Full-Length Genome Analyses of Two New Simian Immunodeficiency Virus (SIV) Strains from Mustached Monkeys (C. Cephus) in Gabon Illustrate a Complex Evolutionary History among the SIVmus/mon/gsn Lineage


 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Non-Human Primate Specimens, Serologic testing, DNA Extractions and SIV PCR Screening
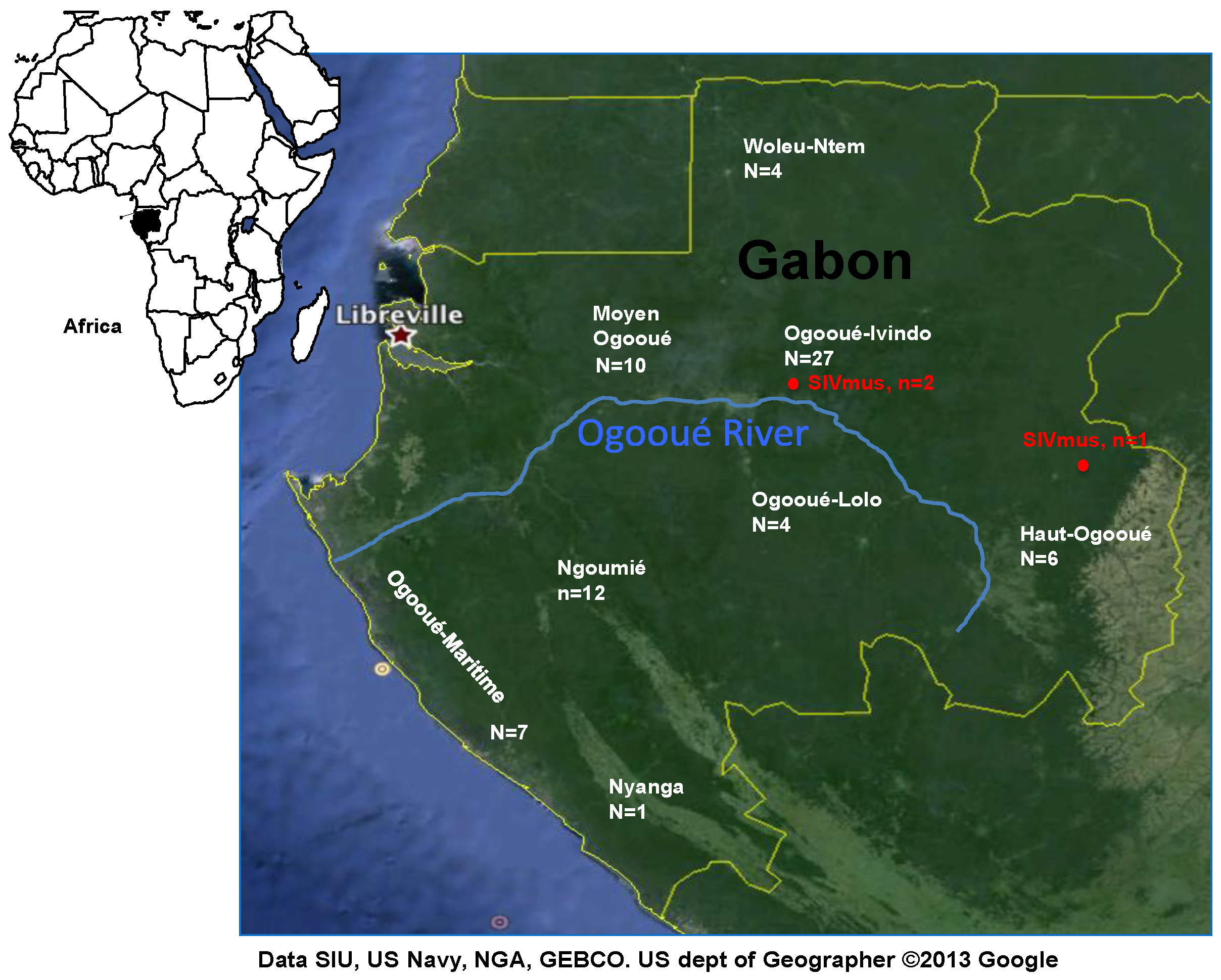
2.2. PCR Amplification and Sequencing of New SIVmus Full-Length Genomes
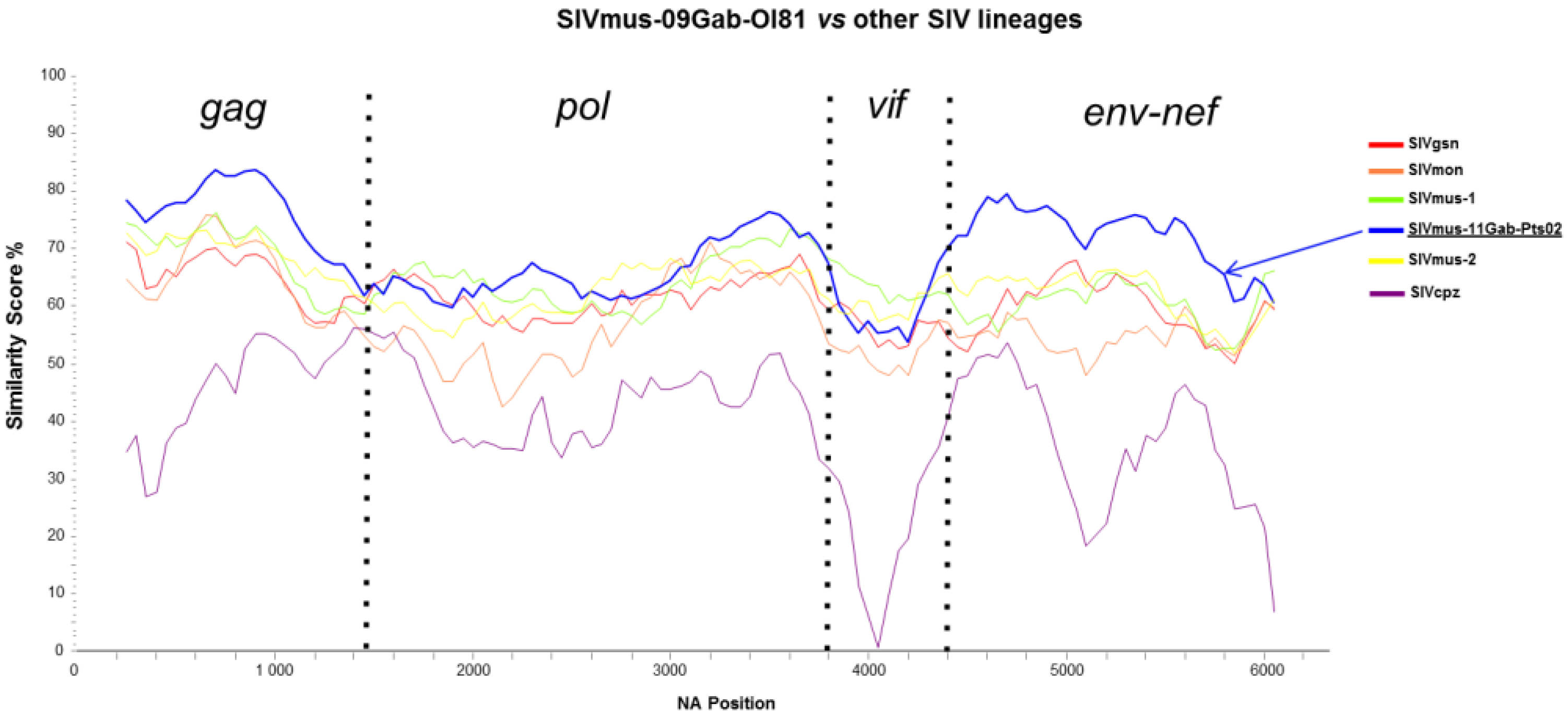
2.3. Sequence Similarity Plots, Bootscan Analyses, and Genetic Identities
2.4. Phylogenetic Analyses
2.5. RNA Secondary Structure and Protein Surface Exposure Predictions
2.6. Nucleotide Sequence Accession Numbers
3. Results
3.1. SIV Antibodies and PCR Screening in Mustached Monkeys
3.2. Molecular Species Confirmation
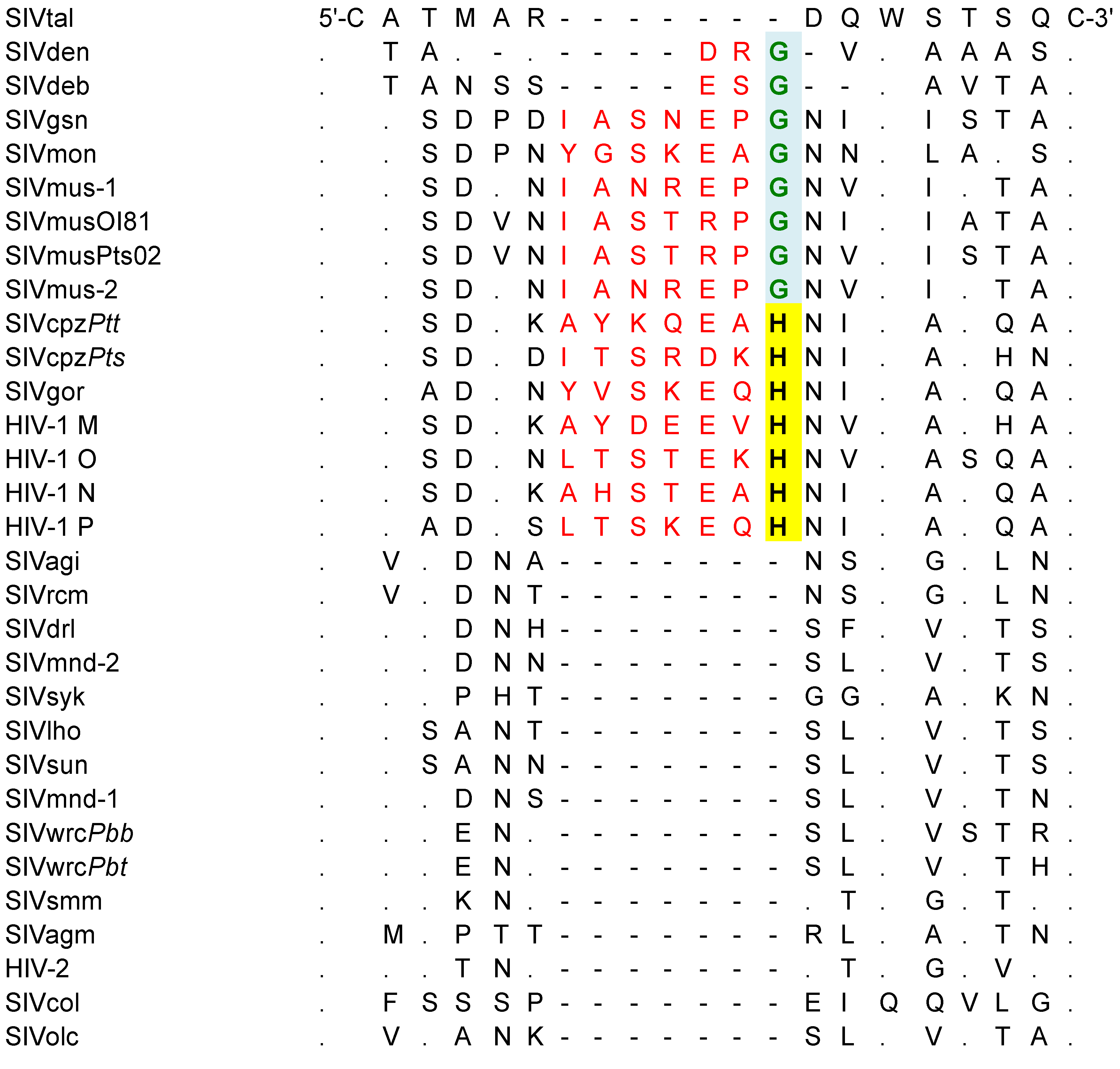
3.3. Genomic Organization and Functional Motifs of New SIVmus Strains
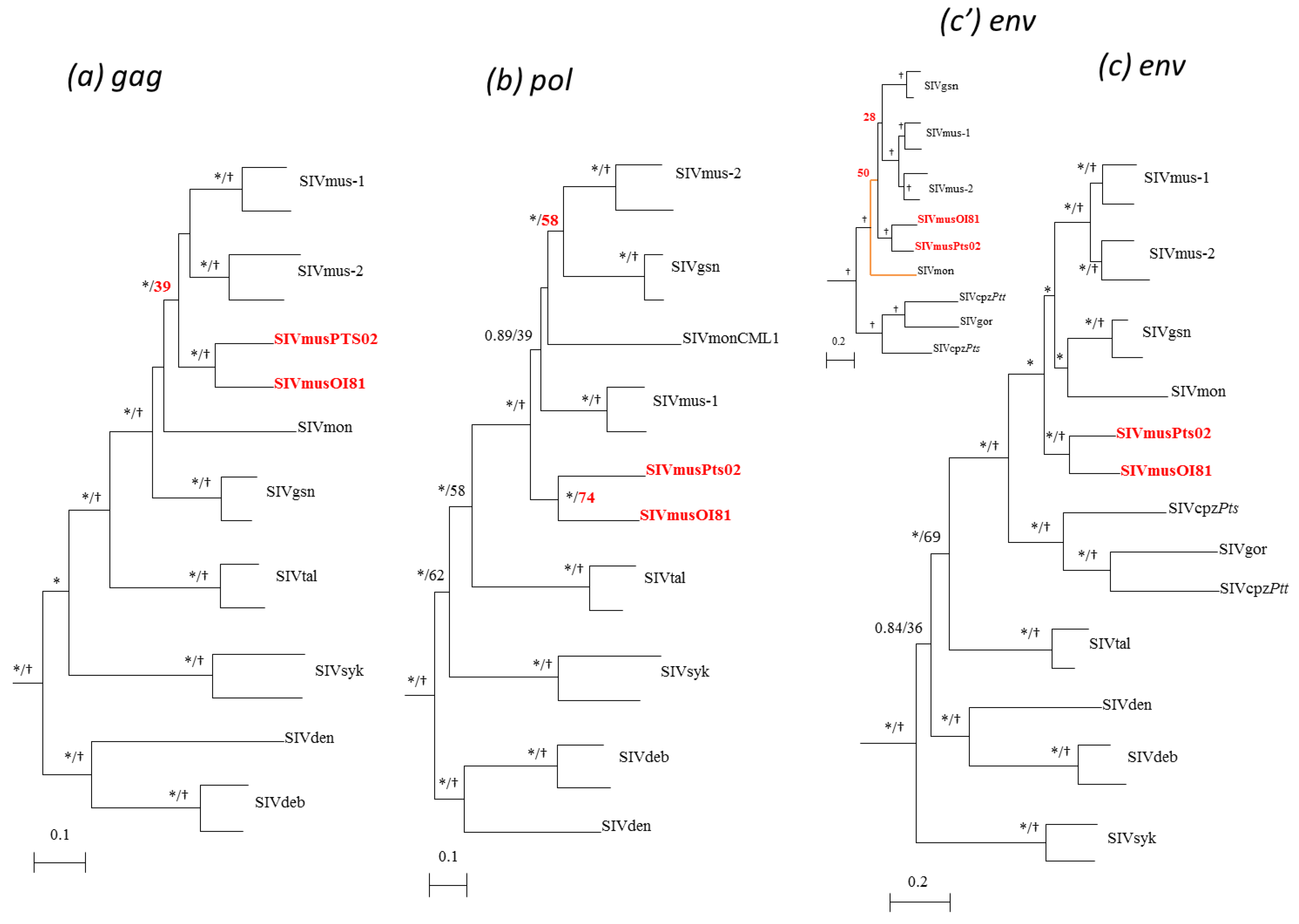
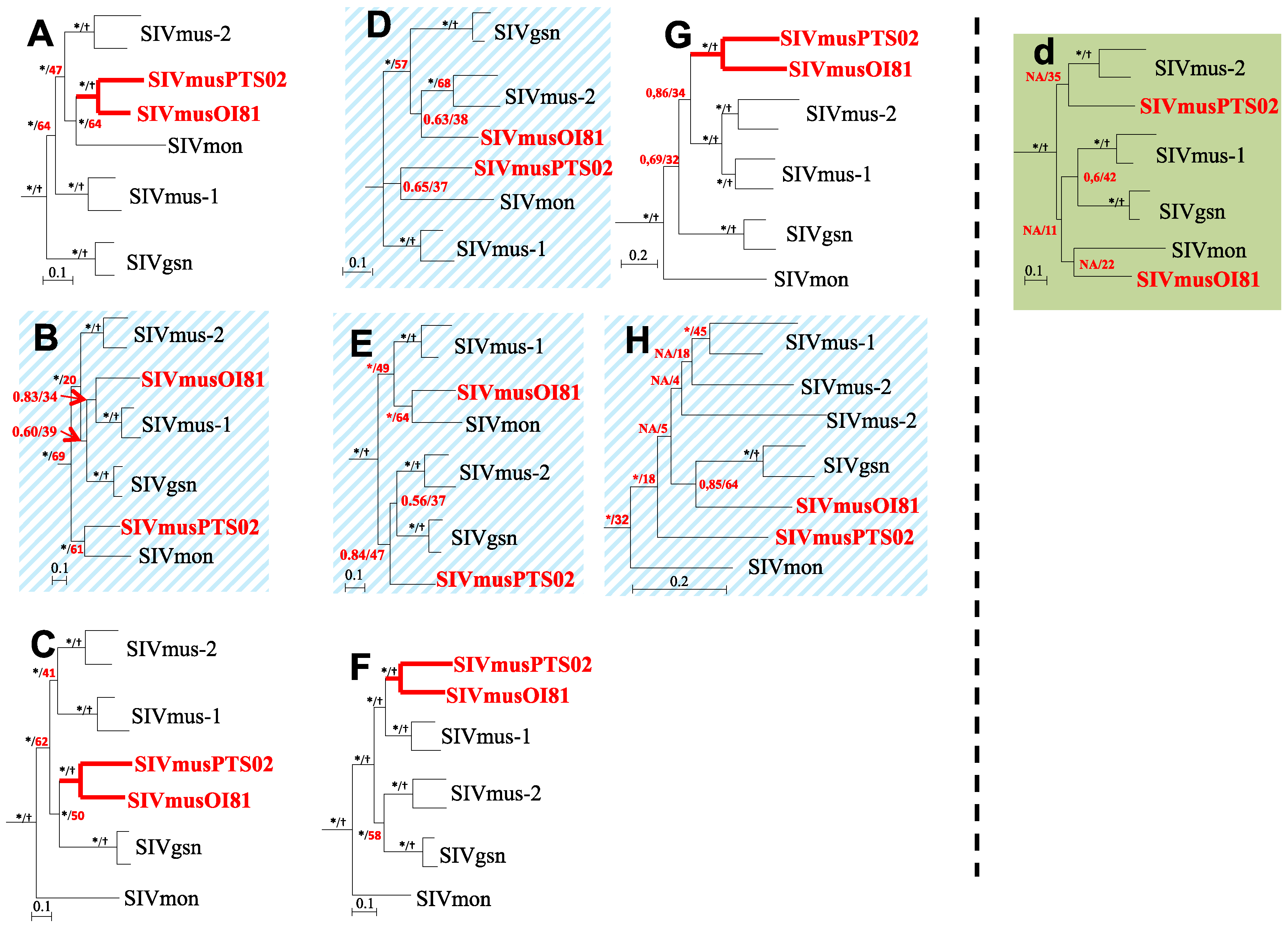
3.4. Phylogenetic Analyses of the New SIVmus Strains
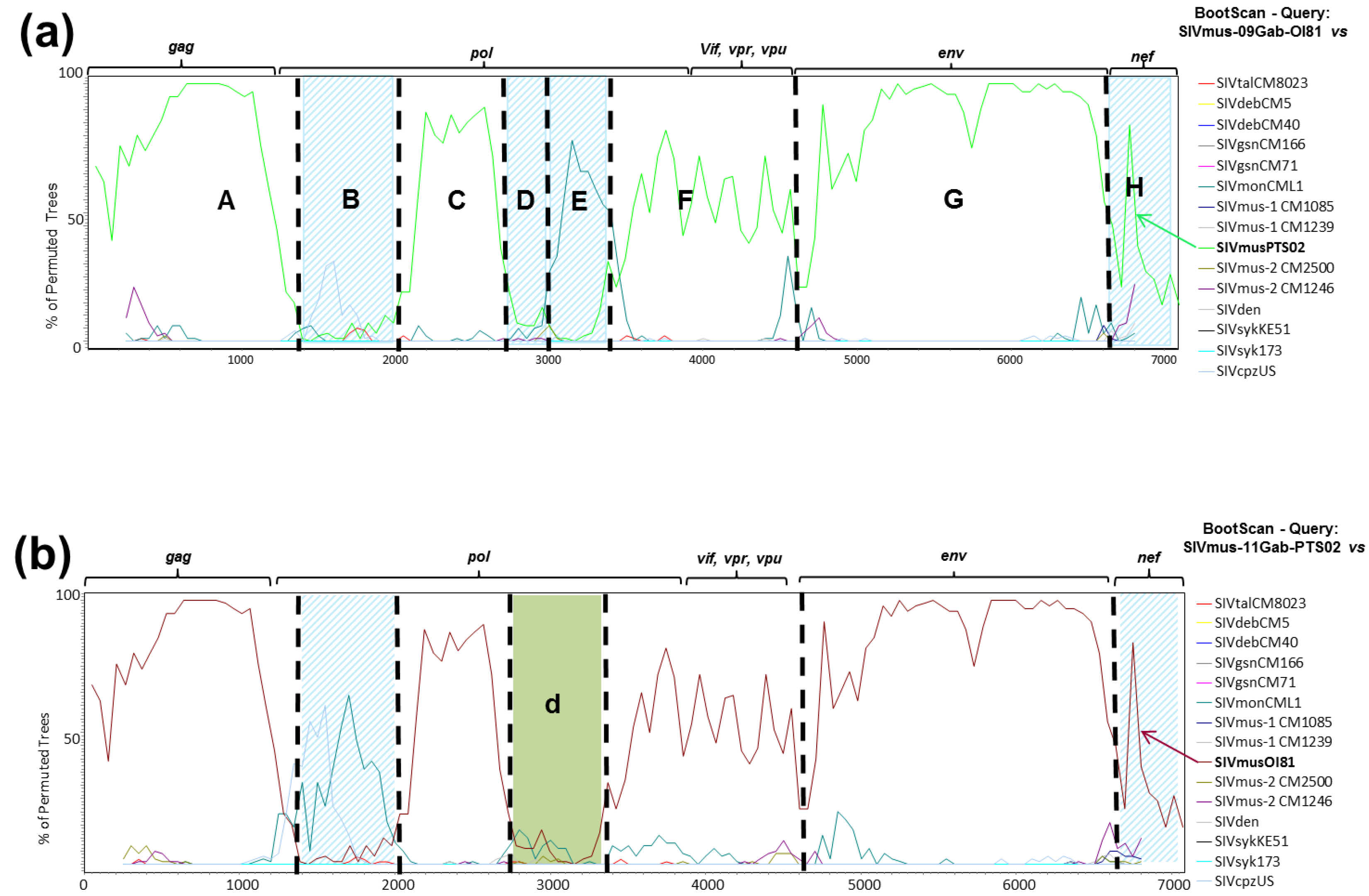

{kind=link}
{kind=link}
{kind=link}
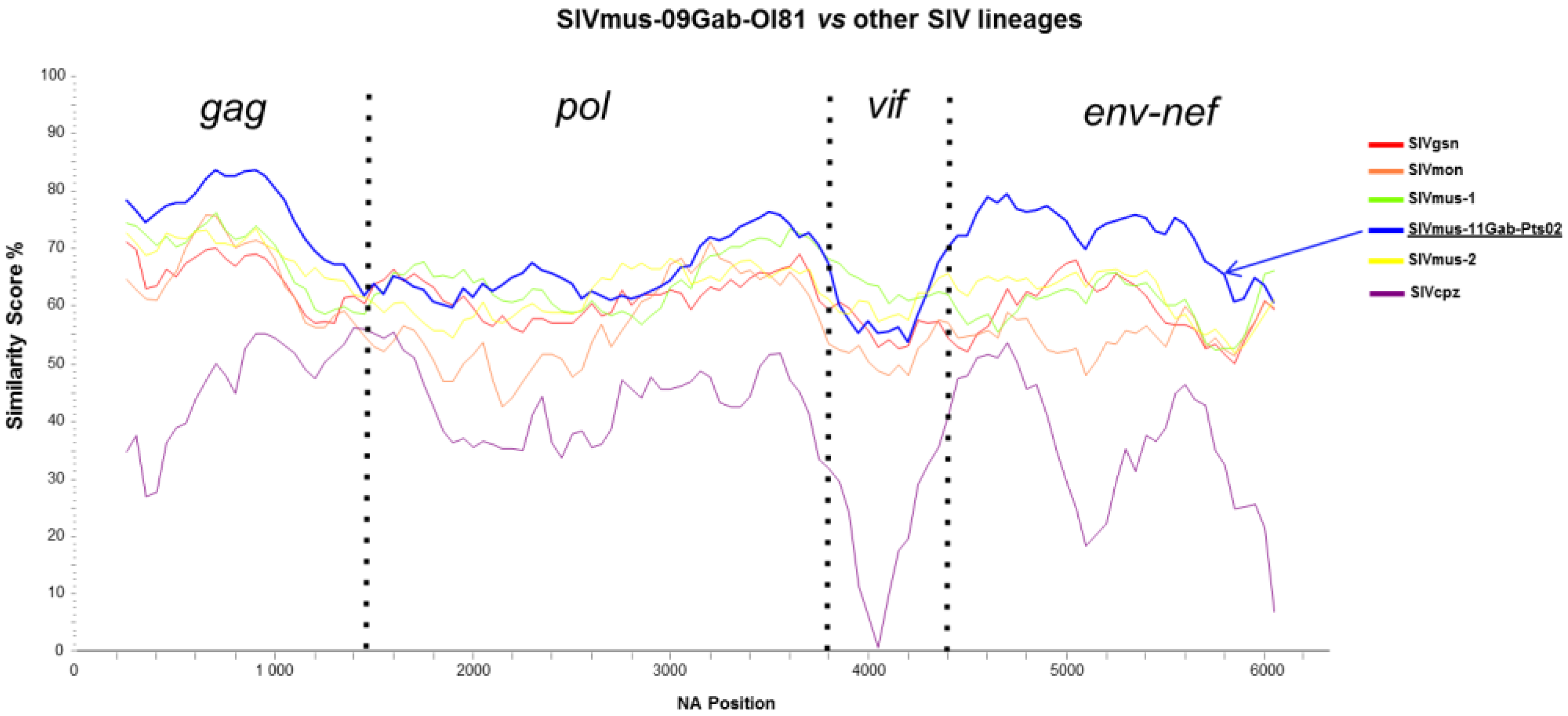{kind=link}
{kind=link}
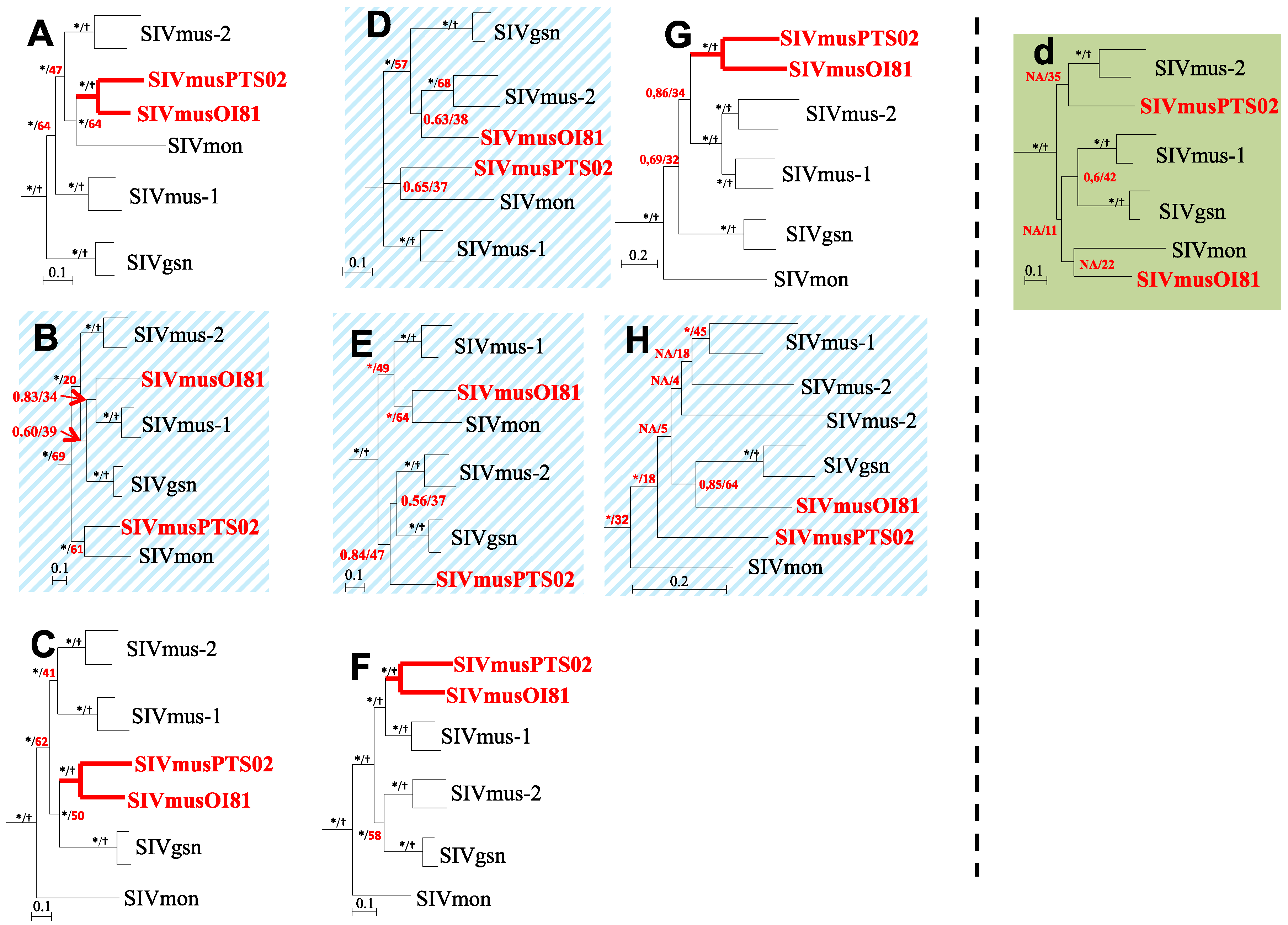{kind=link}
| SIV Strains | Gag | Pol | Env | |||
|---|---|---|---|---|---|---|
