Assessing Proteinase K Resistance of Fish Prion Proteins in a Scrapie-Infected Mouse Neuroblastoma Cell Line

 ,
, 
 and
and
Abstract
:1. Introduction
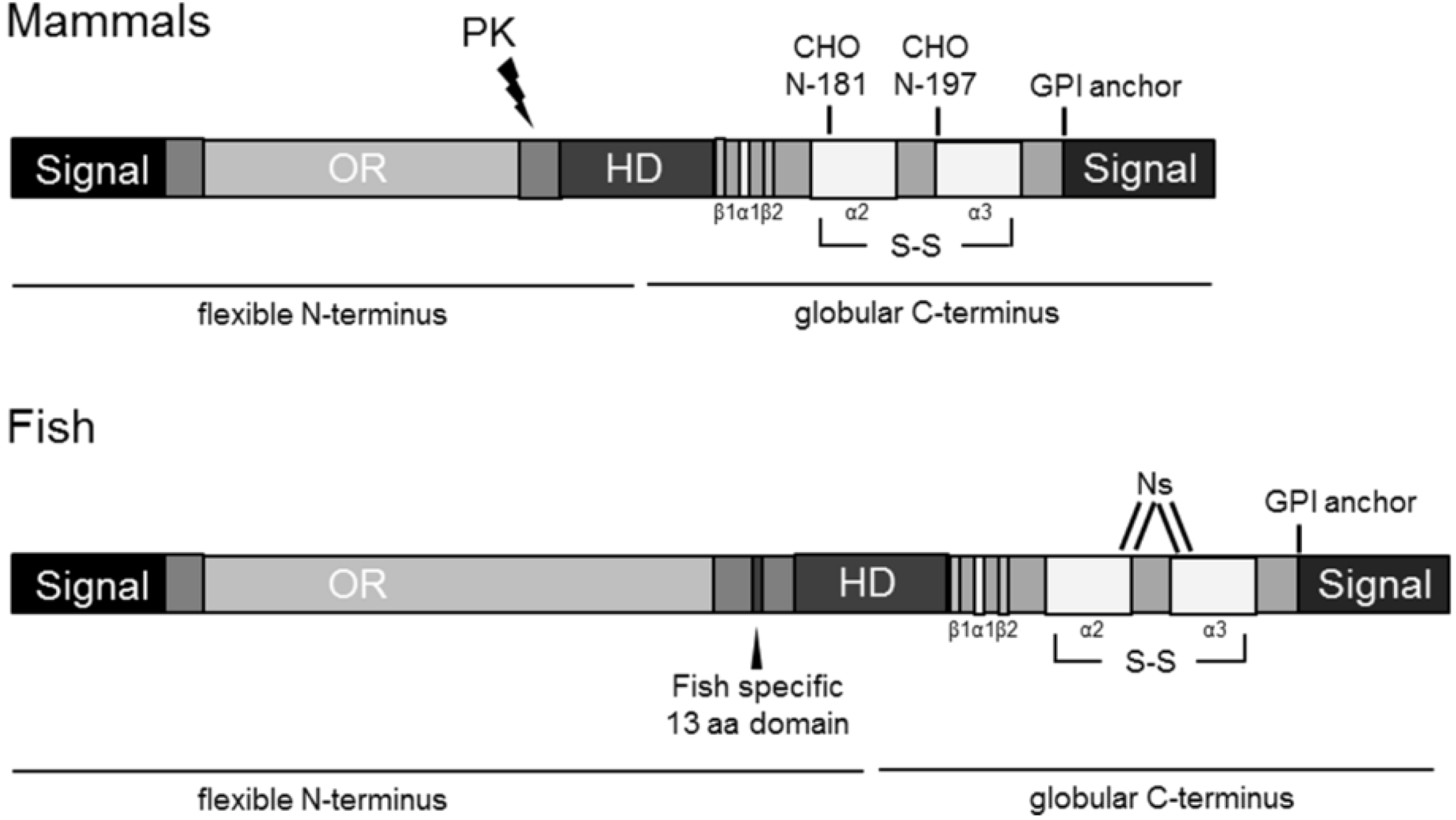
