KSHV LANA—The Master Regulator of KSHV Latency




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Epidemiology
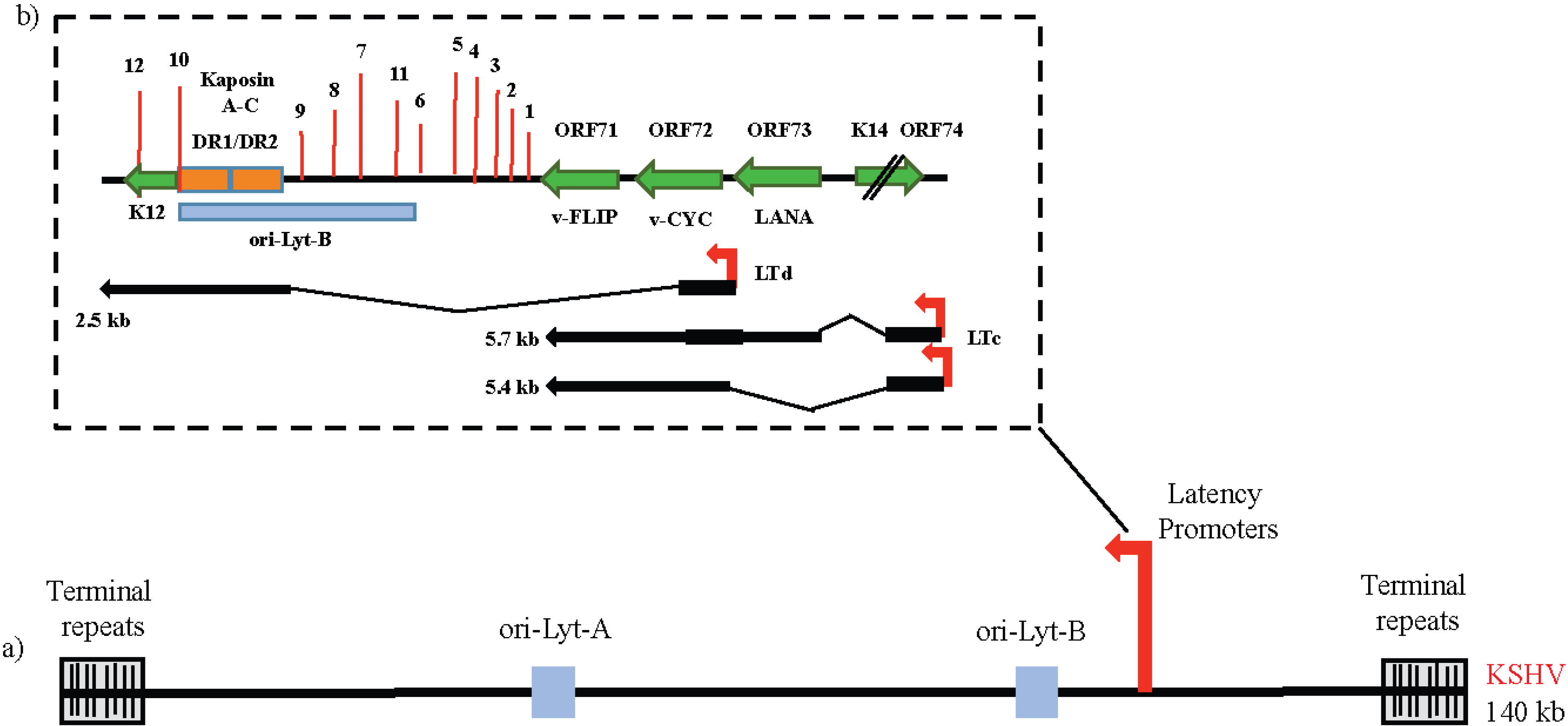
3. Life Cycles of KSHV and Control of Latency
4. The Latency Program of KSHV and the Key Players
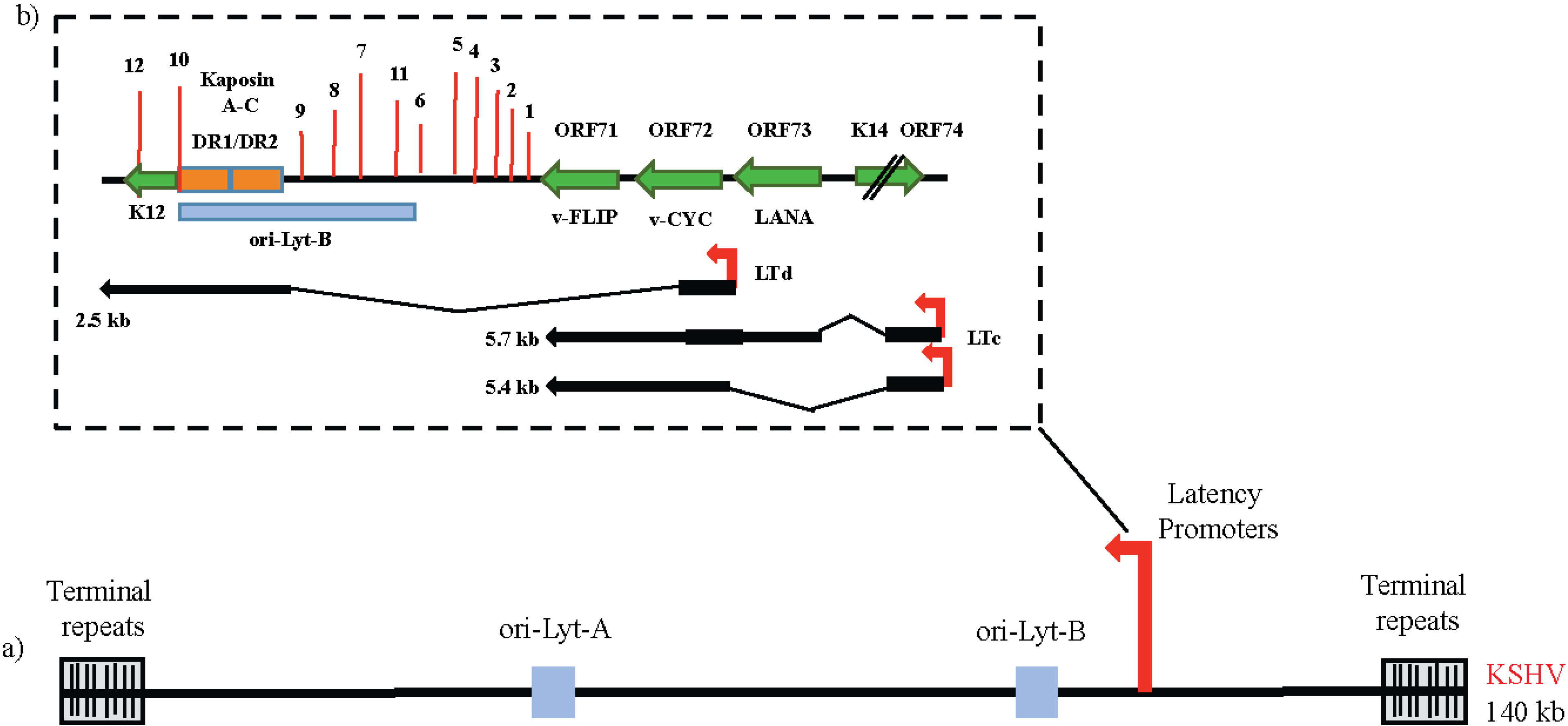
4.1. ORF73/LANA (Enables Replicative Immortality)
4.2. ORF 72/v-Cyclin (Sustains Cell Proliferation)
4.3. ORF71/v-FLIP (Resists Cell Death)
4.4. K12/Kaposins
4.5. Viral microRNAs
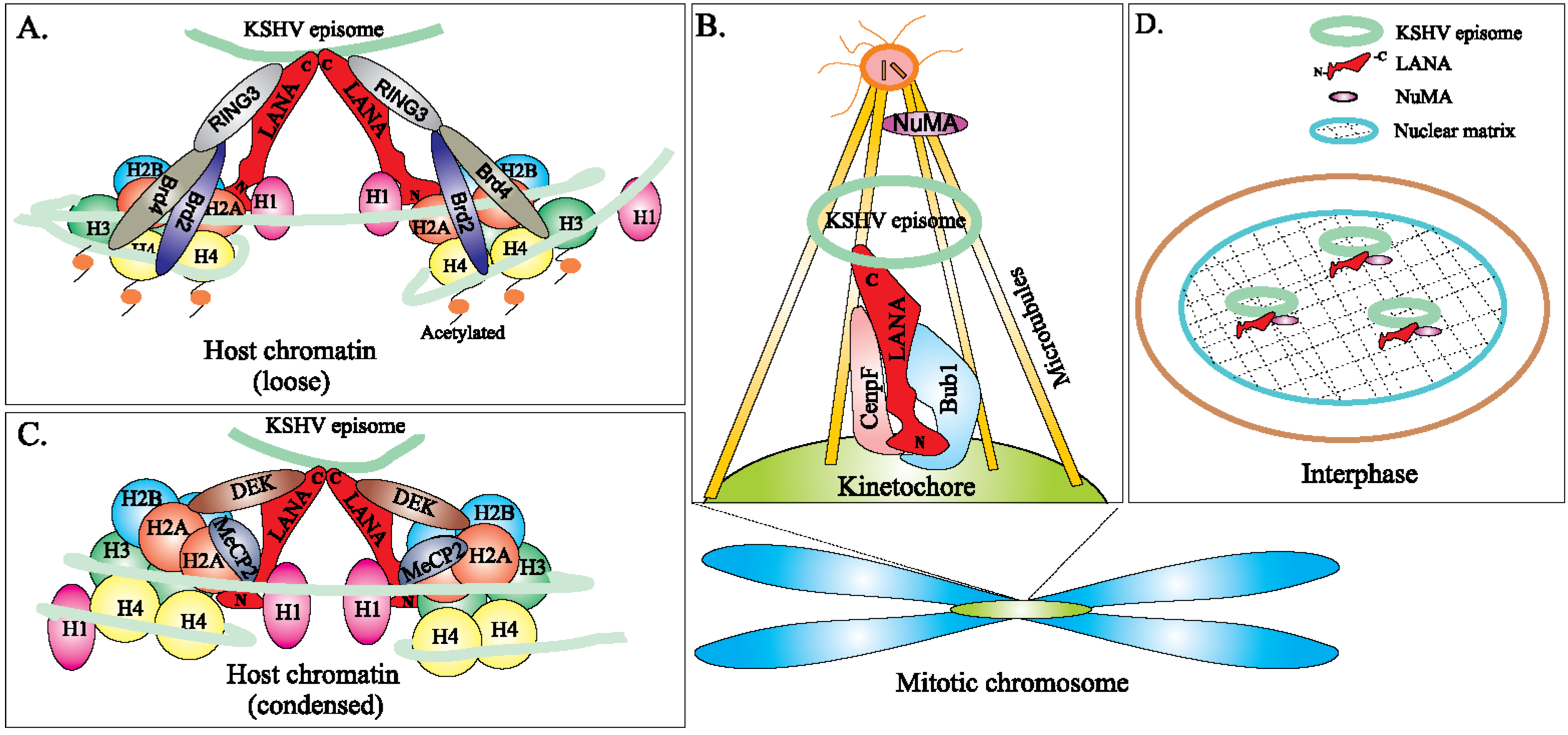
5. Role of LANA in KSHV Episome Maintenance and Partitioning during Cell Division
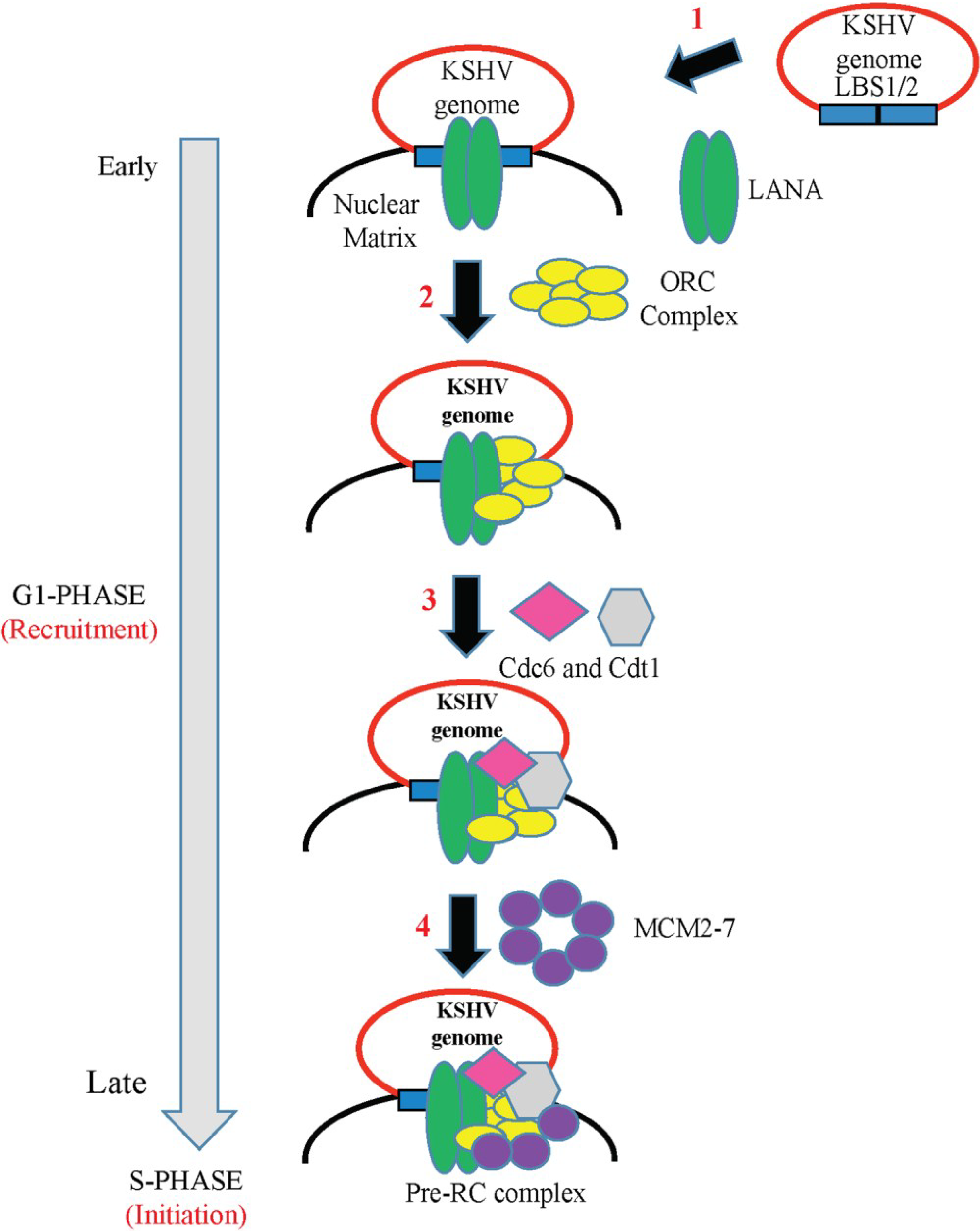
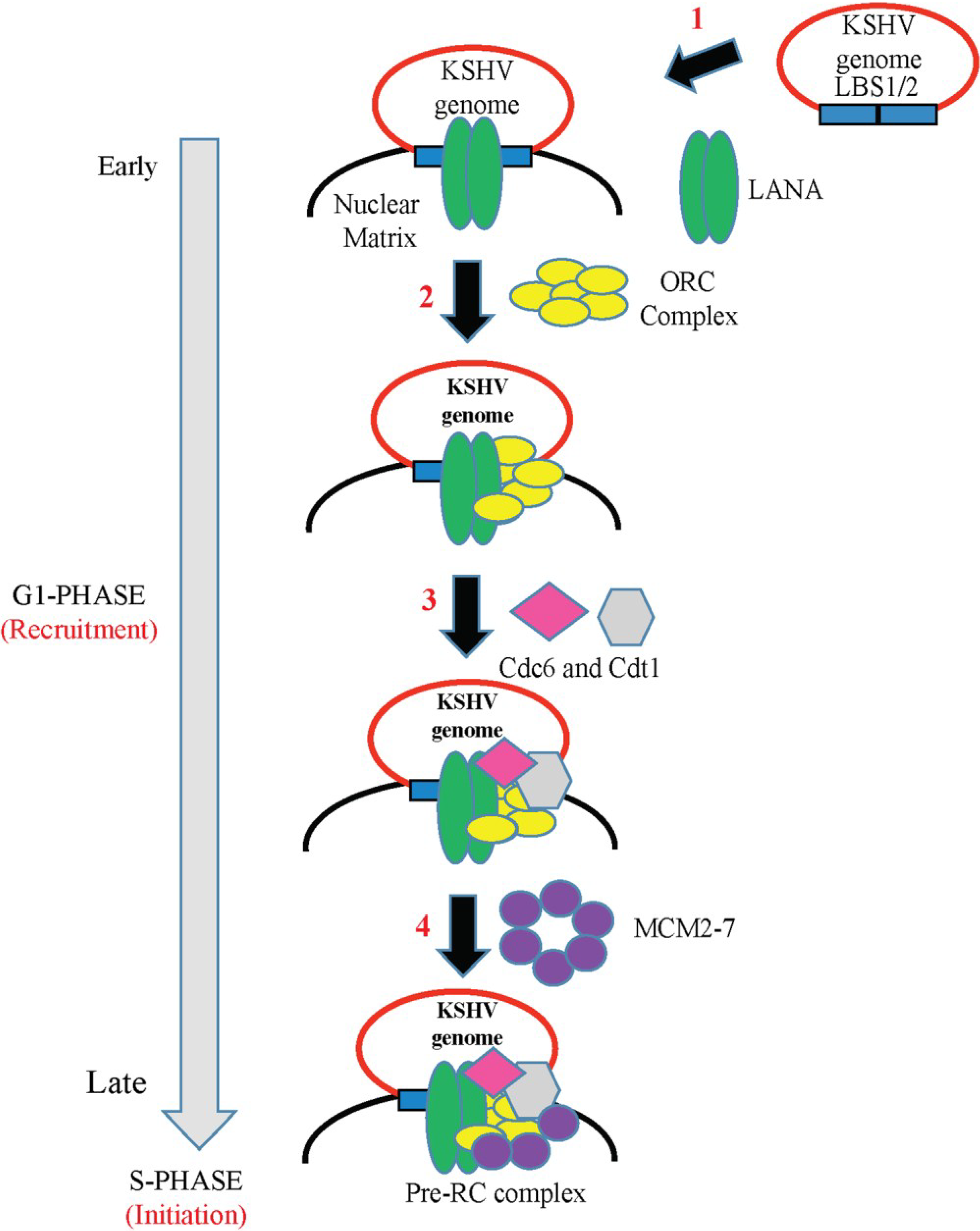
6. Role of LANA in KSHV DNA Replication
6.1. LANA-TR Mediated DNA Replication of KSHV Episome
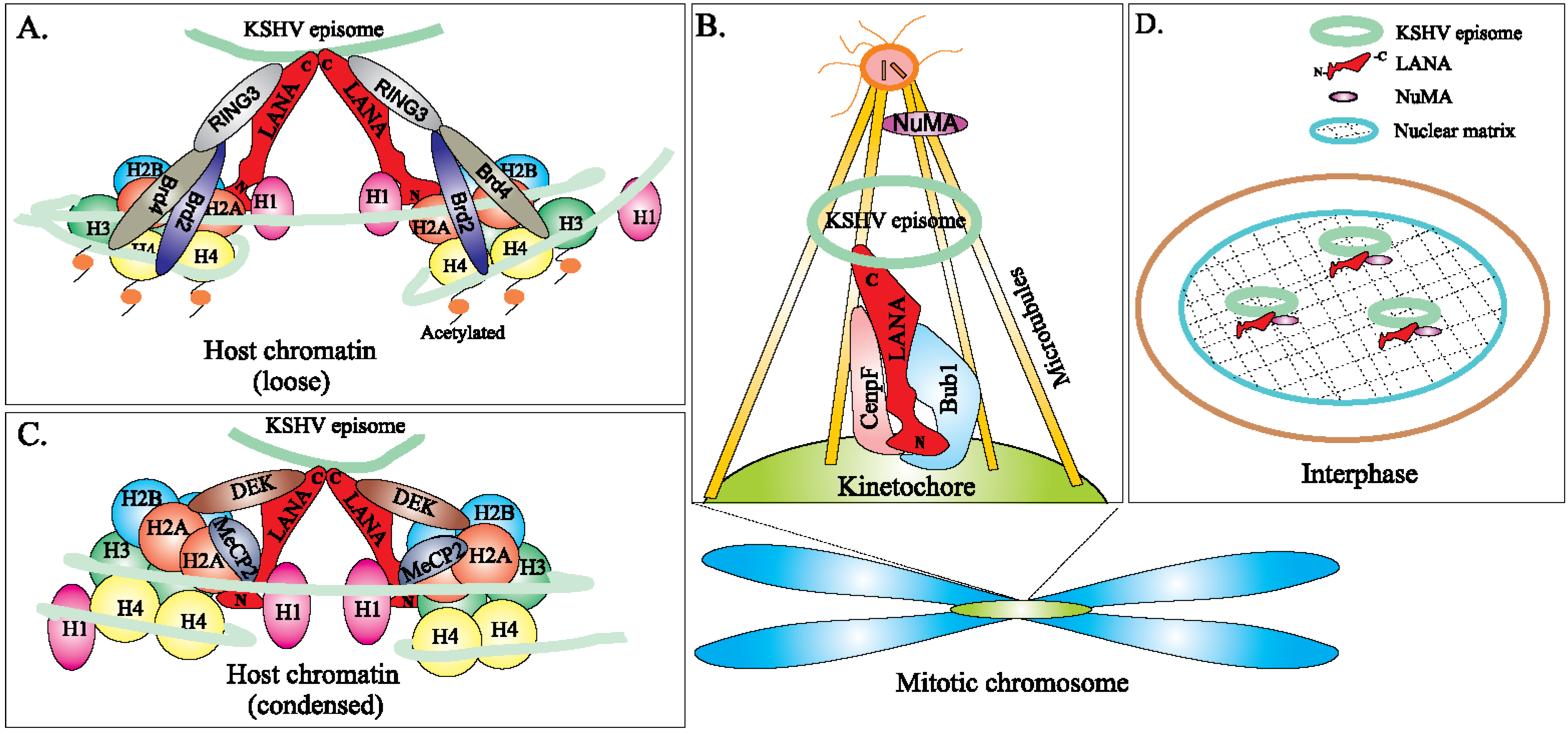
6.2. Epigenetic Regulation of LANA-TR Mediated KSHV DNA Replication
6.3. Non-TR Mediated KSHV DNA Replication
7. LANAs Interaction with Various Cellular and Viral Pathways/Promoters
7.1. Analysis and Implication of Multiple LANA-Binding Sites within the KSHV Genome
7.2. LANA Associates with Several Viral Promoters and Causes Epigenetic Modifications
7.3. LANA Associates with Host Proteins and Signaling Pathways
7.3.1. LANA Interaction with p53 and pRb
7.3.2. LANA Interaction with GSK-3β (Wnt Signaling Pathway)
7.3.3. LANA Interacts with ANG and Promotes Angiogenesis
7.3.4. LANA Interacts with BMP-Activated Smad1
7.3.5. LANA Interacts with Host KAP1 and Facilitates the Establishment of KSHV Latency
7.3.6. LANA Interacts with Daxx and Contributes to VEGFR–Mediated Angiogenesis
8. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Sturzl, M.; Zietz, C.; Monini, P.; Ensoli, B. Human herpesvirus-8 and Kaposi’s sarcoma: Relationship with the multistep concept of tumorigenesis. Adv. Cancer Res. 2001, 81, 125–159. [Google Scholar] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Boshoff, C.; Chang, Y. Kaposi’s sarcoma-associated herpesvirus: A new DNA tumor virus. Annu. Rev. Med. 2001, 52, 453–470. [Google Scholar] [CrossRef] [PubMed]
- Whitby, D.; Howard, M.R.; Tenant-Flowers, M.; Brink, N.S.; Copas, A.; Boshoff, C.; Hatzioannou, T.; Suggett, F.E.; Aldam, D.M.; Denton, A.S.; et al. Detection of Kaposi sarcoma associated herpesvirus in peripheral blood of HIV-infected individuals and progression to Kaposi’s sarcoma. Lancet 1995, 346, 799–802. [Google Scholar] [CrossRef]
- Gao, S.J.; Kingsley, L.; Hoover, D.R.; Spira, T.J.; Rinaldo, C.R.; Saah, A.; Phair, J.; Detels, R.; Parry, P.; Chang, Y.; et al. Seroconversion to antibodies against Kaposi’s sarcoma-associated herpesvirus-related latent nuclear antigens before the development of Kaposi’s sarcoma. N. Engl. J. Med. 1996, 335, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.N.; Ganem, D.E.; Osmond, D.H.; Page-Shafer, K.A.; Macrae, D.; Kedes, D.H. Sexual transmission and the natural history of human herpesvirus 8 infection. N. Engl. J. Med. 1998, 338, 948–954. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar]
- Gill, P.S.; Tsai, Y.C.; Rao, A.P.; Spruck, C.H., 3rd; Zheng, T.; Harrington, W.A., Jr.; Cheung, T.; Nathwani, B.; Jones, P.A. Evidence for multiclonality in multicentric Kaposi’s sarcoma. Proc. Natl. Acad. Sci. USA 1998, 95, 8257–8261. [Google Scholar] [CrossRef]
- Gessain, A.; Duprez, R. Spindle cells and their role in Kaposi’s sarcoma. Int. J. Biochem. Cell Biol. 2005, 37, 2457–2465. [Google Scholar] [CrossRef] [PubMed]
- Ambroziak, J.A.; Blackbourn, D.J.; Herndier, B.G.; Glogau, R.G.; Gullett, J.H.; McDonald, A.R.; Lennette, E.T.; Levy, J.A. Herpes-like sequences in HIV-infected and uninfected Kaposi’s sarcoma patients. Science 1995, 268, 582–583. [Google Scholar] [CrossRef]
- Uldrick, T.S.; Wang, V.; O’Mahony, D.; Aleman, K.; Wyvill, K.M.; Marshall, V.; Steinberg, S.M.; Pittaluga, S.; Maric, I.; Whitby, D.; et al. An Interleukin-6-Related Systemic Inflammatory Syndrome in Patients Co-Infected with Kaposi Sarcoma-Associated Herpesvirus and HIV but without Multicentric Castleman Disease. Clin. Infect. Dis. 2010, 51, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Holkova, B.; Takeshita, K.; Cheng, D.M.; Volm, M.; Wasserheit, C.; Demopoulos, R.; Chanan-Khan, A. Effect of highly active antiretroviral therapy on survival in patients with AIDS-associated pulmonary Kaposi’s sarcoma treated with chemotherapy. J. Clin. Oncol. 2001, 19, 3848–3851. [Google Scholar] [PubMed]
- Martin, J.N. The epidemiology of KSHV and its association with malignant disease. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Dourmishev, L.A.; Dourmishev, A.L.; Palmeri, D.; Schwartz, R.A.; Lukac, D.M. Molecular genetics of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol. Mol. Biol. Rev. 2003, 67, 175–212. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.J.; Kingsley, L.; Li, M.; Zheng, W.; Parravicini, C.; Ziegler, J.; Newton, R.; Rinaldo, C.R.; Saah, A.; Phair, J.; et al. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi’s sarcoma. Nat. Med. 1996, 2, 925–928. [Google Scholar] [CrossRef]
- Kedes, D.H.; Operskalski, E.; Busch, M.; Kohn, R.; Flood, J.; Ganem, D. The seroepidemiology of human herpesvirus 8 (Kaposi Kaposi’s sarcoma-associated herpesvirus): Distribution of infection in KS risk groups and evidence for sexual transmission. Nat. Med. 1996, 2, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Simpson, G.R.; Schulz, T.F.; Whitby, D.; Cook, P.M.; Boshoff, C.; Rainbow, L.; Howard, M.R.; Gao, S.J.; Bohenzky, R.A.; Simmonds, P.; et al. Prevalence of Kaposi’s sarcoma associated herpesvirus infection measured by antibodies to recombinant capsid protein and latent immunofluorescence antigen. Lancet 1996, 348, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Dukers, N.H.; Rezza, G. Human herpesvirus 8 epidemiology: What we do and do not know. AIDS 2003, 17, 1717–1730. [Google Scholar] [CrossRef] [PubMed]
- Hayward, G.S. KSHV strains: The origins and global spread of the virus. Semin. Cancer Biol. 1999, 9, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Hayward, G.S.; Zong, J.C. Modern evolutionary history of the human KSHV genome. Curr. Top. Microbiol. Immunol. 2007, 312, 1–42. [Google Scholar] [PubMed]
- Stebbing, J.; Bourboulia, D.; Johnson, M.; Henderson, S.; Williams, I.; Wilder, N.; Tyrer, M.; Youle, M.; Imami, N.; Kobu, T.; et al. Kaposi’s sarcoma-associated herpesvirus cytotoxic T lymphocytes recognize and target Darwinian positively selected autologous K1 epitopes. J. Virol. 2003, 77, 4306–4314. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.J.; Zong, J.C.; Ciufo, D.M.; Alcendor, D.J.; Cannon, J.S.; Ambinder, R.; Orenstein, J.M.; Reitz, M.S.; Hayward, G.S. Comparison of genetic variability at multiple loci across the genomes of the major subtypes of Kaposi’s sarcoma-associated herpesvirus reveals evidence for recombination and for two distinct types of open reading frame K15 alleles at the right-hand end. J. Virol. 1999, 73, 6646–6660. [Google Scholar] [PubMed]