| SIVmus | SIVmus | SIVmus | SIVmus | SIVmus | SIVmus | |
| 09GabOI81 | 11GabPts02 | 09GabOI81 | 11GabPts02 | 09GabOI81 | 11GabPts02 | |
| SIVgsnCM166 | 80 | 80 | 74 | 75 | 73 | 74 |
| SIVgsnCM71 | 78 | 77 | 75 | 75 | 74 | 76 |
| SIVmonCML1 | 77 | 75 | 71 | 73 | 69 | 70 |
| SIVmus-1 CM1085 | 81 | 82 | 76 | 74 | 76 | 79 |
| SIVmus-1 CM1239 | 82 | 83 | 75 | 73 | 77 | 78 |
| SIVmusOI81 | 100 | 89 | 100 | 76 | 100 | 86 |
| SIVmusPTS02 | 89 | 100 | 76 | 100 | 86 | 100 |
| SIVmus-2 CM2500 | 84 | 84 | 75 | 72 | 77 | 78 |
| SIVmus-2 CM1246 | 83 | 82 | 72 | 71 | 75 | 76 |
| SIVtal | 75 | 74 | 63 | 65 | 54 | 53 |
| SIVdeb | 67 | 66 | 60 | 61 | 45 | 45 |
| SIVden | 65 | 66 | 62 | 63 | 47 | 46 |
| SIVrcm | 65 | 64 | 57 | 59 | 42 | 40 |
| SIVagi | 64 | 63 | 58 | 60 | 40 | 40 |
| SIVdrl | 66 | 65 | 58 | 60 | 37 | 37 |
| SIVmnd-2 | 64 | 63 | 57 | 59 | 38 | 39 |
| SIVsyk | 68 | 66 | 61 | 61 | 49 | 49 |
| SIVcpzPts | 57 | 57 | 57 | 59 | 57 | 56 |
| SIVlho | 52 | 51 | 55 | 55 | 37 | 37 |
| SIVsun | 53 | 54 | 54 | 54 | 36 | 36 |
| SIVmnd-1 | 56 | 56 | 55 | 55 | 36 | 35 |
| SIVwrcPbb | 49 | 50 | 56 | 55 | 35 | 35 |
| SIVwrcPbt | 50 | 50 | 54 | 55 | 36 | 36 |
| SIVsmm | 61 | 62 | 58 | 58 | 42 | 41 |
| SIVagm | 63 | 63 | 56 | 58 | 44 | 44 |
| SIVcpzPtt | 59 | 59 | 59 | 59 | 52 | 52 |
| SIVgor | 48 | 49 | 58 | 59 | 53 | 52 |
| HIV-2 | 62 | 63 | 57 | 57 | 42 | 41 |