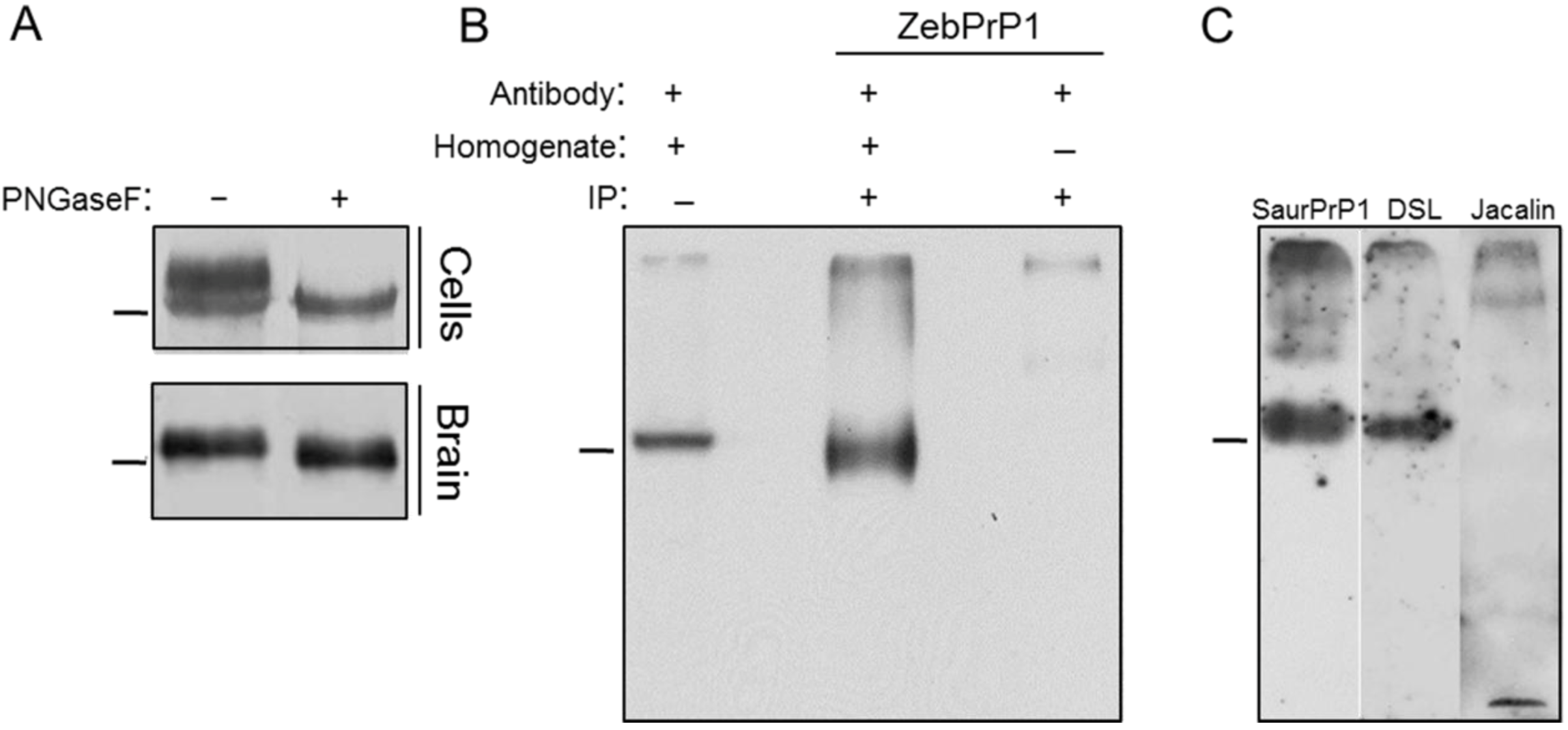
2. Results
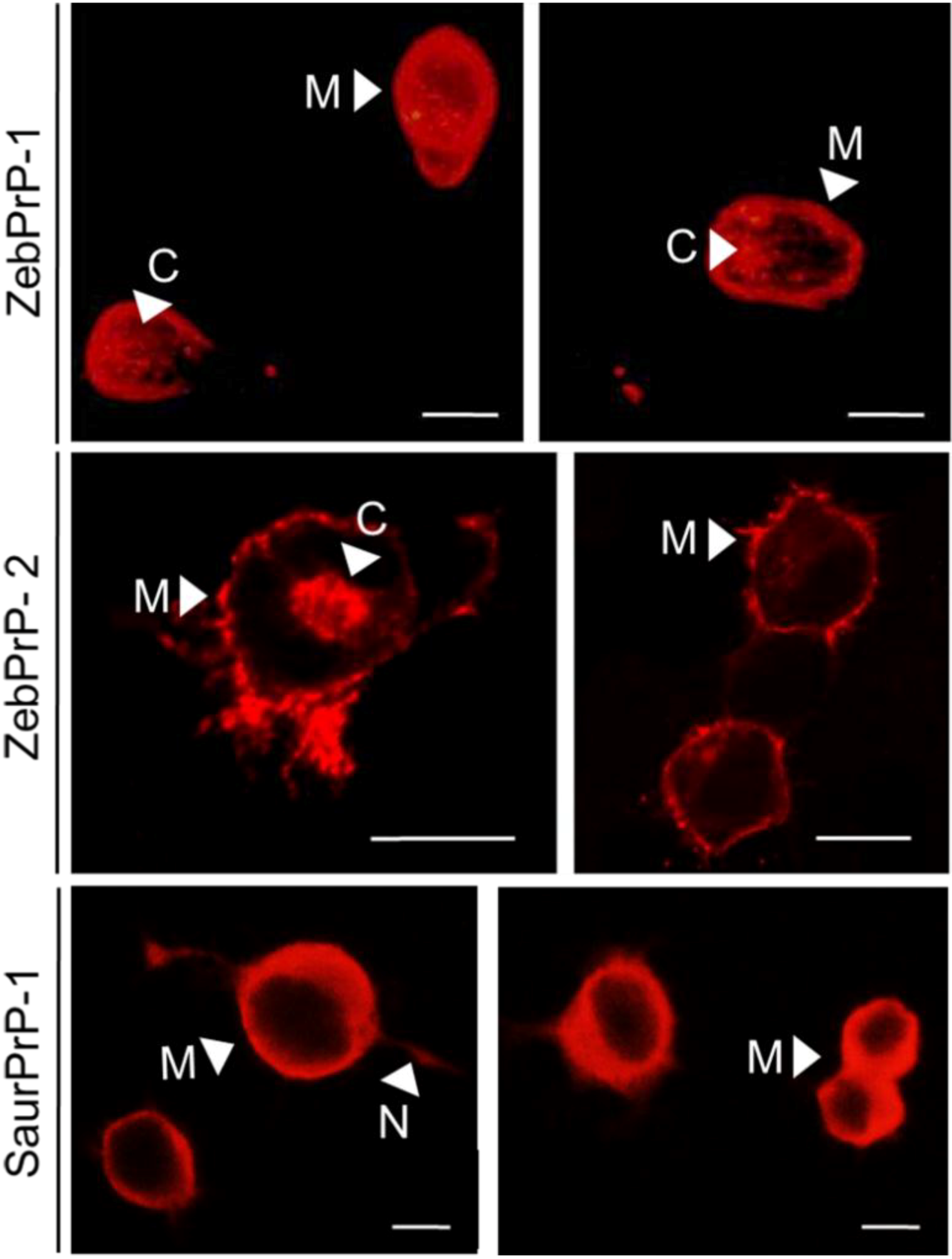
