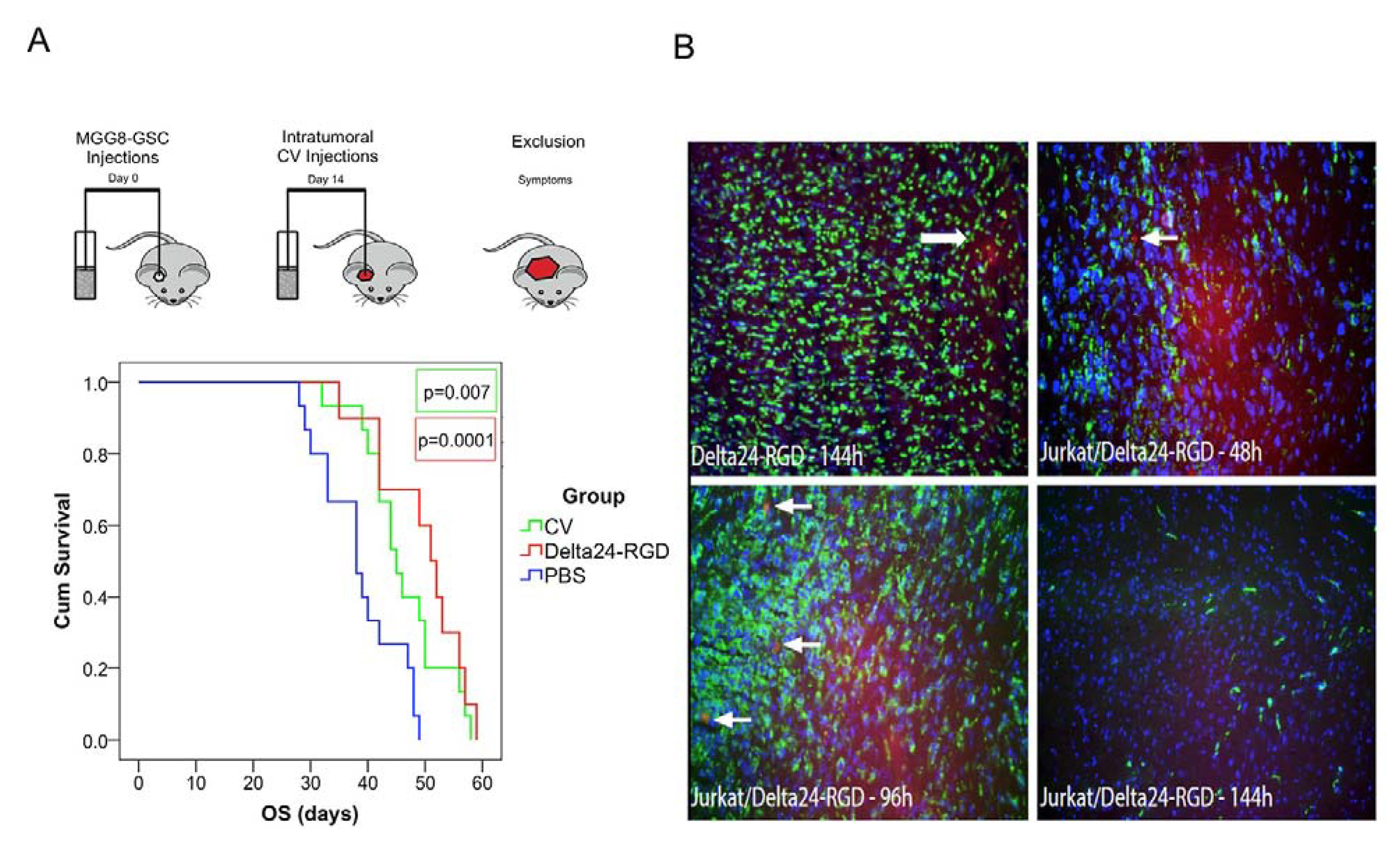
3.5. Intratumoral Delivery of Delta24-RGD by T-Cells Leads to Prolonged Survival
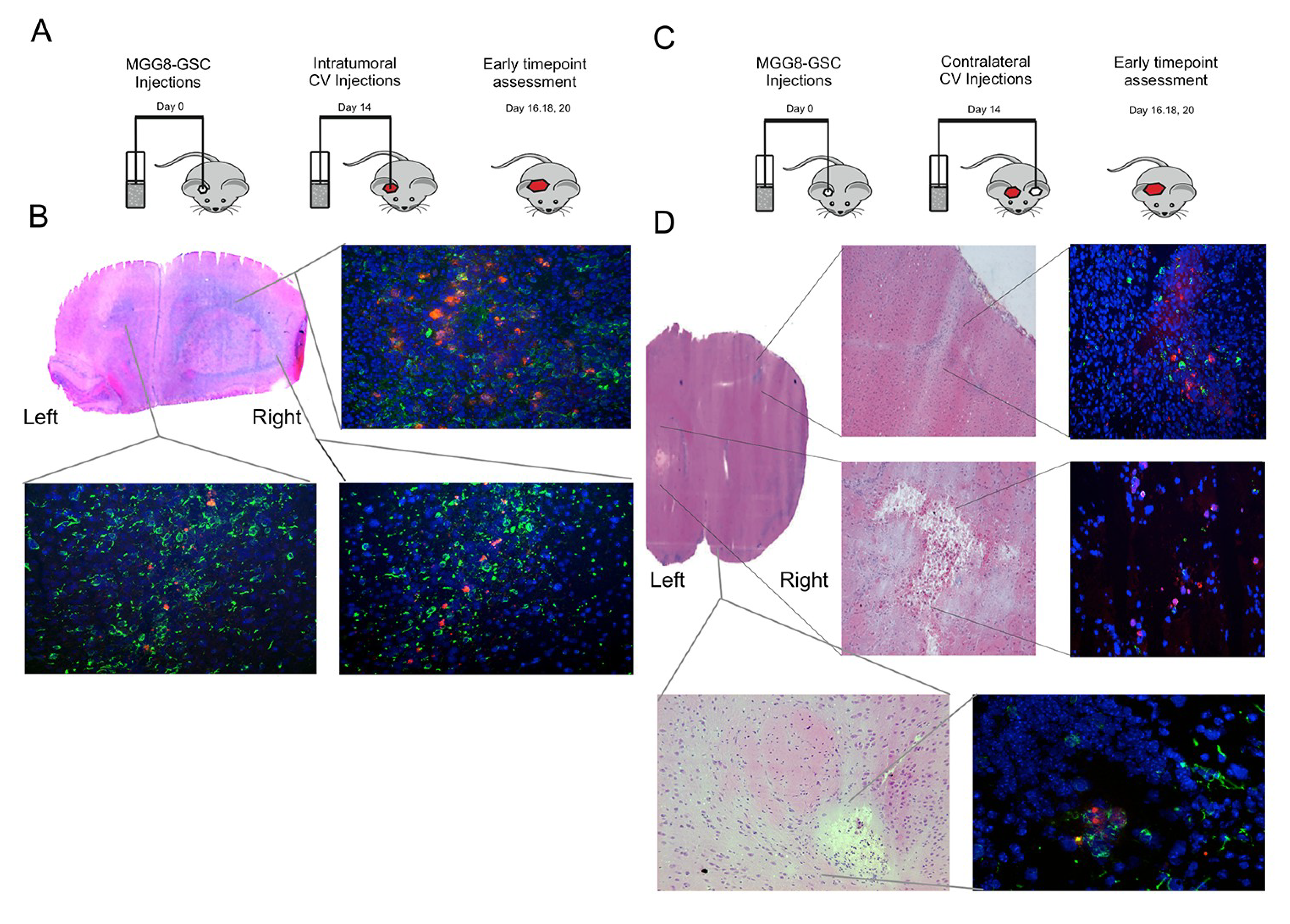
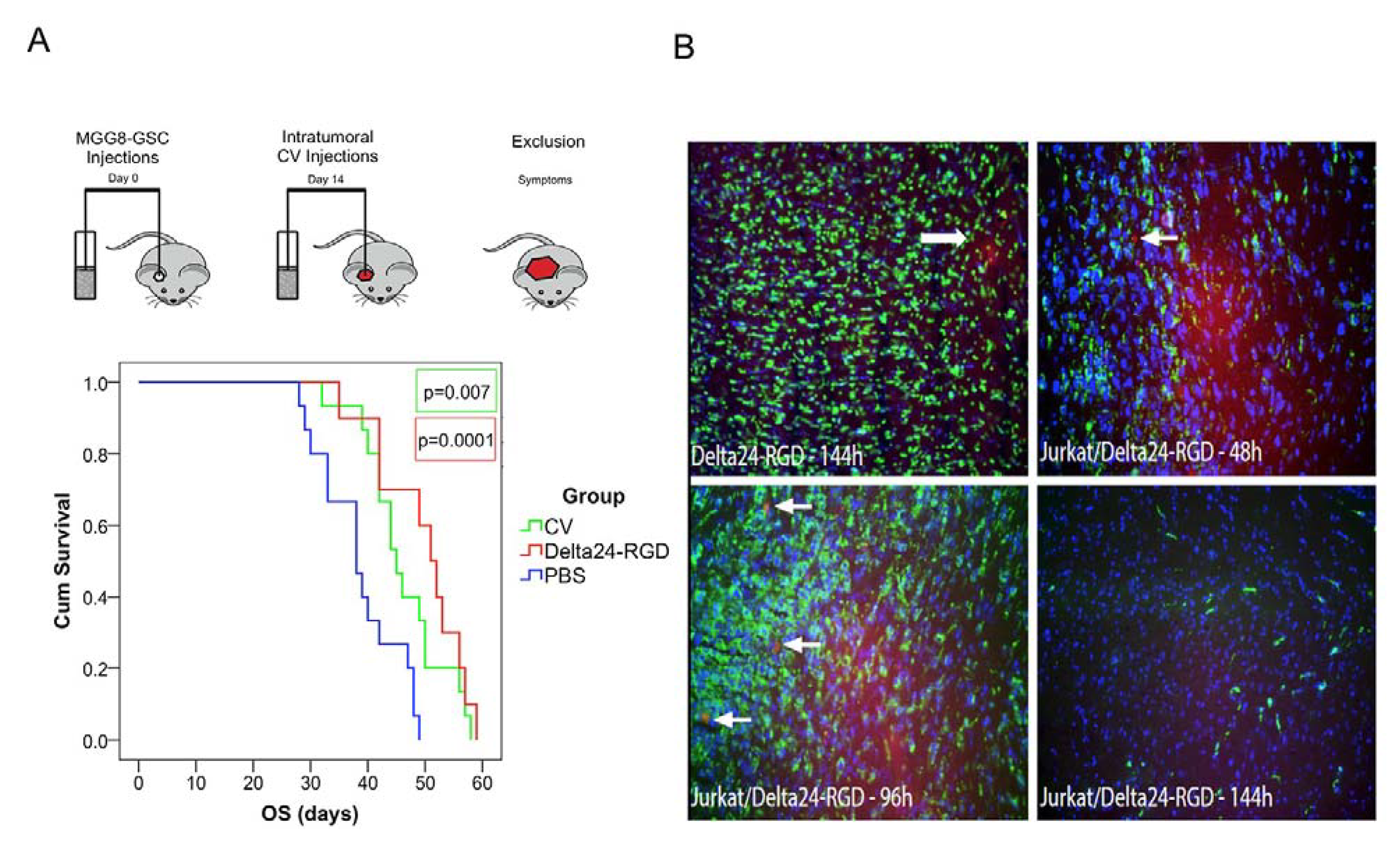
3.6. Delta24-RGD-Loaded T-Cells Demonstrate Tropism for Intracranial Tumors
4. Discussion
Author Contributions
Conflicts of Interests
References
- Huse, J.T.; Holland, E.C. Targeting brain cancer: Advances in the molecular pathology of malignant glioma and medulloblastoma. Nat. Rev. Cancer 2010, 10, 319–331. [Google Scholar] [CrossRef]
- Kroeger, K.M.; Muhammad, A.K.; Baker, G.J.; Assi, H.; Wibowo, M.K.; Xiong, W.; Yagiz, K.; Candolfi, M.; Lowenstein, P.R.; Castro, M.G. Gene therapy and virotherapy: Novel therapeutic approaches for brain tumors. Discov. Med. 2010, 10, 293–304. [Google Scholar]
- Murphy, A.M.; Rabkin, S.D. Current status of gene therapy for brain tumors. Transl. Res. 2013, 161, 339–354. [Google Scholar] [CrossRef]
- Lang, F.F.; Bruner, J.M.; Fuller, G.N.; Aldape, K.; Prados, M.D.; Chang, S.; Berger, M.S.; McDermott, M.W.; Kunwar, S.M.; Junck, L.R.; et al. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: Biological and clinical results. J. Clin. Oncol. 2003, 21, 2508–2518. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Broaddus, W.C.; Gillies, G.T.; Visted, T.; Lamfers, M.L.M. Neurosurgical delivery of chemotherapeutics, targeted toxins, genetic and viral therapies in neuro-oncology. J. Neurooncol. 2004, 69, 101–117. [Google Scholar] [CrossRef]
- Lamfers, M.L.; Grill, J.; Dirven, C.M.; van Beusechem, V.W.; Geoerger, B.; van Den Berg, J.; Alemany, R.; Fueyo, J.; Curiel, D.T.; et al. Potential of the conditionally replicative adenovirus Ad5-Delta24RGD in the treatment of malignant gliomas and its enhanced effect with radiotherapy. Cancer Res. 2002, 62, 5736–5742. [Google Scholar]
- Jiang, H.; Gomez-Manzano, C.; Aoki, H.; Alonso, M.M.; Kondo, S.; McCormick, F.; Xu, J.; Kondo, Y.; Bekele, B.N.; Colman, H.; et al. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: Role of autophagic cell death. J. Natl. Cancer Inst. 2007, 99, 1410–1414. [Google Scholar] [CrossRef]
