The Role of the Coat Protein A-Domain in P22 Bacteriophage Maturation

Abstract
:1. Introduction
2. Materials and Methods
2.1. Producing P22 Procapsids
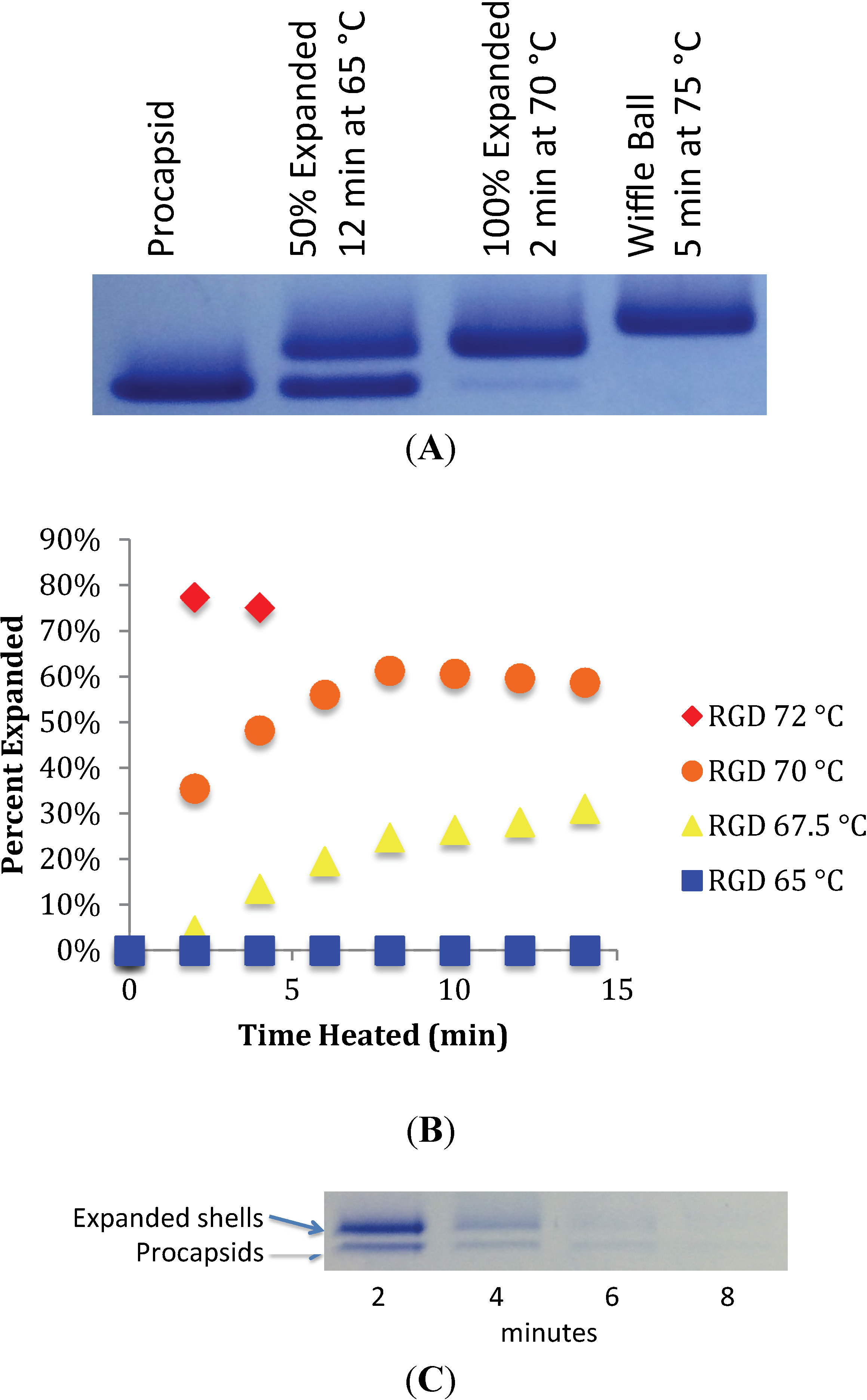
2.2. Heat Expansion Assay
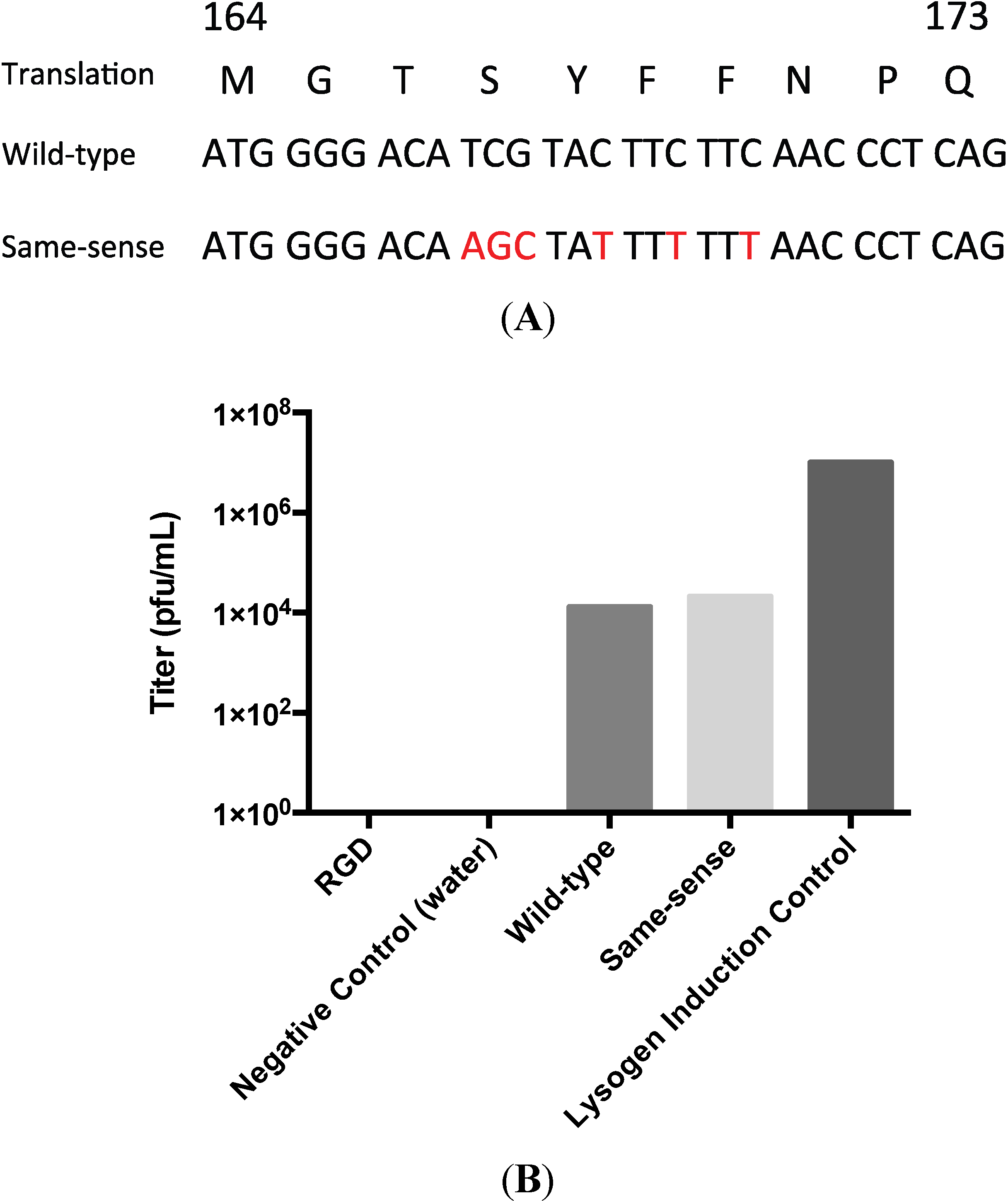
2.3. Recombineering
2.4. Phage Induction and Titering
3. Results
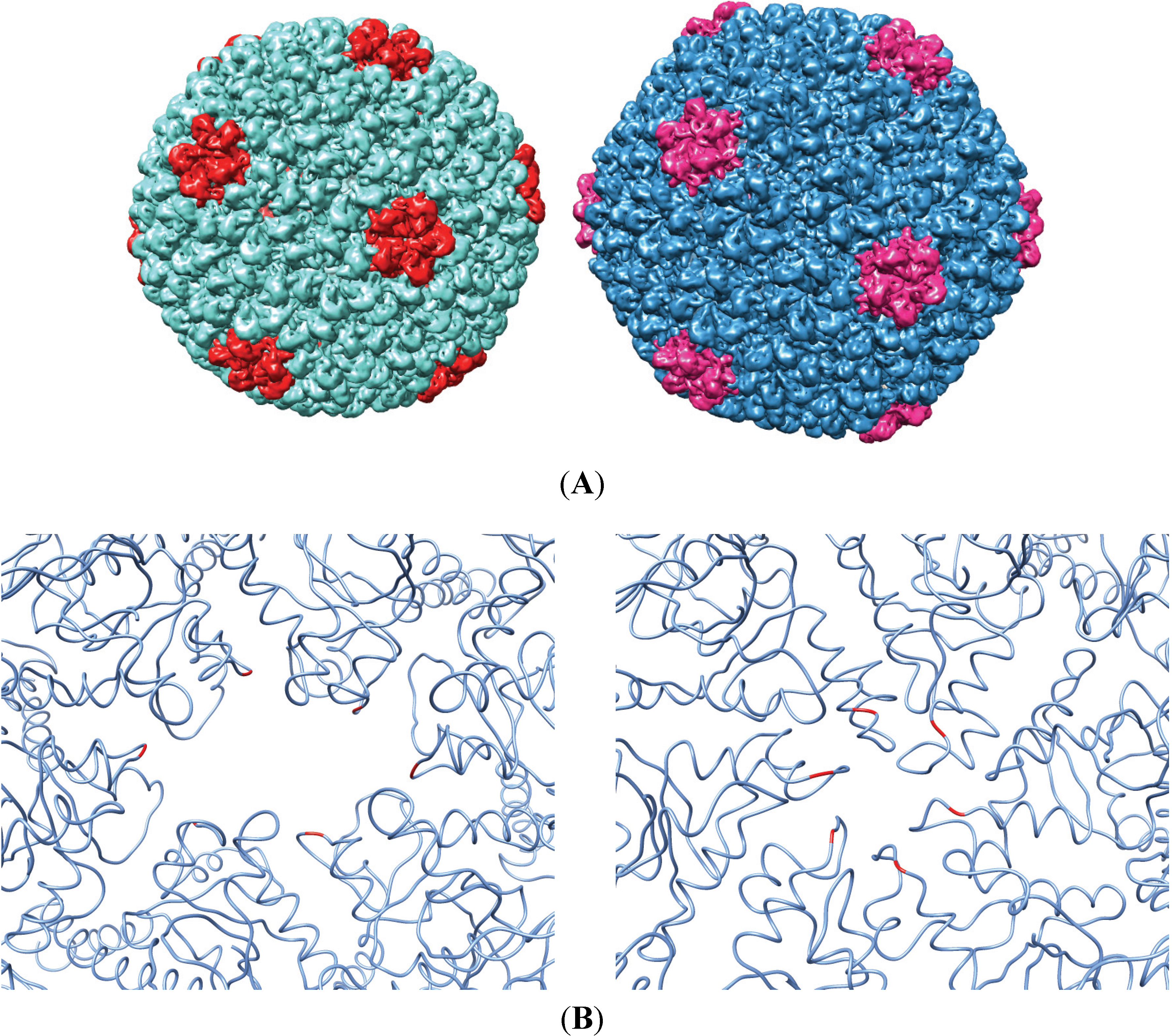
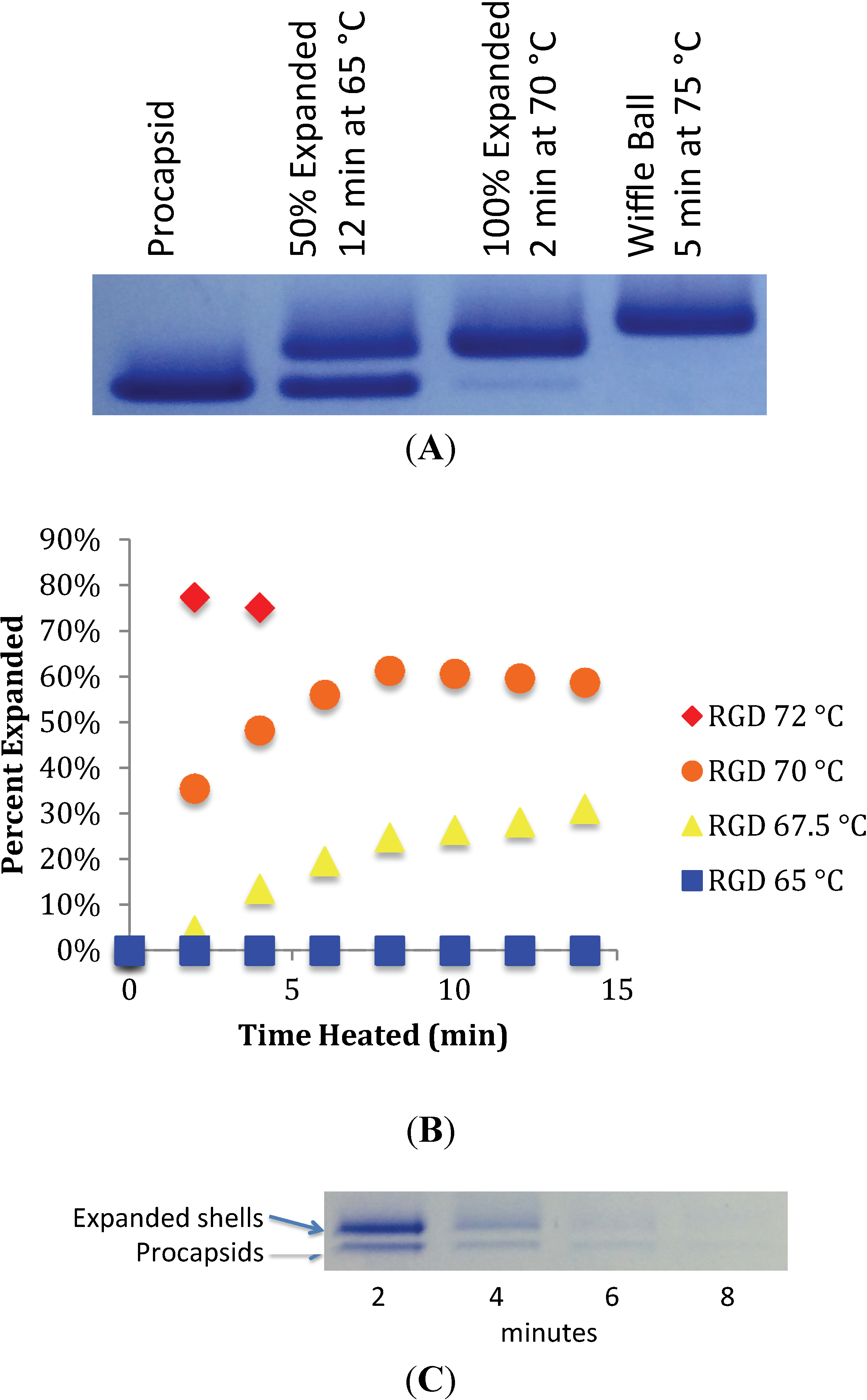
3.1. In Vitro Maturation by Heat Expansion
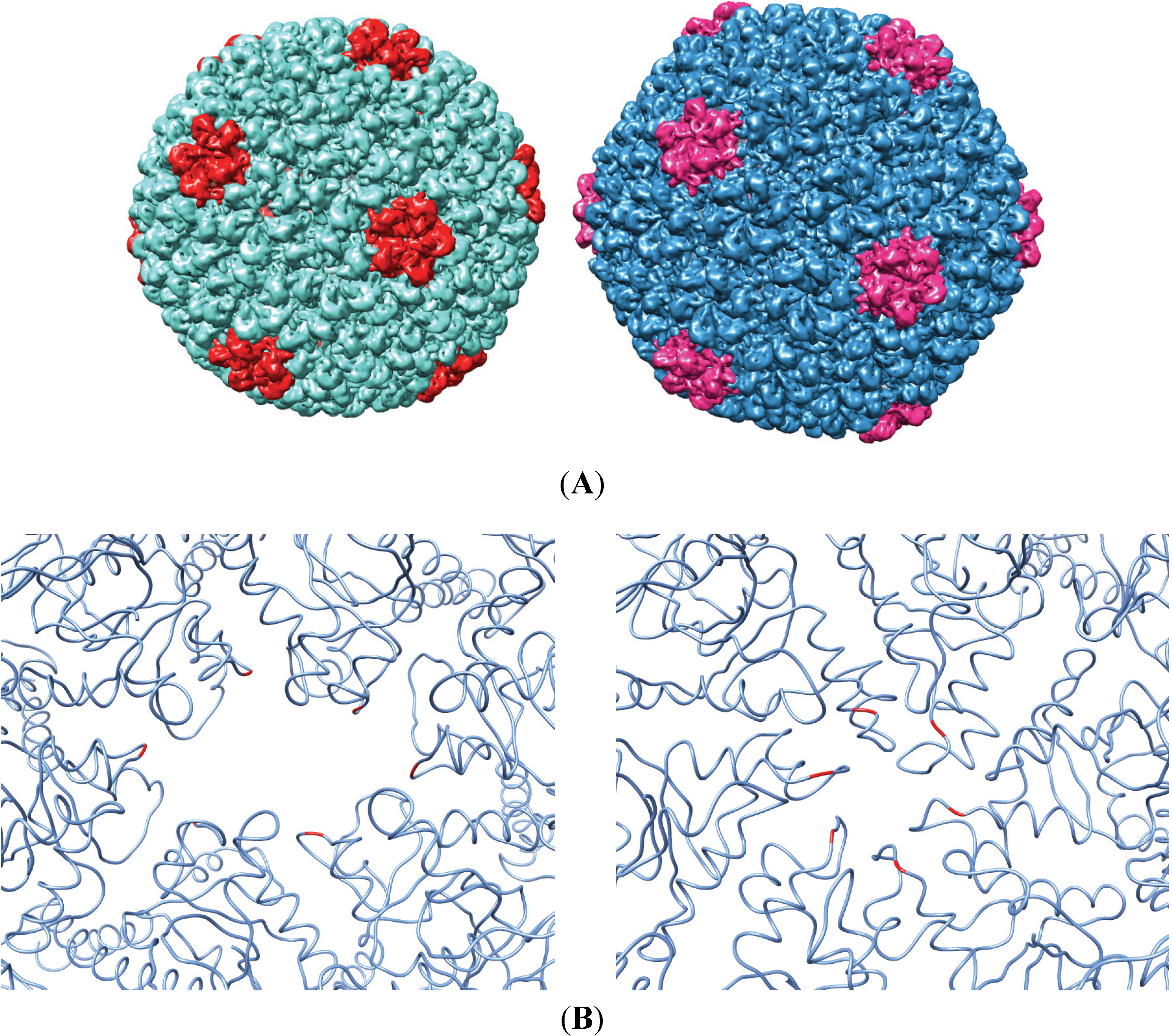
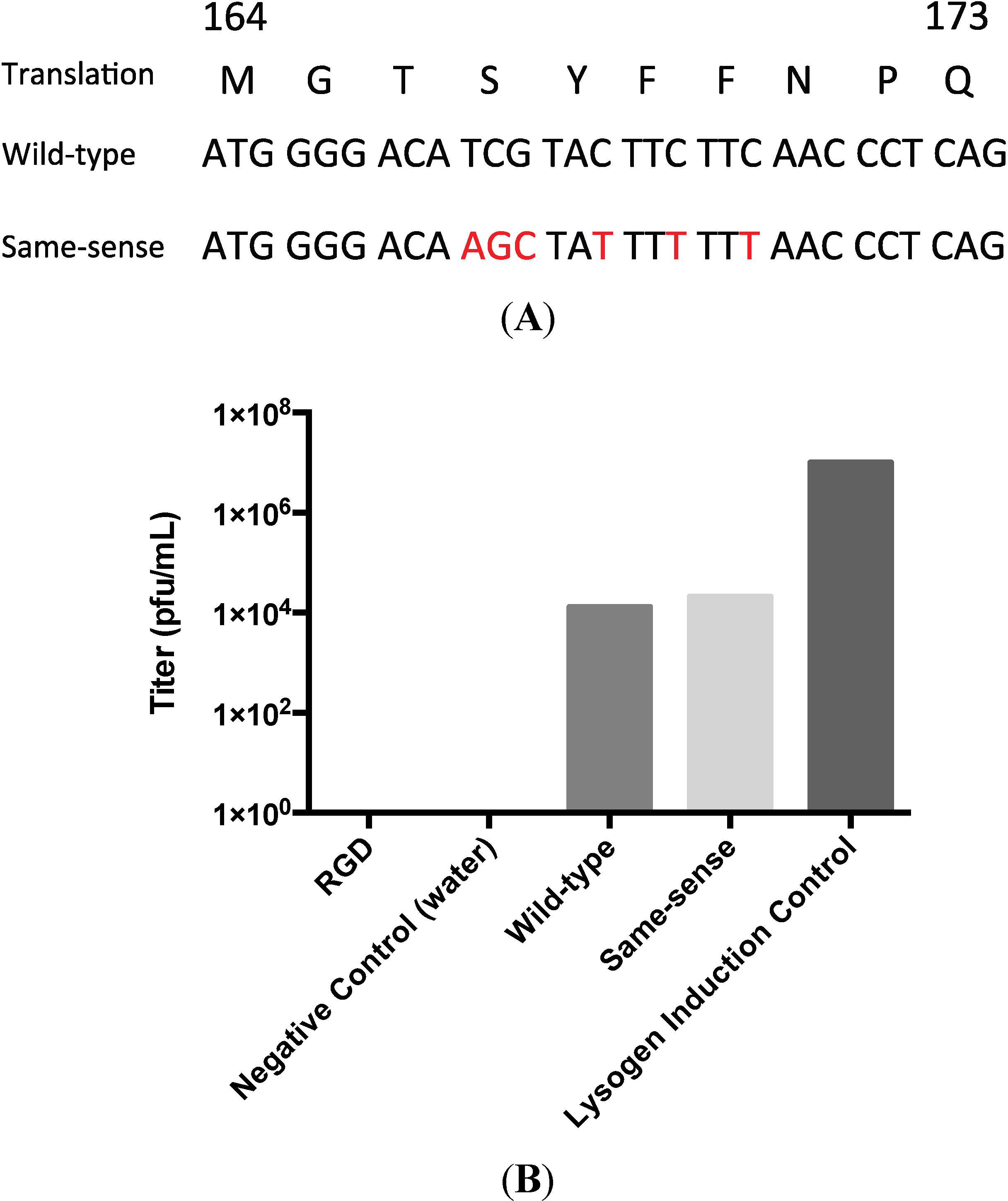
3.2. Testing the A-Loop Insertion in Vivo
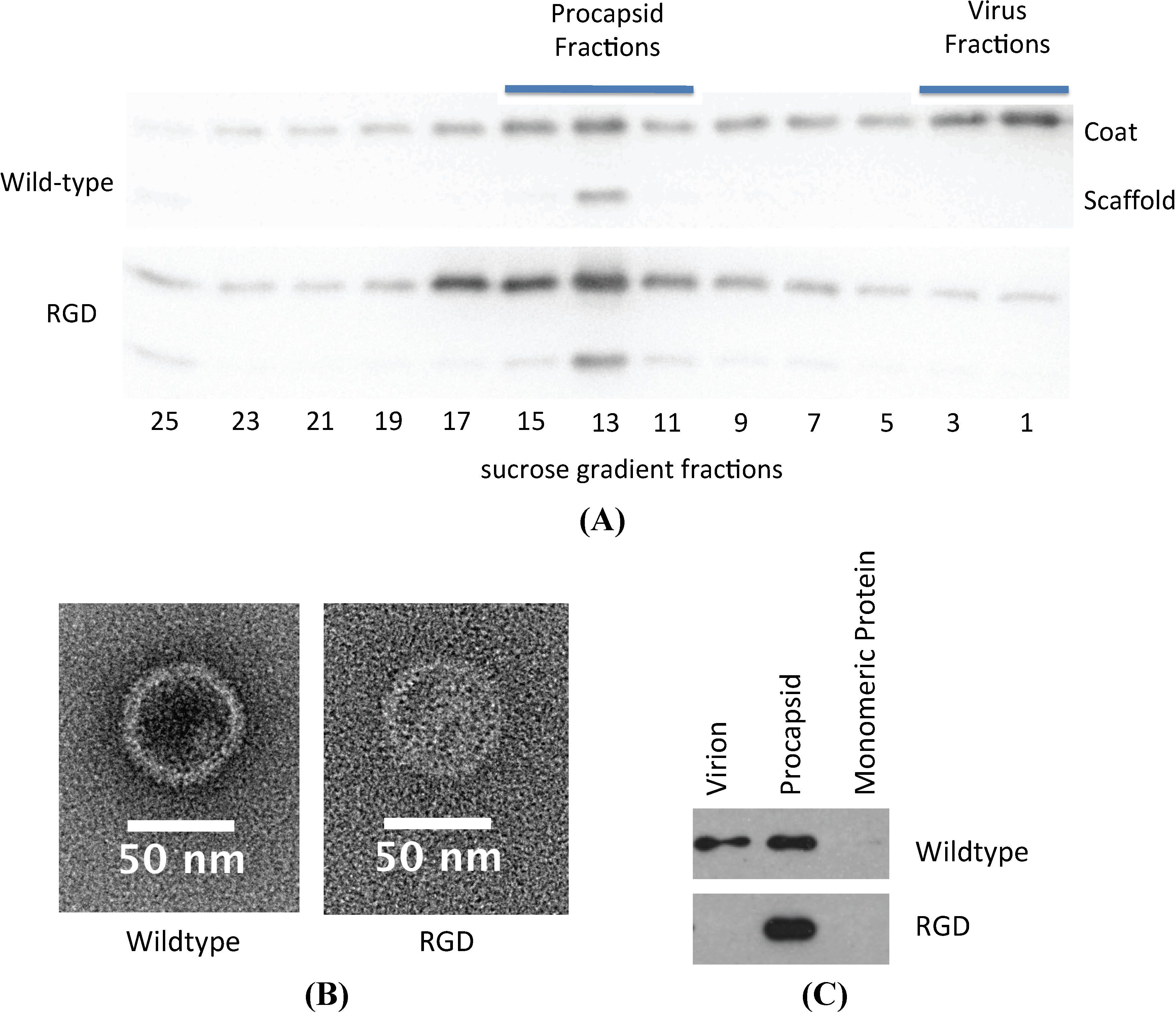
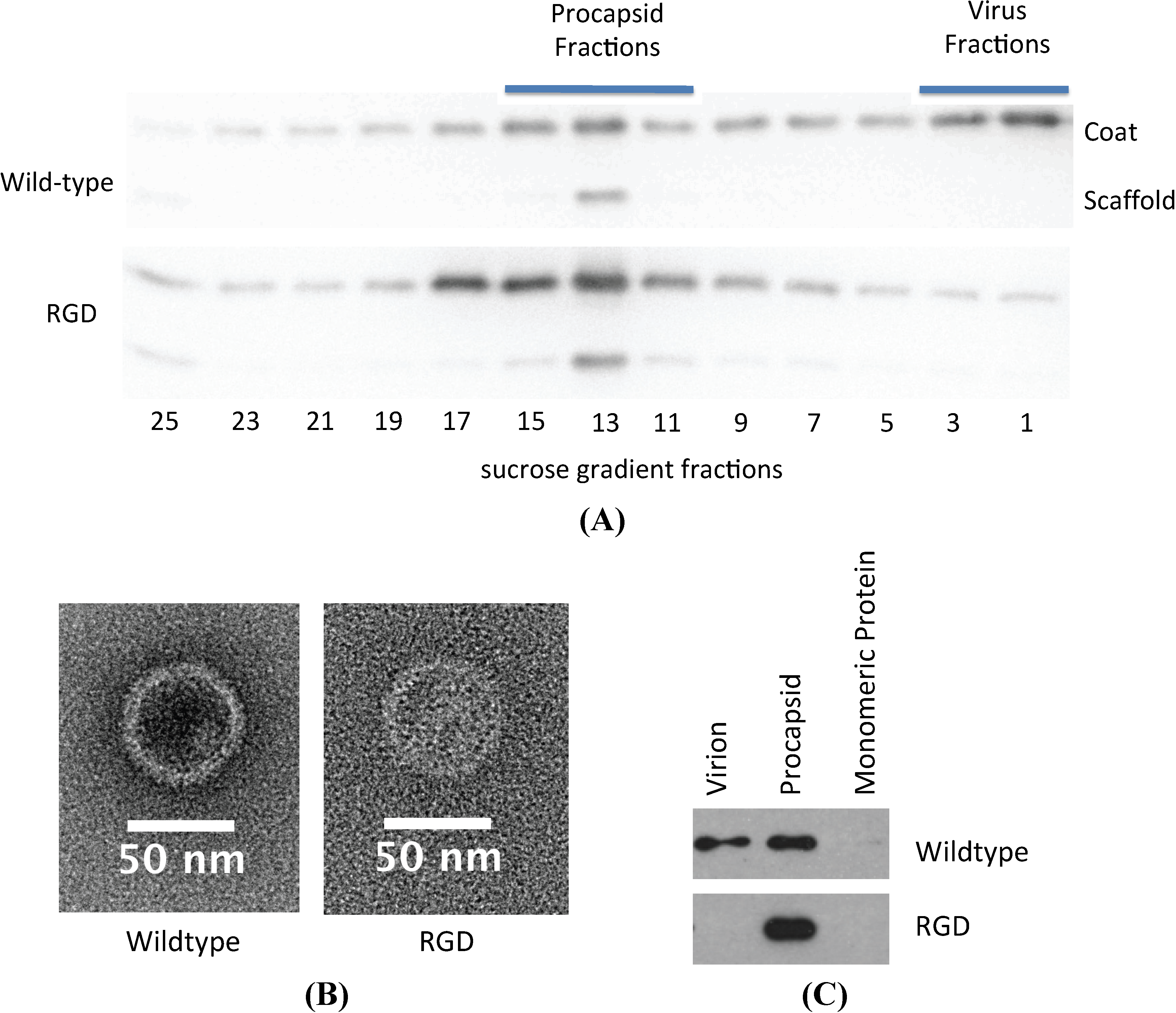
3.3. Biochemical Characterization of the RGD Lysogen
3.4. Additional Manipulations of the A-Domain

{kind=link}
{kind=link}
{kind=link}
{kind=link}
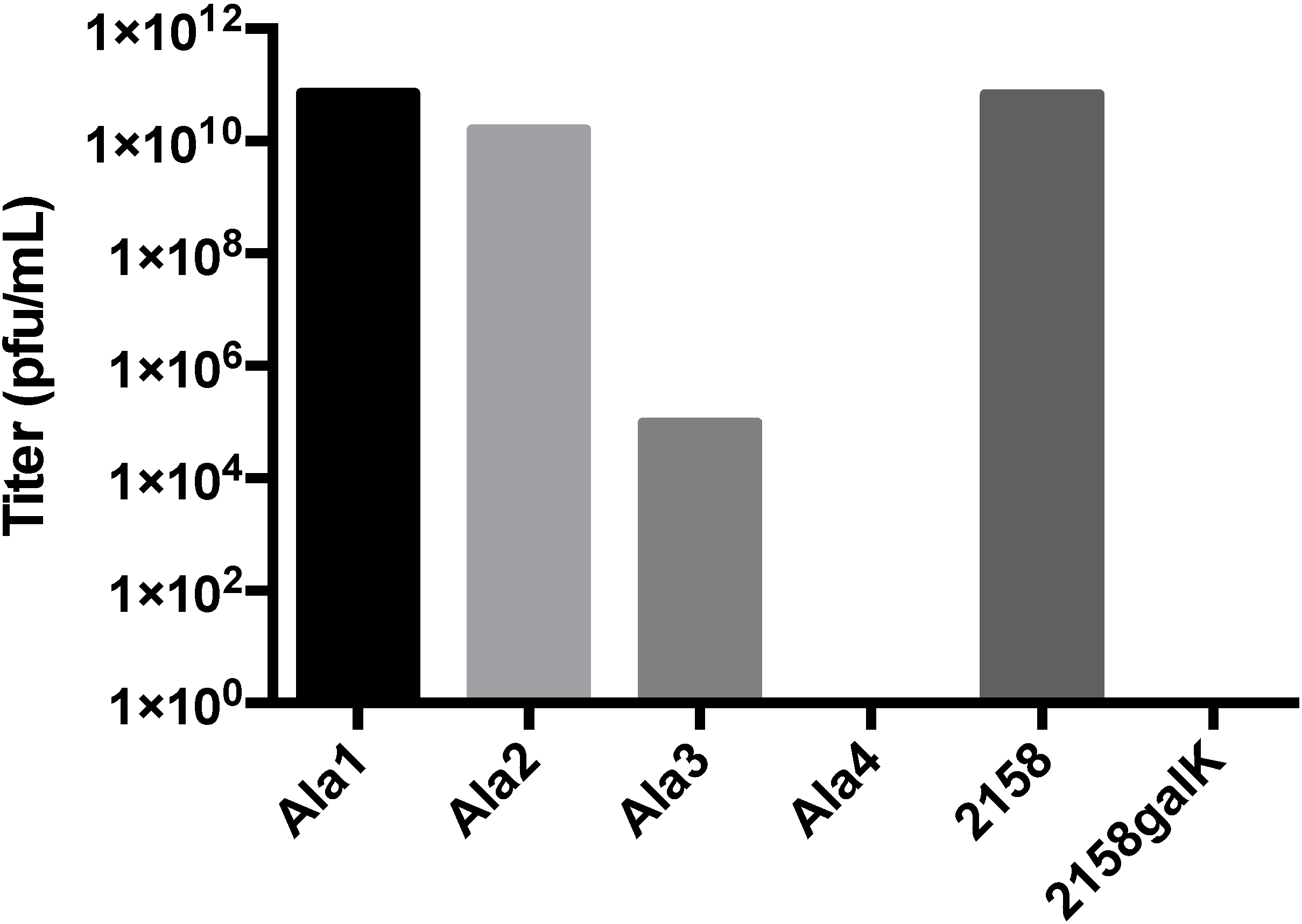{kind=link}
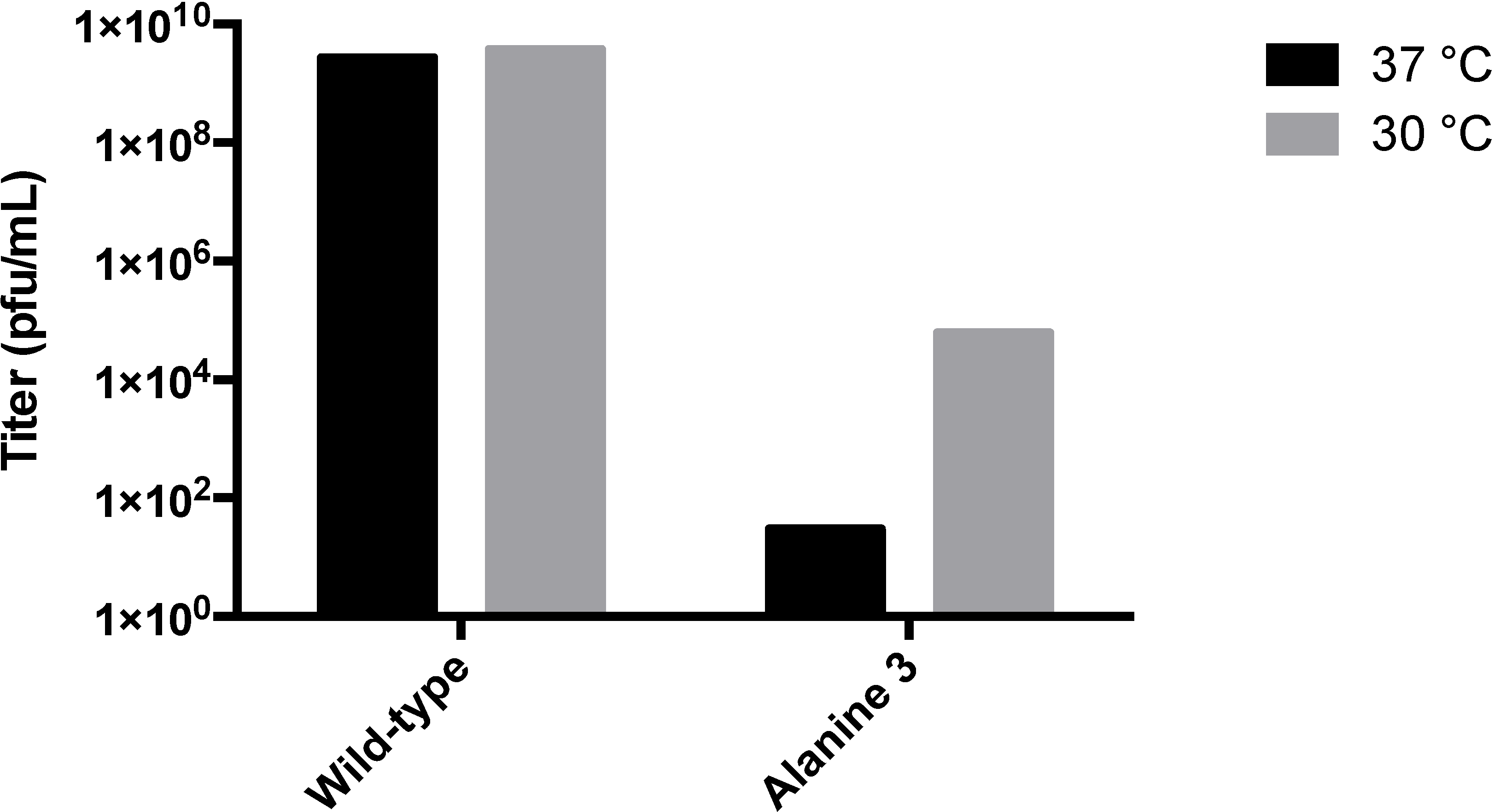{kind=link}
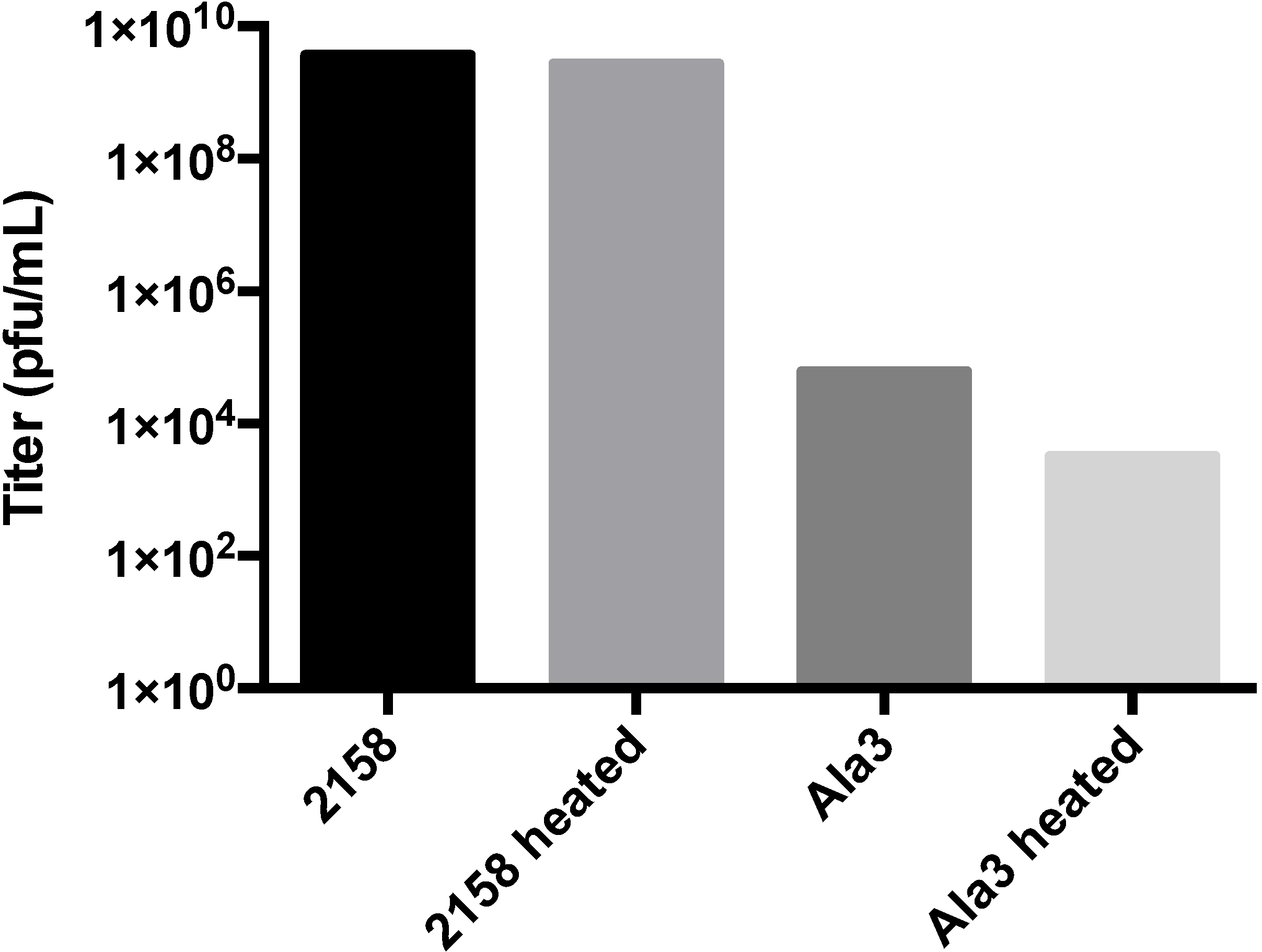{kind=link}
| Mutations Generated in 1757/TetRA | Mutations Generated in 2158/galK |
|---|---|
| T183GGGRGDGGG | T183AAAA |