| SIVcol | 48 | 48 | 52 | 53 | 35 | 35 |
| SIVolc | 50 | 50 | 51 | 50 | 36 | 38 |
| SIV Strains | Fragment | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D * | E * | d ** | F | G | H | |||
| Intra | SIVgsn (CM166 vs. CM71) | 89 | 90 | 89 | 91 | 88 | 89 | 90 | 87 | 85 | |
| host | SIVmus-1 (CM1085 vs. CM1239) | 85 | 83 | 81 | 84 | 82 | 82 | 83 | 82 | 79 | |
| species | SIVmus-2 (CM2500 vs. CM1246) | 81 | 78 | 78 | 76 | 81 | 78 | 80 | 79 | 70 | |
| SIVmus-Gab (GabOI81 vs. GabPts02) | 81 | 70 | 72 | 70 | 73 | 71 | 74 | 76 | 73 | ||
| SIVmus-09Gab-OI81 vs. | |||||||||||
| Inter | SIVgsn | 74 | 72.5 | 67.5 | 72.5 | 72 | 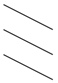 | 71.5 | 71 | 72 | |
| host | SIVmus-1 | 76.5 | 71 | 62 | 70.5 | 73.5 | 75.5 | 70 | 74.5 | ||
| species | SIVmus-2 | 75.5 | 69.5 | 68.5 | 75 | 72.5 | 70.5 | 70.5 | 70 | ||
| SIVmon | 73 | 66 | 64 | 67 | 75 | 69 | 68 | 72 | |||
| SIVmus-11Gab-Pts02 vs. | |||||||||||
| SIVgsn | 74.5 | 71 | 68 | 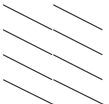 | 74.5 | 71 | 68 | 72 | |||
| SIVmus-1 | 75 | 70 | 66 | 70.5 | 72.5 | 70 | 74.5 | ||||
| SIVmus-2 | 75.5 | 68.5 | 70 | 72 | 71 | 70.5 | 71.5 | ||||
| SIVmon | 72 | 71 | 64 | 68 | 69 | 68 | 70 | ||||
4. Discussion
5. Conclusions
Supplementary Files
Acknowledgments
Authors Contributions
Conflicts of Interest
References and Notes