- Zong, J.; Ciufo, D.M.; Viscidi, R.; Alagiozoglou, L.; Tyring, S.; Rady, P.; Orenstein, J.; Boto, W.; Kalumbuja, H.; Romano, N.; et al. Genotypic analysis at multiple loci across Kaposi’s sarcoma herpesvirus (KSHV) DNA molecules: Clustering patterns, novel variants and chimerism. J. Clin. Virol. 2002, 23, 119–148. [Google Scholar] [CrossRef] [PubMed]
- Vitale, F.; Viviano, E.; Perna, A.M.; Bonura, F.; Mazzola, G.; Ajello, F.; Romano, N. Serological and virological evidence of non-sexual transmission of human herpesvirus type 8 (HHV8). Epidemiol. Infect. 2000, 125, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Sitas, F.; Newton, R. Kaposi’s sarcoma in South Africa. J. Natl. Cancer Inst. Monogr. 2001, 28, 1–4. [Google Scholar] [PubMed]
- Bagni, R.; Whitby, D. Kaposi’s sarcoma-associated herpesvirus transmission and primary infection. Curr. Opin. HIV AIDS 2009, 4, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Vieira, J.; Huang, M.L.; Koelle, D.M.; Corey, L. Transmissible Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in saliva of men with a history of Kaposi’s sarcoma. J. Virol. 1997, 71, 7083–7087. [Google Scholar] [PubMed]
- Farge, D.; Lebbe, C.; Marjanovic, Z.; Tuppin, P.; Mouquet, C.; Peraldi, M.N.; Lang, P.; Hiesse, C.; Antoine, C.; Legendre, C.; et al. Human herpes virus-8 and other risk factors for Kaposi’s sarcoma in kidney transplant recipients. Groupe Cooperatif de Transplantation d’Ile de France (GCIF). Transplantation 1999, 67, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Astudillo, L.A.; Leyva-Cobian, F. Human herpesvirus-8 infection and Kaposi’s sarcoma after liver and kidney transplantation in different geographical areas of Spain. Transpl. Immunol. 2006, 17, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Hladik, W.; Dollard, S.C.; Mermin, J.; Fowlkes, A.L.; Downing, R.; Amin, M.M.; Banage, F.; Nzaro, E.; Kataaha, P.; Dondero, T.J.; et al. Transmission of human herpesvirus 8 by blood transfusion. N. Engl. J. Med. 2006, 355, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef] [PubMed]
- Ohsaki, E.; Ueda, K. Kaposi’s Sarcoma-Associated Herpesvirus Genome Replication, Partitioning, and Maintenance in Latency. Front. Microbiol. 2012, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.; Brulois, K.; Jung, J.U. The chromatin landscape of Kaposi’s sarcoma-associated herpesvirus. Viruses 2013, 5, 1346–1373. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, H.; Kanno, T.; Hasegawa, H.; Katano, H. Pathology of Kaposi’s Sarcoma-Associated Herpesvirus Infection. Front. Microbiol. 2011, 2, 175. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Black, J.B.; Goldsmith, C.S.; Browning, P.J.; Bhalla, K.; Offermann, M.K. Induction of human herpesvirus-8 DNA replication and transcription by butyrate and TPA in BCBL-1 cells. J. Gen. Virol. 1999, 80, 83–90. [Google Scholar] [PubMed]
- Lieberman, P.M. Keeping it quiet: Chromatin control of gammaherpesvirus latency. Nat. Rev. Microbiol. 2013, 11, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Verma, S.C.; Lu, J.; Robertson, E.S. Molecular biology of Kaposi’s sarcoma-associated herpesvirus and related oncogenesis. Adv. Virus Res. 2010, 78, 87–142. [Google Scholar] [PubMed]
- Davis, D.A.; Rinderknecht, A.S.; Zoeteweij, J.P.; Aoki, Y.; Read-Connole, E.L.; Tosato, G.; Blauvelt, A.; Yarchoan, R. Hypoxia induces lytic replication of Kaposi sarcoma-associated herpesvirus. Blood 2001, 97, 3244–3250. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Zhou, F.; Bedolla, R.G.; Jones, T.; Lei, X.; Kang, T.; Guadalupe, M.; Gao, S.J. Reactive oxygen species hydrogen peroxide mediates Kaposi’s sarcoma-associated herpesvirus reactivation from latency. PLoS Pathog. 2011, 7, e1002054. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Ajibade, A.O.; Ye, F.; Kuhne, K.; Gao, S.J. Reactivation of Kaposi’s sarcoma-associated herpesvirus from latency requires MEK/ERK, JNK and p38 multiple mitogen-activated protein kinase pathways. Virology 2008, 371, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; He, M.; Zhou, F.; Ye, F.; Gao, S.J. Activation of Kaposi’s sarcoma-associated herpesvirus (KSHV) by inhibitors of class III histone deacetylases: Identification of sirtuin 1 as a regulator of the KSHV life cycle. J. Virol. 2014, 88, 6355–6367. [Google Scholar] [CrossRef] [PubMed]
- Pantry, S.N.; Medveczky, P.G. Epigenetic regulation of Kaposi’s sarcoma-associated herpesvirus replication. Semin. Cancer Biol. 2009, 19, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Jenner, R.G.; Alba, M.M.; Boshoff, C.; Kellam, P. Kaposi’s sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 2001, 75, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Polizzotto, M.N.; Uldrick, T.S.; Hu, D.; Yarchoan, R. Clinical Manifestations of Kaposi Sarcoma Herpesvirus Lytic Activation: Multicentric Castleman Disease (KSHV-MCD) and the KSHV Inflammatory Cytokine Syndrome. Front. Microbiol. 2012, 3, 73. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Ganem, D. A unique herpesviral transcriptional program in KSHV-infected lymphatic endothelial cells leads to mTORC1 activation and rapamycin sensitivity. Cell Host Microbe 2013, 13, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Yogev, O.; Boshoff, C. Redefining KSHV latency. Cell Host Microbe 2013, 13, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.; Weisburd, B.; Stern-Ginossar, N.; Mercier, A.; Madrid, A.S.; Bellare, P.; Holdorf, M.; Weissman, J.S.; Ganem, D. KSHV 2.0: A comprehensive annotation of the Kaposi’s sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 2014, 10, e1003847. [Google Scholar] [CrossRef] [PubMed]
- Speck, S.H.; Ganem, D. Viral latency and its regulation: Lessons from the gamma-herpesviruses. Cell Host Microbe 2010, 8, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Burysek, L.; Pitha, P.M. Latently expressed human herpesvirus 8-encoded interferon regulatory factor 2 inhibits double-stranded RNA-activated protein kinase. J. Virol. 2001, 75, 2345–2352. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Lagunoff, M.; Renne, R.; Staskus, K.; Haase, A.; Ganem, D. A cluster of latently expressed genes in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1998, 72, 8309–8315. [Google Scholar] [PubMed]