| T183GGGGGGGGG | T183AAA |
| T183RGD | T183AA |
| T183GGG | T183A |

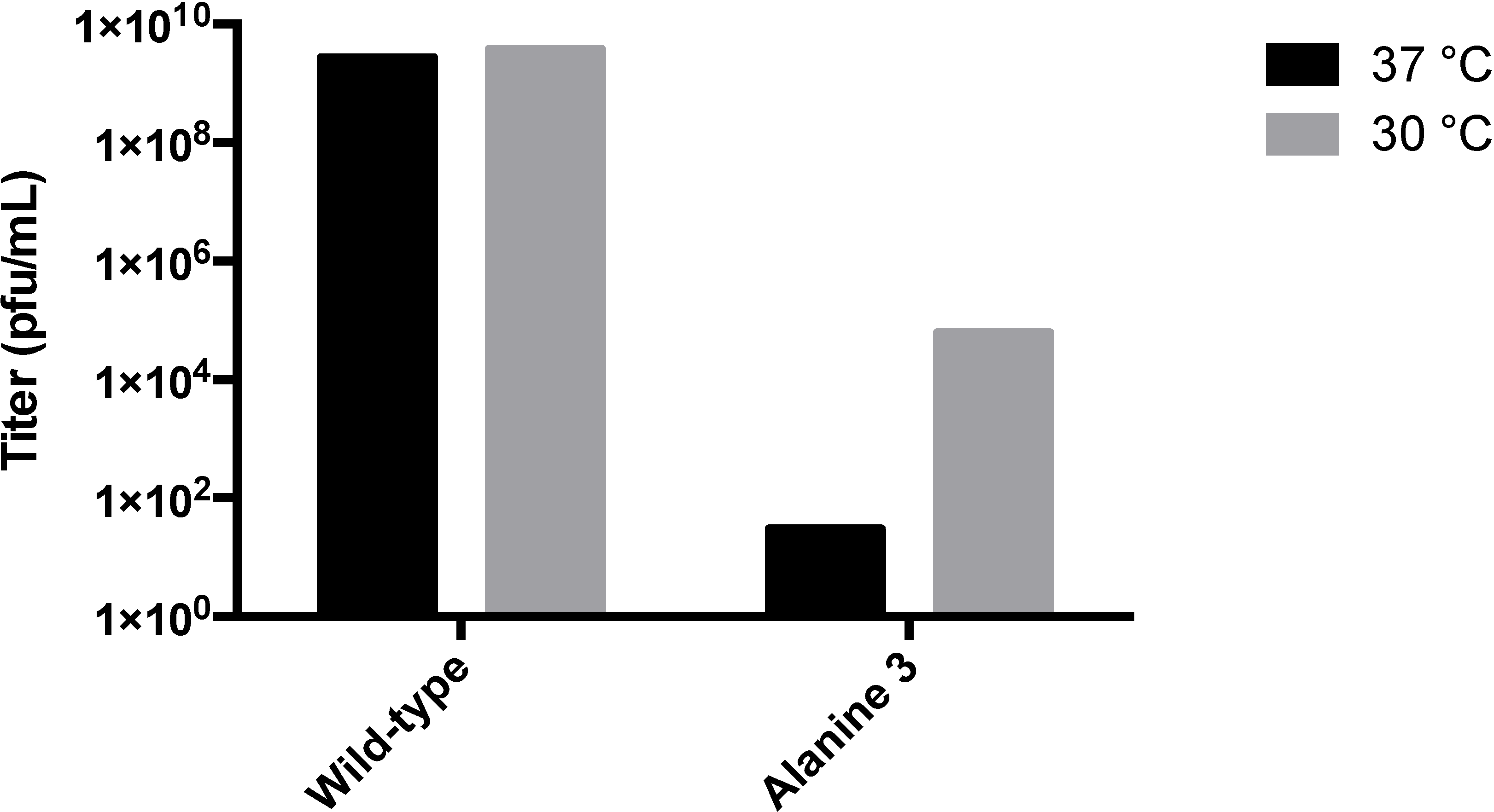
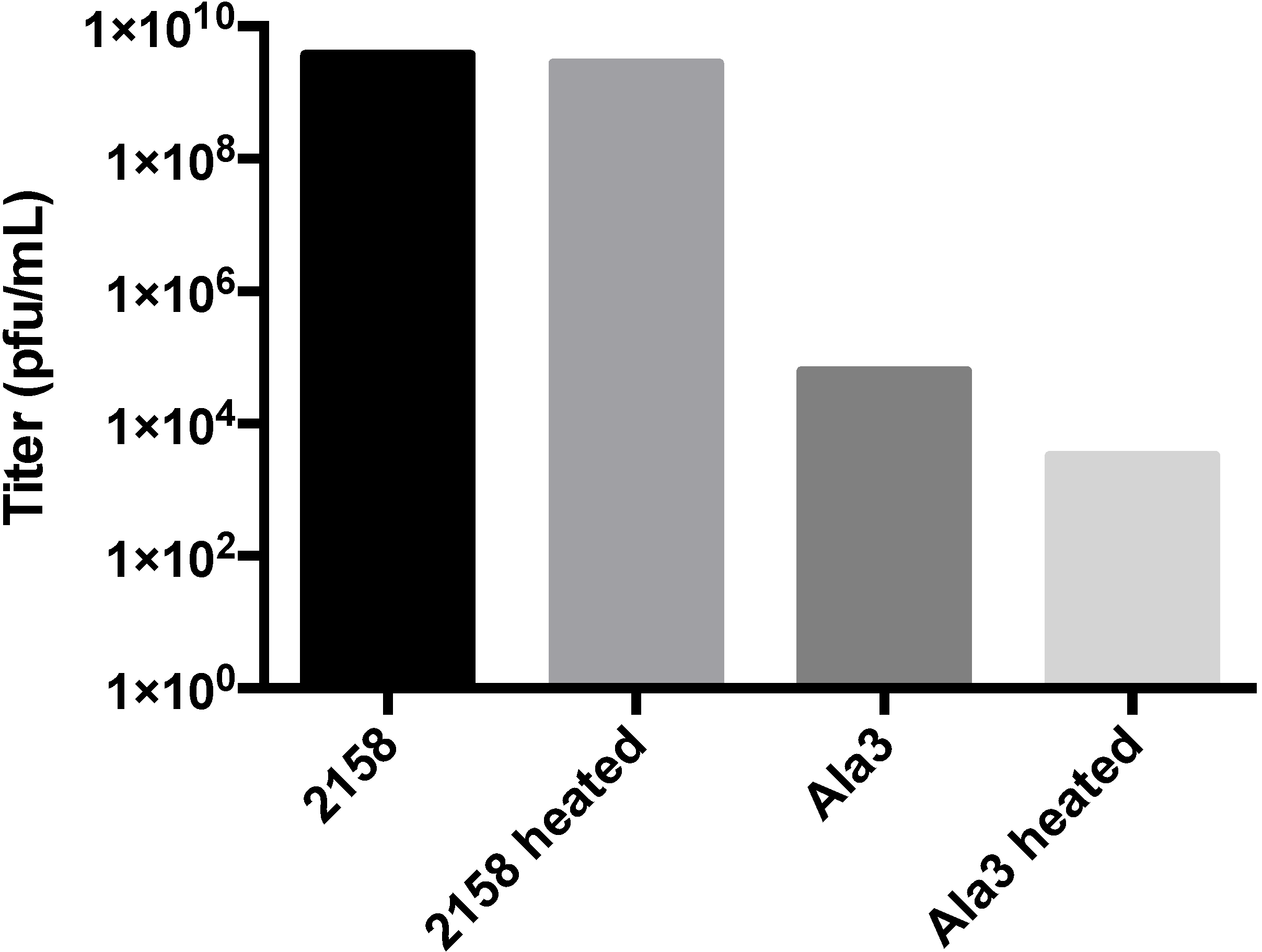
4. Discussion
4.1. Stabilizing Effect of the A-Domain and Restriction of Scaffolding Protein Escape
4.2. A-Domain Has Strong Selection Pressure toward Near-Wild-Type Sequences in Vivo
4.3. A-Domain Manipulations Can Produce Conditionally-Stable Procapsids
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Prevelige, P.E., Jr.; Thomas, D.; King, J.; Towse, S.A.; Thomas, G.J., Jr. Conformational states of the bacteriophage P22 capsid subunit in relation to self-assembly. Biochemistry (Mosc). 1990, 29, 5626–5633. [Google Scholar] [CrossRef]
- Caspar, D.L.; Klug, A. Physical principles in the construction of regular viruses. Cold Spring Harb. Symp. Quant. Biol. 1962, 27, 1–24. [Google Scholar] [CrossRef]
- King, J.; Chiu, W. The procapsid-to-capsid transition in double-stranded DNA bacteriophages; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Kang, S.; Uchida, M. Implementations of P22 Viral Capsids as Nanoplatforms. Biomacromolecules 2010, 11, 2804–2809. [Google Scholar] [CrossRef]