- Hahn, B.H.; Shaw, G.M.; de Cock, K.M.; Sharp, P.M. AIDS as a zoonosis: Scientific and public health implications. Science 2000, 287, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, S.; Peeters, M. Cross-species transmission of simian retroviruses: How and why they could lead to the emergence of new diseases in the human population. AIDS 2012, 26, 659–673. [Google Scholar] [CrossRef] [PubMed]
- Allan, J.S.; Short, M.; Taylor, M.E.; Su, S.; Hirsch, V.M.; Johnson, P.R.; Shaw, G.M.; Hahn, B.H. Species-specific diversity among simian immunodeficiency viruses from african green monkeys. J. Virol. 1991, 65, 2816–2828. [Google Scholar] [PubMed]
- Beer, B.E.; Bailes, E.; Goeken, R.; Dapolito, G.; Coulibaly, C.; Norley, S.G.; Kurth, R.; Gautier, J.P.; Gautier-Hion, A.; Vallet, D.; et al. Simian immunodeficiency virus (SIV) from sun-tailed monkeys (Cercopithecus solatus): Evidence for host-dependent evolution of SIV within the C. Lhoesti superspecies. J. Virol. 1999, 73, 7734–7744. [Google Scholar] [PubMed]
- Bibollet-Ruche, F.; Bailes, E.; Gao, F.; Pourrut, X.; Barlow, K.L.; Clewley, J.P.; Mwenda, J.M.; Langat, D.K.; Chege, G.K.; McClure, H.M.; et al. New simian immunodeficiency virus infecting de brazza’s monkeys (Cercopithecus neglectus): Evidence for a Cercopithecus monkey virus clade. J. Virol. 2004, 78, 7748–7762. [Google Scholar] [CrossRef] [PubMed]