- Fueyo, J.; Alemany, R.; Gomez-Manzano, C.; Fuller, G.N.; Khan, A.; Conrad, C.A.; Liu, T.J.; Jiang, H.; Lemoine, M.G.; Suzuki, K.; et al. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway. J. Natl. Cancer Inst. 2003, 95, 652–660. [Google Scholar] [CrossRef]
- Suzuki, K.; Fueyo, J.; Krasnykh, V.; Reynolds, P.N.; Curiel, D.T.; Alemany, R. A conditionally replicative adenovirus with enhanced infectivity shows improved oncolytic potency. Clin. Cancer Res. 2001, 7, 120–126. [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Grill, J.; van Beusechem, V.W.; van Der Valk, P.; Dirven, C.M.; Leonhart, A.; Pherai, D.S.; Haisma, H.J.; Pinedo, H.M.; Curiel, D.T.; Gerritsen, W.R. Combined targeting of adenoviruses to integrins and epidermal growth factor receptors increases gene transfer into primary glioma cells and spheroids. Clin. Cancer Res. 2001, 7, 641–650. [Google Scholar]
- Jiang, H.; Gomez-Manzano, C.; Lang, F.F.; Alemany, R.; Fueyo, J. Oncolytic adenoviruses for malignant glioma therapy. Front. Biosci. 2003, 8, 577–588. [Google Scholar] [CrossRef]
- Tabatabai, G.; Wick, W.; Weller, M. Stem cell-mediated gene therapies for malignant gliomas: A promising targeted therapeutic approach? Discov. Med. 2011, 11, 529–536. [Google Scholar]
- Willmon, C.; Harrington, K.; Kottke, T.; Prestwich, R.; Melcher, A.; Vile, R. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol. Ther. 2009, 17, 1667–1676. [Google Scholar] [CrossRef]
- Yong, R.L.; Shinojima, N.; Fueyo, J.; Gumin, J.; Vecil, G.G.; Marini, F.C.; Bogler, O.; Andreeff, M.; Lang, F.F. Human bone marrow-derived mesenchymal stem cells for intravascular delivery of oncolytic adenovirus Delta24-RGD to human gliomas. Cancer Res. 2009, 69, 8932–8940. [Google Scholar] [CrossRef]
- Ahmed, A.U.; Tyler, M.A.; Thaci, B.; Alexiades, N.G.; Han, Y.; Ulasov, I.V.; Lesniak, M.S. A comparative study of neural and mesenchymal stem cell-based carriers for oncolytic adenovirus in a model of malignant glioma. Mol. Pharm. 2011, 8, 1559–1572. [Google Scholar] [CrossRef]