- Culpepper, B.K.; Morris, D.S.; Prevelige, P.E.; Bellis, S.L. Engineering nanocages with polyglutamate domains for coupling to hydroxyapatite biomaterials and allograft bone. Biomaterials 2013, 34, 2455–2462. [Google Scholar]
- Maham, A.; Tang, Z.; Wu, H.; Wang, J.; Lin, Y. Protein-based nanomedicine platforms for drug delivery. Small 2009, 5, 1706–1721. [Google Scholar] [CrossRef]
- Kang, S.; Prevelige, P.E., Jr. Domain study of bacteriophage p22 coat protein and characterization of the capsid lattice transformation by hydrogen/deuterium exchange. J. Mol. Biol. 2005, 347, 935–948. [Google Scholar] [CrossRef]
- Kang, S.; Hawkridge, A.M.; Johnson, K.L.; Muddiman, D.C.; Prevelige, P.E., Jr. Identification of subunit-subunit interactions in bacteriophage P22 procapsids by chemical cross-linking and mass spectrometry. J. Proteome Res. 2006, 5, 370–377. [Google Scholar] [CrossRef]
- Svensen, N.; Walton, J.G.; Bradley, M. Peptides for cell-selective drug delivery. Trends Pharmacol. Sci. 2012, 33, 186–192. [Google Scholar] [CrossRef]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef]