- Souquiere, S.; Bibollet-Ruche, F.; Robertson, D.L.; Makuwa, M.; Apetrei, C.; Onanga, R.; Kornfeld, C.; Plantier, J.C.; Gao, F.; Abernethy, K.; et al. Wild Mandrillus sphinx are carriers of two types of lentivirus. J. Virol. 2001, 75, 7086–7096. [Google Scholar] [CrossRef] [PubMed]
- Aghokeng, A.F.; Bailes, E.; Loul, S.; Courgnaud, V.; Mpoudi-Ngolle, E.; Sharp, P.M.; Delaporte, E.; Peeters, M. Full-length sequence analysis of SIVmus in wild populations of mustached monkeys (Cercopithecus cephus) from Cameroon provides evidence for two co-circulating SIVmus lineages. Virology 2007, 360, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Liegeois, F.; Boue, V.; Mouacha, F.; Butel, C.; Ondo, B.M.; Pourrut, X.; Leroy, E.; Peeters, M.; Rouet, F. New STLV-3 strains and a divergent SIVmus strain identified in non-human primate bushmeat in Gabon. Retrovirology 2012, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Bibollet-Ruche, F.; Galat-Luong, A.; Cuny, G.; Sarni-Manchado, P.; Galat, G.; Durand, J.P.; Pourrut, X.; Veas, F. Simian immunodeficiency virus infection in a patas monkey (Erythrocebus patas): Evidence for cross-species transmission from african green monkeys (Cercopithecus aethiops sabaeus) in the wild. J. Gen. Virol. 1996, 77, 773–781. [Google Scholar] [PubMed]