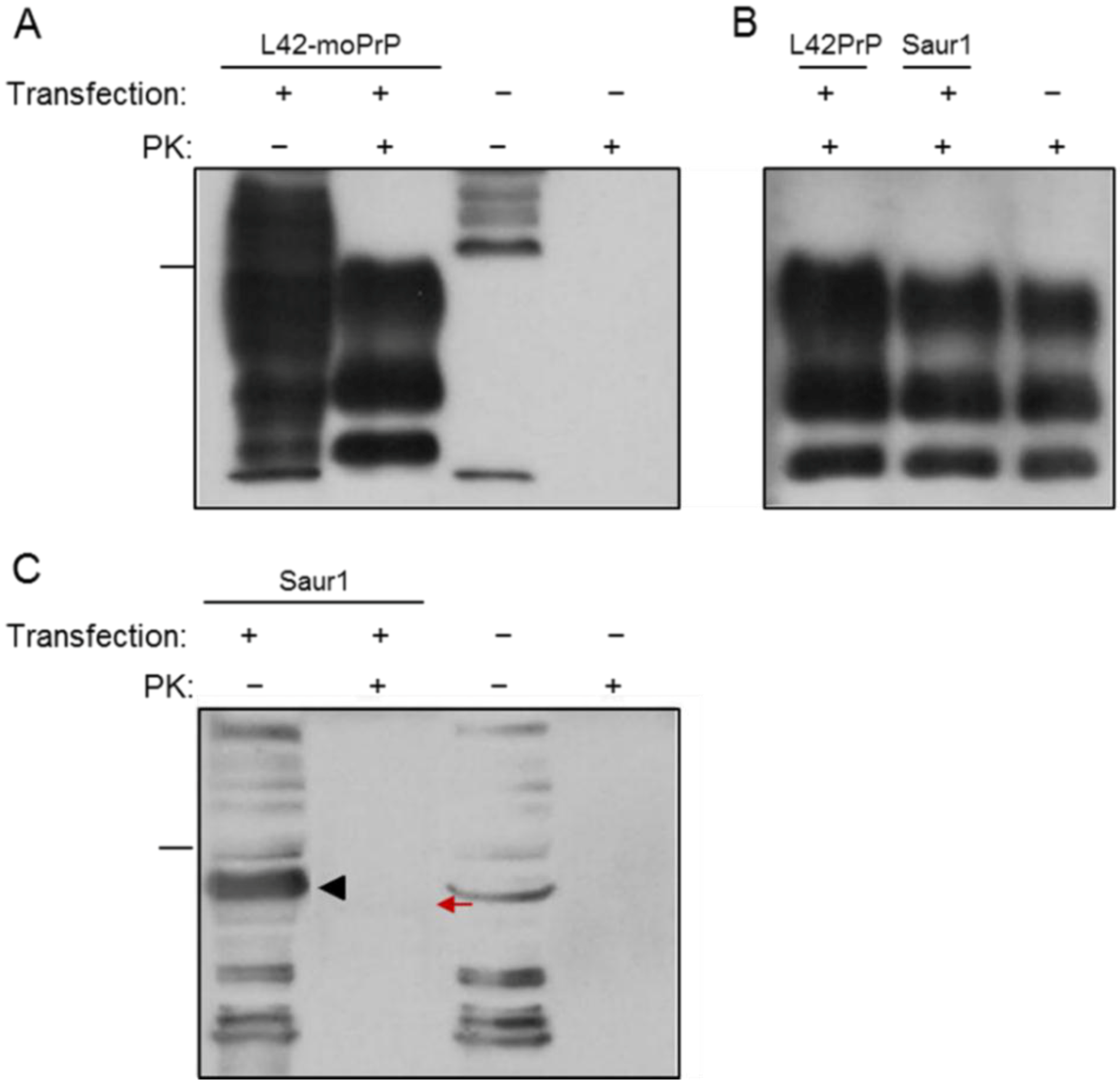
2.1. Expression and Post-Translational Processing of Piscine Prion