- O’Neil, A.; Reichhardt, C.; Johnson, B.; Prevelige, P.E.; Douglas, T. Genetically programmed in vivo packaging of protein cargo and its controlled release from bacteriophage P22. Angew Chem. Int. Ed. Engl. 2011, 50, 7425–7428. [Google Scholar]
- Chen, D.H.; Baker, M.L.; Hryc, C.F.; DiMaio, F.; Jakana, J.; Wu, W.; Dougherty, M.; Haase-Pettingell, C.; Schmid, M.F.; Jiang, W.; et al. Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. Proc. Natl. Acad. Sci. USA 2011, 108, 1355–1360. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 13, 1605–1612. [Google Scholar]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. method. 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Karlinsey, J. λ-Red Genetic Engineering in Salmonella enterica serovar Typhimurium. Method. Enzymol. 2007, 421, 199–209. [Google Scholar] [CrossRef]
- Warming, S.; Costantino, N.; Court, D.L.; Jenkins, N.A.; Copeland, N.G. Simple and highly efficient BAC recombineering using galK selection. Nucl. Acids Res. 2005, 33, e36. [Google Scholar] [CrossRef]
- Galisteo, M.L.; King, J. Conformational transformations in the protein lattice of phage P22 procapsids. Biophys. J. 1993, 65, 227–235. [Google Scholar] [CrossRef]
- Bochner, B.R.; Huang, H.C.; Schieven, G.L.; Ames, B.N. Positive selection for loss of tetracycline resistance. J. Bacteriol. 1980, 143, 926–933. [Google Scholar]
- Maloy, S.R.; Nunn, W.D. Selection for loss of tetracycline resistance by Escherichia coli. J. Bacteriol. 1981, 145, 1110–1111. [Google Scholar]
- King, J.; Lenk, E.V.; Botstein, D. Mechanism of head assembly and DNA encapsulation in Salmonella phage P22. II. Morphogenetic pathway. J. Mol. Biol. 1973, 80, 697–731. [Google Scholar] [CrossRef]
- Lanman, J.; Tuma, R.; Prevelige, P.E., Jr. Identification and characterization of the domain structure of bacteriophage P22 coat protein. Biochemistry (Mosc.) 1999, 38, 14614–14623. [Google Scholar] [CrossRef]
- Tuma, R.; Prevelige, P.E., Jr.; Thomas, G.J., Jr. Mechanism of capsid maturation in a double-stranded DNA virus. Proc. Natl. Acad. Sci. USA 1998, 95, 9885–9890. [Google Scholar] [CrossRef]