- Jin, M.J.; Rogers, J.; Phillips-Conroy, J.E.; Allan, J.S.; Desrosiers, R.C.; Shaw, G.M.; Sharp, P.M.; Hahn, B.H. Infection of a yellow baboon with simian immunodeficiency virus from african green monkeys: Evidence for cross-species transmission in the wild. J. Virol. 1994, 68, 8454–8460. [Google Scholar] [PubMed]
- Van Rensburg, E.J.; Engelbrecht, S.; Mwenda, J.; Laten, J.D.; Robson, B.A.; Stander, T.; Chege, G.K. Simian immunodeficiency viruses (SIVs) from eastern and southern Africa: Detection of a SIVagm variant from a chacma baboon. J. Gen. Virol. 1998, 79, 1809–1814. [Google Scholar] [PubMed]
- Courgnaud, V.; Abela, B.; Pourrut, X.; Mpoudi-Ngole, E.; Loul, S.; Delaporte, E.; Peeters, M. Identification of a new simian immunodeficiency virus lineage with a vpu gene present among different Cercopithecus monkeys (C. mona, C. cephus, and C. nictitans) from Cameroon. J. Virol. 2003, 77, 12523–12534. [Google Scholar] [CrossRef] [PubMed]
- Bailes, E.; Gao, F.; Bibollet-Ruche, F.; Courgnaud, V.; Peeters, M.; Marx, P.A.; Hahn, B.H.; Sharp, P.M. Hybrid origin of SIV in chimpanzees. Science 2003, 300, 1713. [Google Scholar] [PubMed]
- Beer, B.E.; Foley, B.T.; Kuiken, C.L.; Tooze, Z.; Goeken, R.M.; Brown, C.R.; Hu, J.; st Claire, M.; Korber, B.T.; Hirsch, V.M. Characterization of novel simian immunodeficiency viruses from red-capped mangabeys from Nigeria (SIVrcmng409 and -ng411). J. Virol. 2001, 75, 12014–12027. [Google Scholar] [CrossRef] [PubMed]
- Schindler, M.; Munch, J.; Kutsch, O.; Li, H.; Santiago, M.L.; Bibollet-Ruche, F.; Muller-Trutwin, M.C.; Novembre, F.J.; Peeters, M.; Courgnaud, V.; et al. Nef-mediated suppression of T cell activation was lost in a lentiviral lineage that gave rise to HIV-1. Cell 2006, 125, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.; Seharaseyon, J.; Dong, P.; Bour, S.; Marban, E. Mutual functional destruction of HIV-1 vpu and host task-1 channel. Mol. Cell. 2004, 14, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Kingdon, J. The Kingdon Field Guide to African Mammals; Princeton University Press: Princeton, NJ, USA, 1997. [Google Scholar]
- Groves, C. Primate taxonomy. In Smithsonian Series in Comparative Evolutionary Biology; Smithsonian Institution Press: Washington, DC, USA, 2001. [Google Scholar]
- Van der Kuyl, A.C.; Dekker, J.T.; Goudsmit, J. Primate genus Miopithecus: Evidence for the existence of species and subspecies of dwarf guenons based on cellular and endogenous viral sequences. Mol. Phylogenet Evol. 2000, 14, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Basic Local Alignment Search Tool: Blast. Available online: http://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 12 November 2012).