- Xia, X.; Ji, T.; Chen, P.; Li, X.; Fang, Y.; Gao, Q.; Liao, S.; You, L.; Xu, H.; Ma, Q.; et al. Mesenchymal stem cells as carriers and amplifiers in CRAd delivery to tumors. Mol. Cancer 2011, 10, 134. [Google Scholar] [CrossRef]
- Ahmed, A.U.; Thaci, B.; Tobias, A.L.; Auffinger, B.; Zhang, L.; Cheng, Y.; Kim, C.K.; Yunis, C.; Han, Y.; Alexiades, N.G.; et al. A preclinical evaluation of neural stem cell-based cell carrier for targeted antiglioma oncolytic virotherapy. J. Natl. Cancer Inst. 2013, 105, 968–977. [Google Scholar] [CrossRef]
- Tyler, M.A.; Ulasov, I.V.; Sonabend, A.M.; Nandi, S.; Han, Y.; Marler, S.; Roth, J.; Lesniak, M.S. Neural stem cells target intracranial glioma to deliver an oncolytic adenovirus in vivo. Gene Ther. 2009, 16, 262–278. [Google Scholar] [CrossRef]
- Kim, C.K.; Ahmed, A.U.; Auffinger, B.; Ulasov, I.V.; Tobias, A.L.; Moon, K.S.; Lesniak, M.S. N-acetylcysteine amide (NACA) Augments the Therapeutic Effect of Neural Stem Cell.-Based Anti-Glioma Oncolytic Virotherapy. Mol. Ther. 2013, 21, 2063–2073. [Google Scholar] [CrossRef]
- Lamfers, M.; Idema, S.; van Milligen, F.; Schouten, T.; van der Valk, P.; Vandertop, P.; Dirven, C.; Noske, D. Homing properties of adipose-derived stem cells to intracerebral glioma and the effects of adenovirus infection. Cancer Lett. 2009, 274, 78–87. [Google Scholar] [CrossRef]
- Power, A.T.; Wang, J.; Falls, T.J.; Paterson, J.M.; Parato, K.A.; Lichty, B.D.; Stojdl, D.F.; Forsyth, P.A.; Atkins, H.; Bell, J.C. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol. Ther. 2007, 15, 123–130. [Google Scholar] [CrossRef]
- Muthana, M.; Giannoudis, A.; Scott, S.D.; Fang, H.Y.; Coffelt, S.B.; Morrow, F.J.; Murdoch, C.; Burton, J.; Cross, N.; Burke, B.; et al. Use of macrophages to target therapeutic adenovirus to human prostate tumors. Cancer Res. 2011, 71, 1805–1815. [Google Scholar] [CrossRef]