- Parent, K.N.; Khayat, R.; Tu, L.H.; Suhanovsky, M.M.; Cortines, J.R.; Teschke, C.M.; Johnson, J.E.; Baker, T.S. P22 coat protein structures reveal a novel mechanism for capsid maturation: Stability without auxiliary proteins or chemical crosslinks. Structure 2010, 18, 390–401. [Google Scholar] [CrossRef]
- Zandi, R.; Reguera, D. Mechanical properties of viral capsids. Phys. Rev. Stat. Nonlinear Soft Matter Phys. 2005, 72, 021917. [Google Scholar] [CrossRef]
- Teschke, C.M.; McGough, A.; Thuman-Commike, P.A. Penton release from P22 heat-expanded capsids suggests importance of stabilizing penton-hexon interactions during capsid maturation. Biophys. J. 2003, 84, 2585–2592. [Google Scholar] [CrossRef]
- Veesler, D.; Quispe, J.; Grigorieff, N.; Potter, C.S.; Carragher, B.; Johnson, J.E. Maturation in Action: CryoEM Study of a Viral Capsid Caught during Expansion. Structure 2012, 20, 1384–1390. [Google Scholar] [CrossRef]
- Veesler, D.; Khayat, R.; Krishnamurthy, S.; Snijder, J.; Huang, R.K.; Heck, A.J.; Anand, G.S.; Johnson, J.E. Architecture of a dsDNA Viral Capsid in Complex with Its Maturation Protease. Structure 2014, 22, 230–237. [Google Scholar] [CrossRef]
- Gertsman, I.; Fu, C.Y.; Huang, R.; Komives, E.A.; Johnson, J.E. Critical salt bridges guide capsid assembly, stability, and maturation behavior in bacteriophage HK97. Mol. Cell. Proteomics 2010, 9, 1752–1763. [Google Scholar] [CrossRef]
- Huang, R.K.; Khayat, R.; Lee, K.K.; Gertsman, I.; Duda, R.L.; Hendrix, R.W.; Johnson, J.E. The Prohead-I structure of bacteriophage HK97: Implications for scaffold-mediated control of particle assembly and maturation. J. Mol. Biol. 2011, 408, 541–554. [Google Scholar] [CrossRef]
- Gertsman, I.; Gan, L.; Guttman, M.; Lee, K.; Speir, J.A.; Duda, R.L.; Hendrix, R.W.; Komives, E.A.; Johnson, J.E. An unexpected twist in viral capsid maturation. Nature 2009, 458, 646–650. [Google Scholar] [CrossRef]
- Gertsman, I.; Komives, E.A.; Johnson, J.E. HK97 maturation studied by crystallography and H/2H exchange reveals the structural basis for exothermic particle transitions. J. Mol. Biol. 2010, 397, 560–574. [Google Scholar] [CrossRef]
- Greene, B.; King, J. Binding of scaffolding subunits within the P22 procapsid lattice. Virology 1994, 205, 188–197. [Google Scholar] [CrossRef]
- Hendrix, R.W. Bacteriophages: Evolution of the majority. Theor. Popul. Biol. 2002, 61, 471–480. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Morris, D.S.; Prevelige, P.E., Jr. The Role of the Coat Protein A-Domain in P22 Bacteriophage Maturation. Viruses 2014, 6, 2708-2722. https://doi.org/10.3390/v6072708
Morris DS, Prevelige PE Jr. The Role of the Coat Protein A-Domain in P22 Bacteriophage Maturation. Viruses. 2014; 6(7):2708-2722. https://doi.org/10.3390/v6072708
Chicago/Turabian StyleMorris, David S., and Peter E. Prevelige, Jr. 2014. "The Role of the Coat Protein A-Domain in P22 Bacteriophage Maturation" Viruses 6, no. 7: 2708-2722. https://doi.org/10.3390/v6072708
APA StyleMorris, D. S., & Prevelige, P. E., Jr. (2014). The Role of the Coat Protein A-Domain in P22 Bacteriophage Maturation. Viruses, 6(7), 2708-2722. https://doi.org/10.3390/v6072708