Proteins in Neuroblastoma Cells
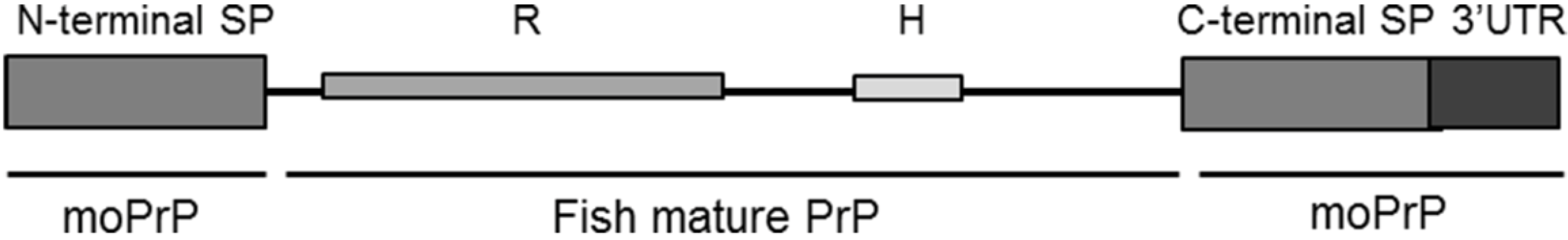
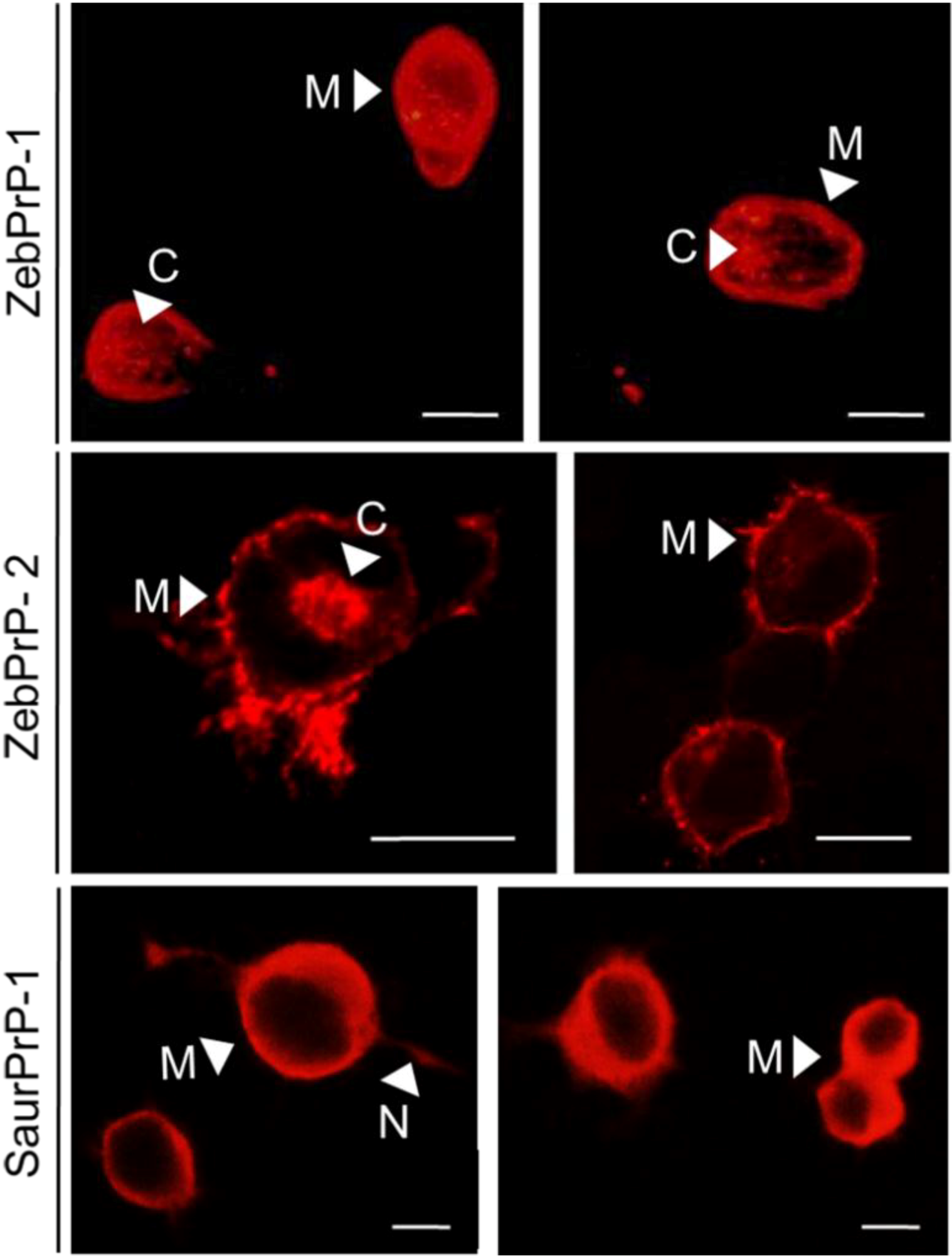