- Kedes, D.H.; Lagunoff, M.; Renne, R.; Ganem, D. Identification of the gene encoding the major latency-associated nuclear antigen of the Kaposi’s sarcoma-associated herpesvirus. J. Clin. Investig. 1997, 100, 2606–2610. [Google Scholar] [CrossRef] [PubMed]
- Rainbow, L.; Platt, G.M.; Simpson, G.R.; Sarid, R.; Gao, S.J.; Stoiber, H.; Herrington, C.S.; Moore, P.S.; Schulz, T.F. The 222- to 234-kilodalton latent nuclear protein (LNA) of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) is encoded by orf73 and is a component of the latency-associated nuclear antigen. J. Virol. 1997, 71, 5915–5921. [Google Scholar] [PubMed]
- Sadler, R.; Wu, L.; Forghani, B.; Renne, R.; Zhong, W.; Herndier, B.; Ganem, D. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1999, 73, 5722–5730. [Google Scholar] [PubMed]
- Wen, K.W.; Damania, B. Kaposi sarcoma-associated herpesvirus (KSHV): Molecular biology and oncogenesis. Cancer Lett. 2010, 289, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Pearce, M.; Matsumura, S.; Wilson, A.C. Transcripts encoding K12, v-FLIP, v-cyclin, and the microRNA cluster of Kaposi’s sarcoma-associated herpesvirus originate from a common promoter. J. Virol. 2005, 79, 14457–14464. [Google Scholar] [CrossRef] [PubMed]
- Wies, E.; Mori, Y.; Hahn, A.; Kremmer, E.; Sturzl, M.; Fleckenstein, B.; Neipel, F. The viral interferon-regulatory factor-3 is required for the survival of KSHV-infected primary effusion lymphoma cells. Blood 2008, 111, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Ganem, D. KSHV and the pathogenesis of Kaposi sarcoma: Listening to human biology and medicine. J. Clin. Investig. 2010, 120, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Guo, J.; Li, M.; Choi, J.K.; deMaria, M.; Rosenzweig, M.; Jung, J.U. Identification of an immunoreceptor tyrosine-based activation motif of K1 transforming protein of Kaposi’s sarcoma-associated herpesvirus. Mol. Cell. Biol. 1998, 18, 5219–5228. [Google Scholar] [PubMed]
- Lagunoff, M.; Majeti, R.; Weiss, A.; Ganem, D. Deregulated signal transduction by the K1 gene product of Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1999, 96, 5704–5709. [Google Scholar] [CrossRef] [PubMed]
- Van Dross, R.; Yao, S.; Asad, S.; Westlake, G.; Mays, D.J.; Barquero, L.; Duell, S.; Pietenpol, J.A.; Browning, P.J. Constitutively active K-cyclin/cdk6 kinase in Kaposi sarcoma-associated herpesvirus-infected cells. J. Natl. Cancer Inst. 2005, 97, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Direkze, S.; Laman, H. Regulation of growth signalling and cell cycle by Kaposi’s sarcoma-associated herpesvirus genes. Int. J. Exp. Pathol. 2004, 85, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Sarek, G.; Jarviluoma, A.; Moore, H.M.; Tojkander, S.; Vartia, S.; Biberfeld, P.; Laiho, M.; Ojala, P.M. Nucleophosmin phosphorylation by v-cyclin-CDK6 controls KSHV latency. PLoS. Pathog. 2010, 6, e1000818. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Paden, C.R.; Morales, F.M.; Powers, R.P.; Jacob, J.; Speck, S.H. Murine gamma-herpesvirus immortalization of fetal liver-derived B cells requires both the viral cyclin D homolog and latency-associated nuclear antigen. PLoS Pathog. 2011, 7, e1002220. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.C.; Zhou, F.C.; Xie, J.P.; Kang, T.; Greene, W.; Kuhne, K.; Lei, X.F.; Li, Q.H.; Gao, S.J. Kaposi’s sarcoma-associated herpesvirus latent gene vFLIP inhibits viral lytic replication through NF-kappaB-mediated suppression of the AP-1 pathway: A novel mechanism of virus control of latency. J. Virol. 2008, 82, 4235–4249. [Google Scholar] [CrossRef] [PubMed]
- Guasparri, I.; Keller, S.A.; Cesarman, E. KSHV vFLIP is essential for the survival of infected lymphoma cells. J. Exp. Med. 2004, 199, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Israel, A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef] [PubMed]
- Field, N.; Low, W.; Daniels, M.; Howell, S.; Daviet, L.; Boshoff, C.; Collins, M. KSHV vFLIP binds to IKK-gamma to activate IKK. J. Cell Sci. 2003, 116, 3721–3728. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Lei, X.; Gao, S.J. Mechanisms of Kaposi’s Sarcoma-Associated Herpesvirus Latency and Reactivation. Adv. Virol. 2011. [Google Scholar] [CrossRef]
- McCormick, C.; Ganem, D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science 2005, 307, 739–741. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Kincaid, R.P.; Arasappan, D.; Dowd, S.E.; Hunicke-Smith, S.P.; Sullivan, C.S. Small RNA profiling reveals antisense transcription throughout the KSHV genome and novel small RNAs. RNA 2010, 16, 1540–1558. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.R.; Kara, M.; Coleman, C.B.; Grau, K.R.; Oko, L.M.; Krueger, B.J.; Renne, R.; van Dyk, L.F.; Tibbetts, S.A. Virus-encoded microRNAs facilitate gammaherpesvirus latency and pathogenesis in vivo. MBIO 2014, 5, e00981–e00914. [Google Scholar] [CrossRef] [PubMed]
- Moody, R.; Zhu, Y.; Huang, Y.; Cui, X.; Jones, T.; Bedolla, R.; Lei, X.; Bai, Z.; Gao, S.J. KSHV microRNAs mediate cellular transformation and tumorigenesis by redundantly targeting cell growth and survival pathways. PLoS Pathog. 2013, 9, e1003857. [Google Scholar] [CrossRef] [PubMed]
- Suffert, G.; Malterer, G.; Hausser, J.; Viiliainen, J.; Fender, A.; Contrant, M.; Ivacevic, T.; Benes, V.; Gros, F.; Voinnet, O.; et al. Kaposi’s sarcoma herpesvirus microRNAs target caspase 3 and regulate apoptosis. PLoS Pathog. 2011, 7, e1002405. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Jakymiw, A.; Findlay, V.; Parsons, C. KSHV-Encoded MicroRNAs: Lessons for Viral Cancer Pathogenesis and Emerging Concepts. Int. J. Cell Biol. 2012, 2012, 603961. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, V.; Judde, J.G.; Bestett, G.; Cadranel, J.; Antoine, M.; Valensi, F.; Delabesse, E.; Macintyre, E.; Gessain, A. Virological and molecular characterisation of a new B lymphoid cell line, established from an AIDS patient with primary effusion lymphoma, harbouring both KSHV/HHV8 and EBV viruses. Leuk. Lymphoma 2000, 38, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Sakakibara, S.; Ohsaki, E.; Yada, K. Lack of a mechanism for faithful partition and maintenance of the KSHV genome. Virus Res. 2006, 122, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.M.; Medveczky, P.G. Genetic requirements for the episomal maintenance of oncogenic herpesvirus genomes. Adv. Cancer Res. 2002, 84, 155–174. [Google Scholar] [PubMed]