- Wakimoto, H.; Kesari, S.; Farrell, C.J.; Curry, W.T., Jr.; Zaupa, C.; Aghi, M.; Kuroda, T.; Stemmer-Rachamimov, A.; Shah, K.; Liu, T.C.; et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009, 69, 3472–3481. [Google Scholar] [CrossRef]
- Wakimoto, H.; Mohapatra, G.; Kanai, R.; Curry, W.T., Jr.; Yip, S.; Nitta, M.; Patel, A.P.; Barnard, Z.R.; Stemmer-Rachamimov, A.O.; Louis, D.N.; et al. Maintenance of primary tumor phenotype and genotype in glioblastoma stem cells. Neuro Oncol. 2012, 14, 132–144. [Google Scholar] [CrossRef]
- Schaft, N.; Willemsen, R.A.; de Vries, J.; Lankiewicz, B.; Essers, B.W.; Gratama, J.W.; Figdor, C.G.; Bolhuis, R.L.; Debets, R.; Adema, G.J. Peptide fine specificity of anti-glycoprotein 100 CTL is preserved following transfer of engineered TCR alpha beta genes into primary human T lymphocytes. J. Immunol. 2003, 170, 2186–2194. [Google Scholar] [CrossRef]
- Willemsen, R.A.; Sebestyén, Z.; Ronteltap, C.; Berrevoets, C.; Drexhage, J.; Debets, R. CD8 alpha coreceptor to improve TCR gene transfer to treat melanoma: Down-regulation of tumor-specific production of IL-4, IL-5, and IL-10. J. Immunol. 2006, 177, 991–998. [Google Scholar] [CrossRef]
- Schaft, N.; Lankiewicz, B.; Gratama, J.W.; Bolhuis, R.L.; Debets, R. Flexible and sensitive method to functionally validate tumor-specific receptors via activation of NFAT. J. Immunol. Methods 2003, 280, 13–24. [Google Scholar] [CrossRef]
- Ono, H.A.; Le, L.P.; Davydova, J.G.; Gavrikova, T.; Yamamoto, M. Noninvasive visualization of adenovirus replication with a fluorescent reporter in the E3 region. Cancer Res. 2005, 65, 10154–10158. [Google Scholar] [CrossRef]
- He, T.C.; Zhou, S.; da Costa, L.T.; Yu, J.; Kinzler, K.W.; Vogelstein, B. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA 1998, 95, 2509–2514. [Google Scholar]
- De Vrij, J.; van den Hengel, S.K.; Uil, T.G.; Koppers-Lalic, D.; Dautzenberg, I.J.; Stassen, O.M.; Bárcena, M.; Yamamoto, M.; de Ridder, C.M.; Kraaij, R. Enhanced transduction of CAR-negative cells by protein IX-gene deleted adenovirus 5 vectors. Virology 2011, 410, 192–200. [Google Scholar] [CrossRef]