- Simon, F.; Souquiere, S.; Damond, F.; Kfutwah, A.; Makuwa, M.; Leroy, E.; Rouquet, P.; Berthier, J.L.; Rigoulet, J.; Lecu, A.; et al. Synthetic peptide strategy for the detection of and discrimination among highly divergent primate lentiviruses. AIDS Res. Hum. Retrovir. 2001, 17, 937–952. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.; Courgnaud, V.; Abela, B.; Auzel, P.; Pourrut, X.; Bibollet-Ruche, F.; Loul, S.; Liegeois, F.; Butel, C.; Koulagna, D.; et al. Risk to human health from a plethora of simian immunodeficiency viruses in primate bushmeat. Emerg. Infect. Dis. 2002, 8, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Liegeois, F.; Courgnaud, V.; Switzer, W.M.; Murphy, H.W.; Loul, S.; Aghokeng, A.; Pourrut, X.; Mpoudi-Ngole, E.; Delaporte, E.; Peeters, M. Molecular characterization of a novel simian immunodeficiency virus lineage (SIVtal) from northern talapoins (Miopithecus ogouensis). Virology 2006, 349, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Liegeois, F.; Lafay, B.; Formenty, P.; Locatelli, S.; Courgnaud, V.; Delaporte, E.; Peeters, M. Full-length genome characterization of a novel simian immunodeficiency virus lineage (SIVolc) from olive colobus (Procolobus verus) and new SIVwrcPbb strains from western red colobus (Piliocolobus badius badius) from the tai forest in Ivory Coast. J. Virol. 2009, 83, 428–439. [Google Scholar] [PubMed]
- Liegeois, F.; Butel, C.; Mouinga-Ondeme, A.; Verrier, D.; Motsch, P.; Gonzalez, J.P.; Peeters, M.; Rouet, F.; Onanga, R. Full-length genome sequence of a simian immunodeficiency virus from a wild-captured sun-tailed monkey in Gabon provides evidence for a species-specific monophyletic SIVsun lineage. AIDS Res. Hum. Retrovir. 2011, 27, 1237–1241. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. Mega5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The clustal_x windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. Mrbayes: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of phyml 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Tracer. v1.4. Available online: http://beast.bio.ed.ac.uk/Tracer (accessed on 15 January 2013).
- Milne, I.; Lindner, D.; Bayer, M.; Husmeier, D.; McGuire, G.; Marshall, D.F.; Wright, F. Topali v2: A rich graphical interface for evolutionary analyses of multiple alignments on HPC clusters and multi-core desktops. Bioinformatics 2009, 25, 126–127. [Google Scholar] [PubMed]
- Xia, X. Dambe5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evolut. 2013, 30, 1720–1728. [Google Scholar] [CrossRef]
- Petersen, B.; Petersen, T.N.; Andersen, P.; Nielsen, M.; Lundegaard, C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 2009, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, S.; Lafay, B.; Liegeois, F.; Ting, N.; Delaporte, E.; Peeters, M. Full molecular characterization of a simian immunodeficiency virus, SIVwrcPbt from temminck's red colobus (Piliocolobus badius temminckii) from Abuko nature reserve, the Gambia. Virology 2008, 376, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Telfer, P.; Souquiere, S.; Hunter, M.; Coleman, C.A.; Metzger, M.J.; Reed, P.; Makuwa, M.; Hearn, G.; Honarvar, S.; et al. Island biogeography reveals the deep history of SIV. Science 2010, 329, 1487. [Google Scholar] [PubMed]
- Suzuki, Y.; Glazko, G.V.; Nei, M. Overcredibility of molecular phylogenies obtained by bayesian phylogenetics. Proc. Natl. Acad. Sci. USA 2002, 99, 16138–16143. [Google Scholar] [CrossRef] [PubMed]
- Aghokeng, A.F.; Ayouba, A.; Mpoudi-Ngole, E.; Loul, S.; Liegeois, F.; Delaporte, E.; Peeters, M. Extensive survey on the prevalence and genetic diversity of SIVs in primate bushmeat provides insights into risks for potential new cross-species transmissions. Infect. Genet. Evolut. 2010, 10, 386–396. [Google Scholar] [CrossRef]
- Gautier-Hion, A.; Colyn, M.; Gauthier, J.P. Histoire Naturelle Des Primates d’Afrique Centrale; Libreville Ecofac; Backhuys Publishers: Netherlands, UK, 1999. [Google Scholar]
- Lauck, M.; Switzer, M.W.; Sibley, D.S.; Hyeroba, D.; Tumukunde, A.; Geoffrey Weny, G.; Taylor, B.; Shankar, A.; Ting, N.; et al. Discovery and full genome characterization of two highly divergent simian immunodeficiency viruses infecting black-and-white colobus monkeys (Colobus. guereza) in Kibale National Park, Uganda. Retrovirology 2013, 10, 107. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liégeois, F.; Schmidt, F.; Boué, V.; Butel, C.; Mouacha, F.; Ngari, P.; Ondo, B.M.; Leroy, E.; Heeney, J.L.; Delaporte, E.; et al. Full-Length Genome Analyses of Two New Simian Immunodeficiency Virus (SIV) Strains from Mustached Monkeys (C. Cephus) in Gabon Illustrate a Complex Evolutionary History among the SIVmus/mon/gsn Lineage. Viruses 2014, 6, 2880-2898. https://doi.org/10.3390/v6072880
Liégeois F, Schmidt F, Boué V, Butel C, Mouacha F, Ngari P, Ondo BM, Leroy E, Heeney JL, Delaporte E, et al. Full-Length Genome Analyses of Two New Simian Immunodeficiency Virus (SIV) Strains from Mustached Monkeys (C. Cephus) in Gabon Illustrate a Complex Evolutionary History among the SIVmus/mon/gsn Lineage. Viruses. 2014; 6(7):2880-2898. https://doi.org/10.3390/v6072880
Chicago/Turabian StyleLiégeois, Florian, Fabian Schmidt, Vanina Boué, Christelle Butel, Fatima Mouacha, Paul Ngari, Bertrand Mve Ondo, Eric Leroy, Jonathan L. Heeney, Eric Delaporte, and et al. 2014. "Full-Length Genome Analyses of Two New Simian Immunodeficiency Virus (SIV) Strains from Mustached Monkeys (C. Cephus) in Gabon Illustrate a Complex Evolutionary History among the SIVmus/mon/gsn Lineage" Viruses 6, no. 7: 2880-2898. https://doi.org/10.3390/v6072880
APA StyleLiégeois, F., Schmidt, F., Boué, V., Butel, C., Mouacha, F., Ngari, P., Ondo, B. M., Leroy, E., Heeney, J. L., Delaporte, E., Peeters, M., & Rouet, F. (2014). Full-Length Genome Analyses of Two New Simian Immunodeficiency Virus (SIV) Strains from Mustached Monkeys (C. Cephus) in Gabon Illustrate a Complex Evolutionary History among the SIVmus/mon/gsn Lineage. Viruses, 6(7), 2880-2898. https://doi.org/10.3390/v6072880