- Ye, F.C.; Zhou, F.C.; Yoo, S.M.; Xie, J.P.; Browning, P.J.; Gao, S.J. Disruption of Kaposi’s sarcoma-associated herpesvirus latent nuclear antigen leads to abortive episome persistence. J. Virol. 2004, 78, 11121–11129. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.Y.; Wilson, A.C. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen induces a strong bend on binding to terminal repeat DNA. J. Virol. 2005, 79, 13829–13836. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Tsai, K.; Chen, H.S.; Wikramasinghe, P.; Davuluri, R.V.; Showe, L.; Domsic, J.; Marmorstein, R.; Lieberman, P.M. Identification of host-chromosome binding sites and candidate gene targets for Kaposi’s sarcoma-associated herpesvirus LANA. J. Virol. 2012, 86, 5752–5762. [Google Scholar] [CrossRef] [PubMed]
- Ballestas, M.E.; Kaye, K.M. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (TR) sequence and specifically binds TR DNA. J. Virol. 2001, 75, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Cotter, M.A., 2nd; Subramanian, C.; Robertson, E.S. The Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen binds to specific sequences at the left end of the viral genome through its carboxy-terminus. Virology 2001, 291, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Barbera, A.J.; Chodaparambil, J.V.; Kelley-Clarke, B.; Joukov, V.; Walter, J.C.; Luger, K.; Kaye, K.M. The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science 2006, 311, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Si, H.; Verma, S.C.; Lampson, M.A.; Cai, Q.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded LANA can interact with the nuclear mitotic apparatus protein to regulate genome maintenance and segregation. J. Virol. 2008, 82, 6734–6746. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Persson, L.M.; Wong, L.; Wilson, A.C. The latency-associated nuclear antigen interacts with MeCP2 and nucleosomes through separate domains. J. Virol. 2010, 84, 2318–2330. [Google Scholar] [CrossRef]
- You, J.; Srinivasan, V.; Denis, G.V.; Harrington, W.J., Jr.; Ballestas, M.E.; Kaye, K.M.; Howley, P.M. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen interacts with bromodomain protein Brd4 on host mitotic chromosomes. J. Virol. 2006, 80, 8909–8919. [Google Scholar] [CrossRef] [PubMed]
- Ottinger, M.; Christalla, T.; Nathan, K.; Brinkmann, M.M.; Viejo-Borbolla, A.; Schulz, T.F. Kaposi’s sarcoma-associated herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J. Virol. 2006, 80, 10772–10786. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Verma, S.C.; Cai, Q.; Kaul, R.; Lu, J.; Saha, A.; Robertson, E.S. Bub1 and CENP-F can contribute to Kaposi’s sarcoma-associated herpesvirus genome persistence by targeting LANA to kinetochores. J. Virol. 2010, 84, 9718–9732. [Google Scholar] [CrossRef] [PubMed]
- Cotter, M.A., 2nd; Robertson, E.S. The latency-associated nuclear antigen tethers the Kaposi’s sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 1999, 264, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.B.; Gerchman, S.E.; Ramakrishnan, V.; Travers, A.; Muyldermans, S. Position and orientation of the globular domain of linker histone H5 on the nucleosome. Nature 1998, 395, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Barbera, A.J.; Chodaparambil, J.V.; Kelley-Clarke, B.; Luger, K.; Kaye, K.M. Kaposi’s sarcoma-associated herpesvirus LANA hitches a ride on the chromosome. Cell Cycle 2006, 5, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Jha, H.C.; Upadhyay, S.K.; MahadeshPrasad, A.J.; Lu, J.; Cai, Q.; Saha, A.; Robertson, E.S. H2AX phosphorylation is important for LANA-mediated Kaposi’s sarcoma-associated herpesvirus episome persistence. J. Virol. 2013, 87, 5255–5269. [Google Scholar] [CrossRef] [PubMed]
- Doxsey, S.; Zimmerman, W.; Mikule, K. Centrosome control of the cell cycle. Trends Cell Biol. 2005, 15, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Guse, A.; Carroll, C.W.; Moree, B.; Fuller, C.J.; Straight, A.F. In vitro centromere and kinetochore assembly on defined chromatin templates. Nature 2011, 477, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.L.; Scott, M.I.; Holt, S.V.; Hussein, D.; Taylor, S.S. Bub1 is required for kinetochore localization of BubR1, Cenp-E, Cenp-F and Mad2, and chromosome congression. J. Cell Sci. 2004, 117, 1577–1589. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, S.; Kobayashi, Y.; Ogiyama, Y.; Takahashi, K. Dual regulation of Mad2 localization on kinetochores by Bub1 and Dam1/DASH that ensure proper spindle interaction. Mol. Biol. Cell 2008, 19, 3885–3897. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Xiao, B.; Jha, H.C.; Lu, J.; Banerjee, S.; Robertson, E.S. Kaposi’s Sarcoma-Associated Herpesvirus-Encoded LANA Can Induce Chromosomal Instability through Targeted Degradation of the Mitotic Checkpoint Kinase Bub1. J. Virol. 2014, 88, 7367–7378. [Google Scholar] [CrossRef] [PubMed]
- Compton, D.A.; Szilak, I.; Cleveland, D.W. Primary structure of NuMA, an intranuclear protein that defines a novel pathway for segregation of proteins at mitosis. J. Cell Biol. 1992, 116, 1395–1408. [Google Scholar] [CrossRef] [PubMed]
- Harborth, J.; Wang, J.; Gueth-Hallonet, C.; Weber, K.; Osborn, M. Self assembly of NuMA: Multiarm oligomers as structural units of a nuclear lattice. EMBO J. 1999, 18, 1689–1700. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Stukenberg, P.T.; Macara, I.G. A mammalian Partner of inscuteable binds NuMA and regulates mitotic spindle organization. Nat. Cell Biol. 2001, 3, 1069–1075. [Google Scholar] [CrossRef]
- Du, Q.; Macara, I.G. Mammalian Pins is a conformational switch that links NuMA to heterotrimeric G proteins. Cell 2004, 119, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Kisurina-Evgenieva, O.; Mack, G.; Du, Q.; Macara, I.; Khodjakov, A.; Compton, D.A. Multiple mechanisms regulate NuMA dynamics at spindle poles. J. Cell Sci. 2004, 117, 6391–6400. [Google Scholar] [CrossRef] [PubMed]
- Gobert, G.N.; Hueser, C.N.; Curran, E.M.; Sun, Q.Y.; Glinsky, V.V.; Welshons, W.V.; Eisenstark, A.; Schatten, H. Immunolocalization of NuMA and phosphorylated proteins during the cell cycle in human breast and prostate cancer cells as analyzed by immunofluorescence and postembedding immunoelectron microscopy. Histochem. Cell Biol. 2001, 115, 381–395. [Google Scholar] [PubMed]
- Sparks, C.A.; Fey, E.G.; Vidair, C.A.; Doxsey, S.J. Phosphorylation of NUMA occurs during nuclear breakdown and not mitotic spindle assembly. J. Cell Sci. 1995, 108, 3389–3396. [Google Scholar] [PubMed]
- Zeng, C.; He, D.; Berget, S.M.; Brinkley, B.R. Nuclear-mitotic apparatus protein: A structural protein interface between the nucleoskeleton and RNA splicing. Proc. Natl. Acad. Sci. USA 1994, 91, 1505–1509. [Google Scholar] [CrossRef] [PubMed]
- Merdes, A.; Cleveland, D.W. The role of NuMA in the interphase nucleus. J. Cell Sci. 1998, 111, 71–79. [Google Scholar] [PubMed]