- Fueyo, J.; Gomez-Manzano, C.; Alemany, R.; Lee, P.S.; McDonnell, T.J.; Mitlianga, P.; Shi, Y.X.; Levin, V.A.; Yung, W.K.; Kyritsis, A.P. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 2000, 19, 2–12. [Google Scholar] [CrossRef]
- Fallaux, F.J.; Kranenburg, O.; Cramer, S.J.; Houweling, A.; van Ormondt, H.; Hoeben, R.C.; Van Der Eb, A.J. Characterization of 911: A new helper cell line for the titration and propagation of early region 1-deleted adenoviral vectors. Hum. Gene Ther. 1996, 7, 215–222. [Google Scholar] [CrossRef]
- Lamfers, M.L.; Fulci, L.G.; Chiocca, E.A. Cyclophosphamide increases transgene expression mediated by an oncolytic adenovirus in glioma-bearing mice monitored by bioluminescence imaging. Mol. Ther. 2006, 14, 779–788. [Google Scholar] [CrossRef]
- Mentel, R.; Döpping, G.; Wegner, U.; Seidel, W.; Liebermann, H.; Döhner, L. Adenovirus-receptor interaction with human lymphocytes. J. Med. Virol. 1997, 51, 252–257. [Google Scholar] [CrossRef]
- Leon, R.P.; Hedlund, T.; Meech, S.J.; Li, S.; Schaack, J.; Hunger, S.P.; Duke, R.C.; Degregori, A.J. Adenoviral-mediated gene transfer in lymphocytes. Proc. Natl. Acad. Sci. USA 1998, 95, 13159–13164. [Google Scholar]
- Yotnda, P.; Onishi, H.; Heslop, H.E.; Shayakhmetov, D.; Lieber, A.; Brenner, M.; Davis, A. Efficient infection of primitive hematopoietic stem cells by modified adenovirus. Gene Ther. 2001, 8, 930–937. [Google Scholar] [CrossRef]
- Yotnda, P.; Zompeta, C.; Heslop, H.E.; Andreeff, M.; Brenner, M.K.; Marini, F. Comparison of the efficiency of transduction of leukemic cells by fiber-modified adenoviruses. Hum. Gene Ther. 2004, 15, 1229–1242. [Google Scholar] [CrossRef]
- Shinojima, N.; Hossain, A.; Takezaki, T.; Fueyo, J.; Gumin, J.; Gao, F.; Nwajei, F.; Marini, F.C.; Andreeff, M.; Kuratsu, J.; et al. TGF-beta mediates homing of bone marrow-derived human mesenchymal stem cells to glioma stem cells. Cancer Res. 2013, 73, 2333–2344. [Google Scholar] [CrossRef]
- Gondi, C.S.; Veeravalli, K.K.; Gorantla, B.; Dinh, D.H.; Fassett, D.; Klopfenstein, J.D.; Gujrati, M.; Rao, J.S. Human umbilical cord blood stem cells show PDGF-D-dependent glioma cell tropism in vitro and in vivo. Neuro Oncol. 2010, 12, 453–465. [Google Scholar]
- Lohr, J.; Ratliff, T.; Huppertz, A.; Ge, Y.; Dictus, C.; Ahmadi, R.; Grau, S.; Hiraoka, N.; Eckstein, V.; Ecker, R.C.; et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin. Cancer Res. 2011, 17, 4296–308. [Google Scholar] [CrossRef]
- Huang, J.Y.; Cheng, Y.J.; Lin, Y.P.; Lin, H.C.; Su, C.C.; Juliano, R.; Yang, B.C. Extracellular matrix of glioblastoma inhibits polarization and transmigration of T cells: The role of tenascin-C in immune suppression. J. Immunol. 2010, 185, 1450–1459. [Google Scholar] [CrossRef]
- Qiao, J.; Wang, H.; Kottke, T.; Diaz, R.M.; Willmon, C.; Hudacek, A.; Thompson, J.; Parato, K.; Bell, J.; Naik, J.; et al. Loading of oncolytic vesicular stomatitis virus onto antigen-specific T cells enhances the efficacy of adoptive T-cell therapy of tumors. Gene Ther. 2008, 15, 604–616. [Google Scholar] [CrossRef]
- Kottke, T.; Diaz, R.M.; Kaluza, M.; Pulido, J.; Galivo, F.; Wongthida, P.; Thompson, J.; Willmon, C.; Barber, G.N.; Cheste, J.; et al. Use of biological therapy to enhance both virotherapy and adoptive T-cell therapy for cancer. Mol. Ther. 2008, 16, 1910–1918. [Google Scholar] [CrossRef]
- Zhang, J.G.; Kruse, C.A.; Driggers, L.; Hoa, N.; Wisoff, J.; Allen, J.C.; Zagzag, D.; Newcomb, E.W.; Jadus, M.R. Tumor antigen precursor protein profiles of adult and pediatric brain tumors identify potential targets for immunotherapy. J. Neurooncol. 2008, 88, 65–76. [Google Scholar] [CrossRef]
- Zhang, J.G.; Eguchi, J.; Kruse, C.A.; Gomez, G.G.; Fakhrai, H.; Schroter, S.; Ma, W.; Hoa, N.; Minev, B.; Delgado, C.; et al. Antigenic profiling of glioma cells to generate allogeneic vaccines or dendritic cell-based therapeutics. Clin. Cancer Res. 2007, 13, 566–575. [Google Scholar] [CrossRef]