- Luderus, M.E.; den Blaauwen, J.L.; de Smit, O.J.; Compton, D.A.; van Driel, R. Binding of matrix attachment regions to lamin polymers involves single-stranded regions and the minor groove. Mol. Cell. Biol. 1994, 14, 6297–6305. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Taylor, L.; Compton, D.A.; Macara, I.G. LGN blocks the ability of NuMA to bind and stabilize microtubules. A mechanism for mitotic spindle assembly regulation. Curr. Biol. 2002, 12, 1928–1933. [Google Scholar] [CrossRef] [PubMed]
- Haren, L.; Merdes, A. Direct binding of NuMA to tubulin is mediated by a novel sequence motif in the tail domain that bundles and stabilizes microtubules. J. Cell Sci 2002, 115, 1815–1824. [Google Scholar] [PubMed]
- Merdes, A.; Ramyar, K.; Vechio, J.D.; Cleveland, D.W. A complex of NuMA and cytoplasmic dynein is essential for mitotic spindle assembly. Cell 1996, 87, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Snyder, M. The nuclear-mitotic apparatus protein is important in the establishment and maintenance of the bipolar mitotic spindle apparatus. Mol. Biol. Cell 1992, 3, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Gueth-Hallonet, C.; Weber, K.; Osborn, M. NuMA: A bipartite nuclear location signal and other functional properties of the tail domain. Exp. Cell Res. 1996, 225, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Robertson, E.S. Molecular biology and pathogenesis of Kaposi sarcoma-associated herpesvirus. FEMS Microbiol. Lett. 2003, 222, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Landsman, D. AT-hook motifs identified in a wide variety of DNA-binding proteins. Nucleic Acids Res. 1998, 26, 4413–4421. [Google Scholar] [CrossRef] [PubMed]
- Meehan, R.R.; Lewis, J.D.; Bird, A.P. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 1992, 20, 5085–5092. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Campoy, F.J.; Bird, A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 1997, 88, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Young, J.I.; Hong, E.P.; Castle, J.C.; Crespo-Barreto, J.; Bowman, A.B.; Rose, M.F.; Kang, D.; Richman, R.; Johnson, J.M.; Berget, S.; et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. USA 2005, 102, 17551–17558. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Yasui, D.H.; Peddada, S.; Bieda, M.C.; Vallero, R.O.; Hogart, A.; Nagarajan, R.P.; Thatcher, K.N.; Farnham, P.J.; Lasalle, J.M. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc. Natl. Acad. Sci. USA 2007, 104, 19416–19421. [Google Scholar] [CrossRef] [PubMed]
- Georgel, P.T.; Horowitz-Scherer, R.A.; Adkins, N.; Woodcock, C.L.; Wade, P.A.; Hansen, J.C. Chromatin compaction by human MeCP2. Assembly of novel secondary chromatin structures in the absence of DNA methylation. J. Biol. Chem. 2003, 278, 32181–32188. [Google Scholar] [CrossRef] [PubMed]
- Nikitina, T.; Ghosh, R.P.; Horowitz-Scherer, R.A.; Hansen, J.C.; Grigoryev, S.A.; Woodcock, C.L. MeCP2-chromatin interactions include the formation of chromatosome-like structures and are altered in mutations causing Rett syndrome. J. Biol. Chem. 2007, 282, 28237–28245. [Google Scholar] [CrossRef] [PubMed]
- Krithivas, A.; Fujimuro, M.; Weidner, M.; Young, D.B.; Hayward, S.D. Protein interactions targeting the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus to cell chromosomes. J. Virol. 2002, 76, 11596–11604. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.; Scholten, I.; Kappes, F.; Hu, H.G.; Knippers, R. The DEK protein—An abundant and ubiquitous constituent of mammalian chromatin. Gene 2004, 343, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Von Lindern, M.; Fornerod, M.; van Baal, S.; Jaegle, M.; de Wit, T.; Buijs, A.; Grosveld, G. The translocation (6;9), associated with a specific subtype of acute myeloid leukemia, results in the fusion of two genes, dek and can, and the expression of a chimeric, leukemia-specific dek-can mRNA. Mol. Cell. Biol. 1992, 12, 1687–1697. [Google Scholar] [PubMed]
- Faulkner, N.E.; Hilfinger, J.M.; Markovitz, D.M. Protein phosphatase 2A activates the HIV-2 promoter through enhancer elements that include the pets site. J. Biol. Chem. 2001, 276, 25804–25812. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.K.; Markovitz, D.M. Purification of the pets factor. A nuclear protein that binds to the inducible TG-rich element of the human immunodeficiency virus type 2 enhancer. J. Biol. Chem. 1996, 271, 19599–19605. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.K.; Grosveld, G.; Markovitz, D.M. DEK, an autoantigen involved in a chromosomal translocation in acute myelogenous leukemia, binds to the HIV-2 enhancer. Proc. Natl. Acad. Sci. USA 1997, 94, 1811–1815. [Google Scholar] [CrossRef] [PubMed]
- McGarvey, T.; Rosonina, E.; McCracken, S.; Li, Q.; Arnaout, R.; Mientjes, E.; Nickerson, J.A.; Awrey, D.; Greenblatt, J.; Grosveld, G.; et al. The acute myeloid leukemia-associated protein, DEK, forms a splicing-dependent interaction with exon-product complexes. J. Cell Biol. 2000, 150, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Alexiadis, V.; Waldmann, T.; Andersen, J.; Mann, M.; Knippers, R.; Gruss, C. The protein encoded by the proto-oncogene DEK changes the topology of chromatin and reduces the efficiency of DNA replication in a chromatin-specific manner. Genes Dev. 2000, 14, 1308–1312. [Google Scholar] [PubMed]
- Chua, P.; Roeder, G.S. Bdf1, a yeast chromosomal protein required for sporulation. Mol. Cell. Biol. 1995, 15, 3685–3696. [Google Scholar] [PubMed]
- Florence, B.; Faller, D.V. You bet-cha: A novel family of transcriptional regulators. Front. Biosci. 2001, 6, D1008–D1018. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Faller, D.V.; Denis, G.V. Bromodomain analysis of Brd2-dependent transcriptional activation of cyclin A. Biochem. J. 2005, 387, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Houzelstein, D.; Bullock, S.L.; Lynch, D.E.; Grigorieva, E.F.; Wilson, V.A.; Beddington, R.S. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol. Cell. Biol. 2002, 22, 3794–3802. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Croyle, J.L.; Nishimura, A.; Ozato, K.; Howley, P.M. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 2004, 117, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Pamblanco, M.; Poveda, A.; Sendra, R.; Rodriguez-Navarro, S.; Perez-Ortin, J.E.; Tordera, V. Bromodomain factor 1 (Bdf1) protein interacts with histones. FEBS Lett. 2001, 496, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Kanno, Y.; Siegel, R.M.; Jang, M.K.; Lenardo, M.J.; Ozato, K. Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Mol. Cell 2004, 13, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Chitsaz, F.; Abbasi, A.; Misteli, T.; Ozato, K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8758–8763. [Google Scholar] [CrossRef] [PubMed]