- Beier, C.P.; Kumar, P.; Meyer, K.; Leukel, P.; Bruttel, V.; Aschenbrenner, I.; Riemenschneider, M.J.; Fragoulis, A.; Rümmele, P.; Lamszus, K.; et al. The cancer stem cell subtype determines immune infiltration of glioblastoma. Stem Cells Dev. 2012, 21, 2753–2761. [Google Scholar] [CrossRef]
- Rutledge, W.C.; Kong, J.; Gao, J.; Gutman, D.A.; Cooper, L.A.; Appin, C.; Park, Y.; Scarpace, L.; Mikkelsen, T.; Cohen, M.L.; Aldape, K.D.; et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin. Cancer Res. 2013, 19, 4951–4960. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Balvers, R.K.; Belcaid, Z.; Van den Hengel, S.K.; Kloezeman, J.; De Vrij, J.; Wakimoto, H.; Hoeben, R.C.; Debets, R.; Leenstra, S.; Dirven, C.; et al. Locally-Delivered T-Cell-Derived Cellular Vehicles Efficiently Track and Deliver Adenovirus Delta24-RGD to Infiltrating Glioma. Viruses 2014, 6, 3080-3096. https://doi.org/10.3390/v6083080
Balvers RK, Belcaid Z, Van den Hengel SK, Kloezeman J, De Vrij J, Wakimoto H, Hoeben RC, Debets R, Leenstra S, Dirven C, et al. Locally-Delivered T-Cell-Derived Cellular Vehicles Efficiently Track and Deliver Adenovirus Delta24-RGD to Infiltrating Glioma. Viruses. 2014; 6(8):3080-3096. https://doi.org/10.3390/v6083080
Chicago/Turabian StyleBalvers, Rutger K., Zineb Belcaid, Sanne K. Van den Hengel, Jenneke Kloezeman, Jeroen De Vrij, Hiroaki Wakimoto, Rob C. Hoeben, Reno Debets, Sieger Leenstra, Clemens Dirven, and et al. 2014. "Locally-Delivered T-Cell-Derived Cellular Vehicles Efficiently Track and Deliver Adenovirus Delta24-RGD to Infiltrating Glioma" Viruses 6, no. 8: 3080-3096. https://doi.org/10.3390/v6083080
APA StyleBalvers, R. K., Belcaid, Z., Van den Hengel, S. K., Kloezeman, J., De Vrij, J., Wakimoto, H., Hoeben, R. C., Debets, R., Leenstra, S., Dirven, C., & Lamfers, M. L. M. (2014). Locally-Delivered T-Cell-Derived Cellular Vehicles Efficiently Track and Deliver Adenovirus Delta24-RGD to Infiltrating Glioma. Viruses, 6(8), 3080-3096. https://doi.org/10.3390/v6083080