- Denis, G.V.; Vaziri, C.; Guo, N.; Faller, D.V. RING3 kinase transactivates promoters of cell cycle regulatory genes through E2F. Cell Growth Differ. 2000, 11, 417–424. [Google Scholar] [PubMed]
- Platt, G.M.; Simpson, G.R.; Mittnacht, S.; Schulz, T.F. Latent nuclear antigen of Kaposi’s sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J. Virol. 1999, 73, 9789–9795. [Google Scholar] [PubMed]
- Mattsson, K.; Kiss, C.; Platt, G.M.; Simpson, G.R.; Kashuba, E.; Klein, G.; Schulz, T.F.; Szekely, L. Latent nuclear antigen of Kaposi’s sarcoma herpesvirus/human herpesvirus-8 induces and relocates RING3 to nuclear heterochromatin regions. J. Gen. Virol. 2002, 83, 179–188. [Google Scholar] [PubMed]
- Haynes, S.R.; Dollard, C.; Winston, F.; Beck, S.; Trowsdale, J.; Dawid, I.B. The bromodomain: A conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res. 1992, 20, 2603. [Google Scholar] [CrossRef] [PubMed]
- Nomura, N.; Nagase, T.; Miyajima, N.; Sazuka, T.; Tanaka, A.; Sato, S.; Seki, N.; Kawarabayasi, Y.; Ishikawa, K.; Tabata, S. Prediction of the coding sequences of unidentified human genes. II. The coding sequences of 40 new genes (KIAA0041-KIAA0080) deduced by analysis of cDNA clones from human cell line KG-1. DNA Res. 1994, 1, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.H.; Numata, M.; Shimane, M. Identification and characterization of BRDT: A testis-specific gene related to the bromodomain genes RING3 and Drosophila fsh. Genomics 1997, 45, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Stedman, W.; Deng, Z.; Lu, F.; Lieberman, P.M. ORC, MCM, and histone hyperacetylation at the Kaposi’s sarcoma-associated herpesvirus latent replication origin. J. Virol. 2004, 78, 12566–12575. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Choudhuri, T.; Kaul, R.; Robertson, E.S. Latency-associated nuclear antigen (LANA) of Kaposi’s sarcoma-associated herpesvirus interacts with origin recognition complexes at the LANA binding sequence within the terminal repeats. J. Virol. 2006, 80, 2243–2256. [Google Scholar] [CrossRef] [PubMed]
- Dheekollu, J.; Chen, H.S.; Kaye, K.M.; Lieberman, P.M. Timeless-dependent DNA replication-coupled recombination promotes Kaposi’s Sarcoma-associated herpesvirus episome maintenance and terminal repeat stability. J. Virol. 2013, 87, 3699–3709. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, P.; McDowell, M.E.; McGuinness, J.; Salas, R.; Rumjahn, S.M.; Verma, S.C. Kaposi’s Sarcoma-Associated Herpesvirus-Encoded LANA Recruits Topoisomerase IIbeta for Latent DNA Replication of the Terminal Repeats. J. Virol. 2012, 86, 9983–9994. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Tsurimoto, T.; Juillard, F.; Li, L.; Li, S.; de Leon Vazquez, E.; Chen, S.; Kaye, K. Kaposi’s sarcoma-associated herpesvirus LANA recruits the DNA polymerase clamp loader to mediate efficient replication and virus persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 11816–11821. [Google Scholar] [CrossRef] [PubMed]
- Green, C.M.; Erdjument-Bromage, H.; Tempst, P.; Lowndes, N.F. A novel Rad24 checkpoint protein complex closely related to replication factor C. Curr. Biol. 2000, 10, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Zhou, J.; Wiedmer, A.; Madden, K.; Yuan, Y.; Lieberman, P.M. Chromatin remodeling of the Kaposi’s sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J. Virol. 2003, 77, 11425–11435. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Jha, H.C.; Verma, S.C.; Sun, Z.; Banerjee, S.; Dzeng, R.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded LANA contributes to viral latent replication by activating phosphorylation of survivin. J. Virol. 2014, 88, 4204–4217. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Verma, S.C.; Murakami, M.; Cai, Q.; Kumar, P.; Xiao, B.; Robertson, E.S. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus (KSHV) upregulates survivin expression in KSHV-associated B-lymphoma cells and contributes to their proliferation. J. Virol. 2009, 83, 7129–7141. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Lu, J.; Cai, Q.; Kosiyatrakul, S.; McDowell, M.E.; Schildkraut, C.L.; Robertson, E.S. Single molecule analysis of replicated DNA reveals the usage of multiple KSHV genome regions for latent replication. PLoS Pathog. 2011, 7, e1002365. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Lan, K.; Choudhuri, T.; Cotter, M.A.; Robertson, E.S. An autonomous replicating element within the KSHV genome. Cell Host Microbe 2007, 2, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hilton, I.B.; Staudt, M.R.; Burd, C.E.; Dittmer, D.P. Distinct p53, p53: LANA, and LANA complexes in Kaposi’s Sarcoma-associated Herpesvirus Lymphomas. J. Virol. 2010, 84, 3898–3908. [Google Scholar] [CrossRef] [PubMed]
- Friborg, J., Jr.; Kong, W.; Hottiger, M.O.; Nabel, G.J. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 1999, 402, 889–894. [Google Scholar] [PubMed]
- Krithivas, A.; Young, D.B.; Liao, G.; Greene, D.; Hayward, S.D. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J. Virol. 2000, 74, 9637–9645. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Kuppers, D.A.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J. Virol. 2005, 79, 3468–3478. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Lee, D.; Seo, T.; Choi, C.; Choe, J. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus functionally interacts with heterochromatin protein 1. J. Biol. Chem. 2003, 278, 7397–7405. [Google Scholar] [CrossRef] [PubMed]
- Ohsaki, E.; Ueda, K.; Sakakibara, S.; Do, E.; Yada, K.; Yamanishi, K. Poly(ADP-ribose) polymerase 1 binds to Kaposi’s sarcoma-associated herpesvirus (KSHV) terminal repeat sequence and modulates KSHV replication in latency. J. Virol. 2004, 78, 9936–9946. [Google Scholar] [CrossRef] [PubMed]
- Shamay, M.; Krithivas, A.; Zhang, J.; Hayward, S.D. Recruitment of the de novo DNA methyltransferase Dnmt3a by Kaposi’s sarcoma-associated herpesvirus LANA. Proc. Natl. Acad. Sci. USA 2006, 103, 14554–14559. [Google Scholar] [CrossRef] [PubMed]
- Si, H.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen induces chromosomal instability through inhibition of p53 function. J. Virol. 2006, 80, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Viejo-Borbolla, A.; Kati, E.; Sheldon, J.A.; Nathan, K.; Mattsson, K.; Szekely, L.; Schulz, T.F. A Domain in the C-terminal region of latency-associated nuclear antigen 1 of Kaposi’s sarcoma-associated Herpesvirus affects transcriptional activation and binding to nuclear heterochromatin. J. Virol. 2003, 77, 7093–7100. [Google Scholar] [CrossRef] [PubMed]
- Viejo-Borbolla, A.; Ottinger, M.; Bruning, E.; Burger, A.; Konig, R.; Kati, E.; Sheldon, J.A.; Schulz, T.F. Brd2/RING3 interacts with a chromatin-binding domain in the Kaposi’s Sarcoma-associated herpesvirus latency-associated nuclear antigen 1 (LANA-1) that is required for multiple functions of LANA-1. J. Virol. 2005, 79, 13618–13629. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Yang, Y.; Turner, P.C.; Jain, V.; McIntyre, L.M.; Renne, R. LANA binds to multiple active viral and cellular promoters and associates with the H3K4methyltransferase hSET1 complex. PLoS Pathog. 2014, 10, e1004240. [Google Scholar] [CrossRef] [PubMed]
- Mercier, A.; Arias, C.; Madrid, A.S.; Holdorf, M.M.; Ganem, D. Site-Specific Association with Host and Viral Chromatin by Kaposi’s Sarcoma-Associated Herpesvirus LANA and Its Reversal during Lytic Reactivation. J. Virol. 2014, 88, 6762–6777. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, M.; del Cid, N.; Rizvi, S.M.; Peters, L.R. MHC class I assembly: Out and about. Trends Immunol. 2008, 29, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Gordon, G.M.; Muller, M.G.; Dahiya, M.; Foreman, K.E. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen induces expression of the helix-loop-helix protein Id-1 in human endothelial cells. J. Virol. 2003, 77, 5975–5984. [Google Scholar] [CrossRef] [PubMed]
- Guito, J.; Lukac, D.M. KSHV Rta Promoter Specification and Viral Reactivation. Front. Microbiol. 2012, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Lin, S.F.; Gradoville, L.; Yuan, Y.; Zhu, F.; Miller, G. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1998, 95, 10866–10871. [Google Scholar] [CrossRef] [PubMed]
- Bu, W.; Palmeri, D.; Krishnan, R.; Marin, R.; Aris, V.M.; Soteropoulos, P.; Lukac, D.M. Identification of direct transcriptional targets of the Kaposi’s sarcoma-associated herpesvirus Rta lytic switch protein by conditional nuclear localization. J. Virol. 2008, 82, 10709–10723. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Young, A.; Sun, R. Auto-activation of the rta gene of human herpesvirus-8/Kaposi’s sarcoma-associated herpesvirus. J. Gen. Virol. 2000, 81, 3043–3048. [Google Scholar] [PubMed]
- Li, Q.; Zhou, F.; Ye, F.; Gao, S.J. Genetic disruption of KSHV major latent nuclear antigen LANA enhances viral lytic transcriptional program. Virology 2008, 379, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.; Izumiya, Y. Post-Translational Modifications of Kaposi’s Sarcoma-Associated Herpesvirus Regulatory Proteins—SUMO and KSHV. Front. Microbiol. 2012, 3, 31. [Google Scholar] [PubMed]
- Bachman, K.E.; Rountree, M.R.; Baylin, S.B. Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J. Biol. Chem. 2001, 276, 32282–32287. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Cai, Q.; Kreider, E.; Lu, J.; Robertson, E.S. Comprehensive analysis of LANA interacting proteins essential for viral genome tethering and persistence. PLoS One 2013, 8, e74662. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, S.; Ueda, K.; Nishimura, K.; Do, E.; Ohsaki, E.; Okuno, T.; Yamanishi, K. Accumulation of heterochromatin components on the terminal repeat sequence of Kaposi’s sarcoma-associated herpesvirus mediated by the latency-associated nuclear antigen. J. Virol. 2004, 78, 7299–7310. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Huerta, S.B.; Izumiya, C.; Wang, D.H.; Martinez, A.; Shevchenko, B.; Kung, H.J.; Campbell, M.; Izumiya, Y. Kaposi’s sarcoma-associated herpesvirus (KSHV) latency-associated nuclear antigen regulates the KSHV epigenome by association with the histone demethylase KDM3A. J. Virol. 2013, 87, 6782–6793. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Verma, S.C.; Cai, Q.; Robertson, E.S. The single RBP-Jkappa site within the LANA promoter is crucial for establishing Kaposi’s sarcoma-associated herpesvirus latency during primary infection. J. Virol. 2011, 85, 6148–6161. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Verma, S.C.; Cai, Q.; Saha, A.; Dzeng, R.K.; Robertson, E.S. The RBP-Jkappa binding sites within the RTA promoter regulate KSHV latent infection and cell proliferation. PLoS Pathog. 2012, 8, e1002479. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Gray, K.S.; Forrest, J.C.; Speck, S.H. The de novo methyltransferases DNMT3a and DNMT3b target the murine gammaherpesvirus immediate-early gene 50 promoter during establishment of latency. J. Virol. 2010, 84, 4946–4959. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Day, L.; Gao, S.J.; Lieberman, P.M. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi’s sarcoma-associated herpesvirus lytic transcription. J. Virol. 2006, 80, 5273–5282. [Google Scholar] [CrossRef] [PubMed]
- Fujimuro, M.; Liu, J.; Zhu, J.; Yokosawa, H.; Hayward, S.D. Regulation of the interaction between glycogen synthase kinase 3 and the Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen. J. Virol. 2005, 79, 10429–10441. [Google Scholar] [CrossRef]
- Cha, S.; Lim, C.; Lee, J.Y.; Song, Y.J.; Park, J.; Choe, J.; Seo, T. DNA-PK/Ku complex binds to latency-associated nuclear antigen and negatively regulates Kaposi’s sarcoma-associated herpesvirus latent replication. Biochem. Biophys. Res. Commun. 2010, 394, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Weidner-Glunde, M.; Varjosalo, M.; Rainio, E.M.; Lehtonen, A.; Schulz, T.F.; Koskinen, P.J.; Taipale, J.; Ojala, P.M. KSHV reactivation from latency requires Pim-1 and Pim-3 kinases to inactivate the latency-associated nuclear antigen LANA. PLoS Pathog. 2009, 5, e1000324. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.; Chang, P.C.; Huerta, S.; Izumiya, C.; Davis, R.; Tepper, C.G.; Kim, K.Y.; Shevchenko, B.; Wang, D.H.; Jung, J.U.; et al. Protein arginine methyltransferase 1-directed methylation of Kaposi sarcoma-associated herpesvirus latency-associated nuclear antigen. J. Biol. Chem. 2012, 287, 5806–5818. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Lieberman, P.M.; Jung, J.U.; McBride, A.A.; Morris, K.V.; Ott, M.; Margolis, D.; Nieto, A.; Nevels, M.; Parks, R.J.; et al. Snapshots: Chromatin control of viral infection. Virology 2013, 435, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Gunther, T.; Grundhoff, A. The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog. 2010, 6, e1000935. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.; Maglinte, D.T.; Lee, S.H.; Lee, H.R.; Wong, L.Y.; Brulois, K.F.; Lee, S.; Buckley, J.D.; Laird, P.W.; Marquez, V.E.; et al. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 2010, 6, e1001013. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.; Brulois, K.; Lee, H.R.; Izumiya, Y.; Tepper, C.; Kung, H.J.; Jung, J.U. Biphasic euchromatin-to-heterochromatin transition on the KSHV genome following de novo infection. PLoS Pathog. 2013, 9, e1003813. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Pari, G. KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLoS Pathog. 2012, 8, e1002680. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Zhang, W.; Bakken, T.; Schutten, M.; Toth, Z.; Jung, J.U.; Gill, P.; Cannon, M.; Gao, S.J. Cancer angiogenesis induced by Kaposi sarcoma-associated herpesvirus is mediated by EZH2. Cancer Res. 2012, 72, 3582–3592. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Lan, K.; Robertson, E. Structure and function of latency-associated nuclear antigen. Curr. Top. Microbiol. Immunol. 2007, 312, 101–136. [Google Scholar] [PubMed]
- Liang, D.; Hu, H.; Li, S.; Dong, J.; Wang, X.; Wang, Y.; He, L.; He, Z.; Gao, Y.; Gao, S.J.; et al. Oncogenic herpesvirus KSHV Hijacks BMP-Smad1-Id signaling to promote tumorigenesis. PLoS Pathog. 2014, 10, e1004253. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Borah, S.; Robertson, E.S. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J. Virol. 2004, 78, 10348–10359. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Sohn, H.; Gwack, Y.; Choe, J. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) binds ATF4/CREB2 and inhibits its transcriptional activation activity. J. Gen. Virol. 2000, 81, 2645–2652. [Google Scholar] [PubMed]
- Murakami, Y.; Yamagoe, S.; Noguchi, K.; Takebe, Y.; Takahashi, N.; Uehara, Y.; Fukazawa, H. Ets-1-dependent expression of vascular endothelial growth factor receptors is activated by latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus through interaction with Daxx. J. Biol. Chem. 2006, 281, 28113–28121. [Google Scholar] [CrossRef] [PubMed]
- Radkov, S.A.; Kellam, P.; Boshoff, C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 2000, 6, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Fujimuro, M.; Hayward, S.D. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus manipulates the activity of glycogen synthase kinase-3beta. J. Virol. 2003, 77, 8019–8030. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Gwack, Y.; Hwang, S.; Kim, S.; Choe, J. The transcriptional activity of cAMP response element-binding protein-binding protein is modulated by the latency associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus. J. Biol. Chem. 2001, 276, 31016–31022. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.L.; Knight, J.S.; Verma, S.C.; Zald, P.; Robertson, E.S. EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog. 2006, 2, e116. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Xiao, B.; Si, H.; Cervini, A.; Gao, J.; Lu, J.; Upadhyay, S.K.; Verma, S.C.; Robertson, E.S. Kaposi’s sarcoma herpesvirus upregulates Aurora A expression to promote p53 phosphorylation and ubiquitylation. PLoS Pathog. 2012, 8, e1002566. [Google Scholar] [CrossRef] [PubMed]
- Hume, A.J.; Kalejta, R.F. Regulation of the retinoblastoma proteins by the human herpesviruses. Cell Div. 2009, 4, e1. [Google Scholar] [CrossRef]
- Cousins, E.; Nicholas, J. Molecular biology of human herpesvirus 8: Novel functions and virus-host interactions implicated in viral pathogenesis and replication. Recent Results Cancer Res. 2014, 193, 227–268. [Google Scholar] [PubMed]
- Liu, J.; Martin, H.; Shamay, M.; Woodard, C.; Tang, Q.Q.; Hayward, S.D. Kaposi’s sarcoma-associated herpesvirus LANA protein downregulates nuclear glycogen synthase kinase 3 activity and consequently blocks differentiation. J. Virol. 2007, 81, 4722–4731. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Martin, H.J.; Liao, G.; Hayward, S.D. The Kaposi’s sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc. J. Virol. 2007, 81, 10451–10459. [Google Scholar] [CrossRef] [PubMed]
- Paudel, N.; Sadagopan, S.; Chakraborty, S.; Sarek, G.; Ojala, P.M.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen interacts with multifunctional angiogenin to utilize its antiapoptotic functions. J. Virol. 2012, 86, 5974–5991. [Google Scholar] [CrossRef] [PubMed]
- Paudel, N.; Sadagopan, S.; Balasubramanian, S.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen and angiogenin interact with common host proteins, including annexin A2, which is essential for survival of latently infected cells. J. Virol. 2012, 86, 1589–1607. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, X.; Li, Y.; Southwood, M.; Ye, L.; Long, L.; Al-Lamki, R.S.; Morrell, N.W. Id proteins are critical downstream effectors of BMP signaling in human pulmonary arterial smooth muscle cells. American J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L312–L321. [Google Scholar] [CrossRef]
- Cai, Q.; Cai, S.; Zhu, C.; Verma, S.C.; Choi, J.Y.; Robertson, E.S. A Unique SUMO-2-Interacting Motif within LANA Is Essential for KSHV Latency. PLoS Pathog. 2013, 9, e1003750. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Liang, D.; Gao, Y.; Lan, K. Kaposi’s sarcoma-associated herpesvirus-encoded LANA interacts with host KAP1 to facilitate establishment of viral latency. J. Virol. 2014, 88, 7331–7344. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhu, C.; Guo, Y.; Wei, F.; Lu, J.; Qin, J.; Banerjee, S.; Wang, J.; Shang, H.; Verma, S.C.; et al. Inhibition of KAP1 enhances hypoxia-induced Kaposi’s sarcoma-associated herpesvirus reactivation through RBP-Jkappa. J. Virol. 2014, 88, 6873–6884. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.C.; Fitzgerald, L.D.; van Geelen, A.; Izumiya, Y.; Ellison, T.J.; Wang, D.H.; Ann, D.K.; Luciw, P.A.; Kung, H.J. Kruppel-associated box domain-associated protein-1 as a latency regulator for Kaposi’s sarcoma-associated herpesvirus and its modulation by the viral protein kinase. Cancer Res. 2009, 69, 5681–5689. [Google Scholar] [CrossRef] [PubMed]
- Drane, P.; Ouararhni, K.; Depaux, A.; Shuaib, M.; Hamiche, A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010, 24, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uppal, T.; Banerjee, S.; Sun, Z.; Verma, S.C.; Robertson, E.S. KSHV LANA—The Master Regulator of KSHV Latency. Viruses 2014, 6, 4961-4998. https://doi.org/10.3390/v6124961
Uppal T, Banerjee S, Sun Z, Verma SC, Robertson ES. KSHV LANA—The Master Regulator of KSHV Latency. Viruses. 2014; 6(12):4961-4998. https://doi.org/10.3390/v6124961
Chicago/Turabian StyleUppal, Timsy, Sagarika Banerjee, Zhiguo Sun, Subhash C. Verma, and Erle S. Robertson. 2014. "KSHV LANA—The Master Regulator of KSHV Latency" Viruses 6, no. 12: 4961-4998. https://doi.org/10.3390/v6124961
APA StyleUppal, T., Banerjee, S., Sun, Z., Verma, S. C., & Robertson, E. S. (2014). KSHV LANA—The Master Regulator of KSHV Latency. Viruses, 6(12), 4961-4998. https://doi.org/10.3390/v6124961