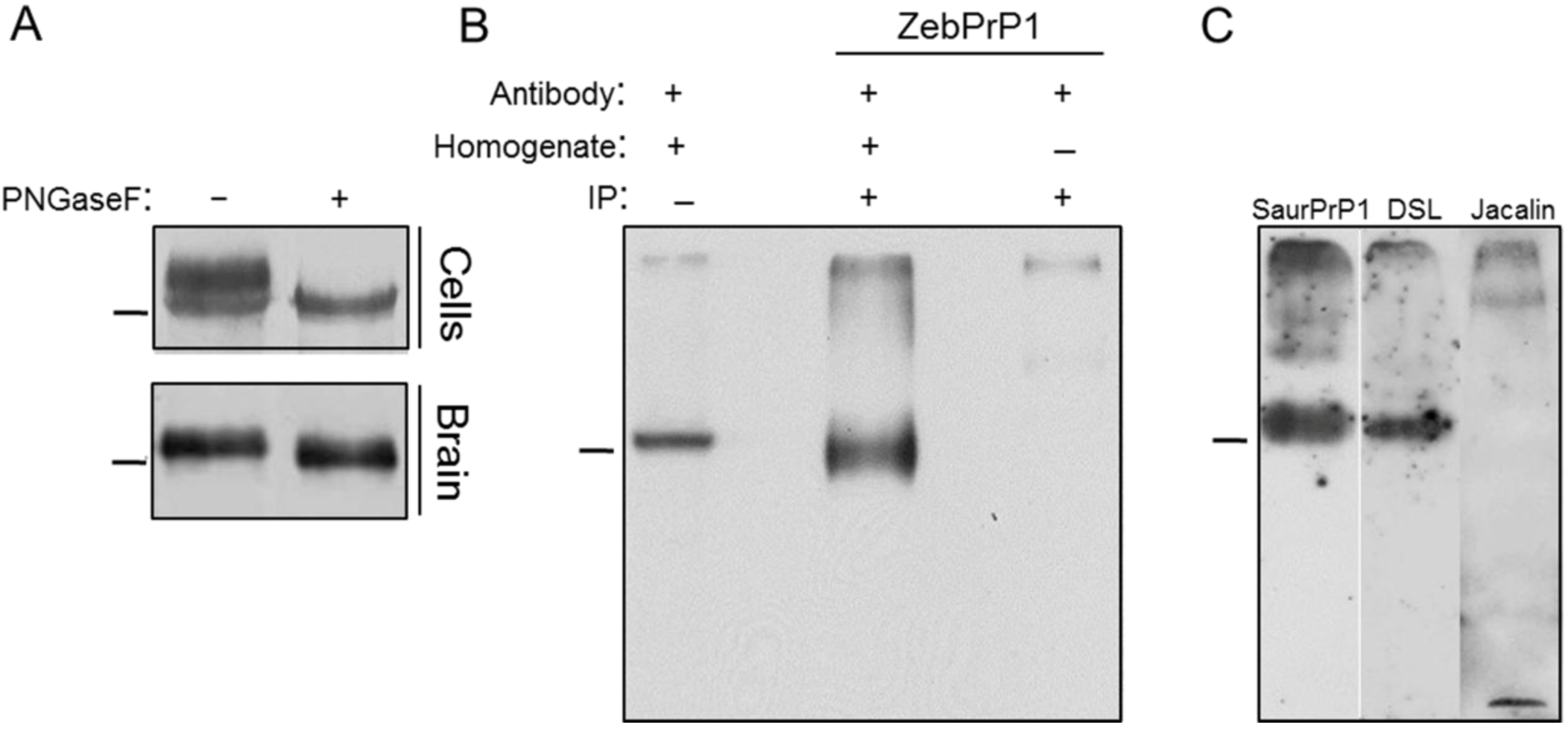
2.2. Evaluation of Piscine PrP Convertibility
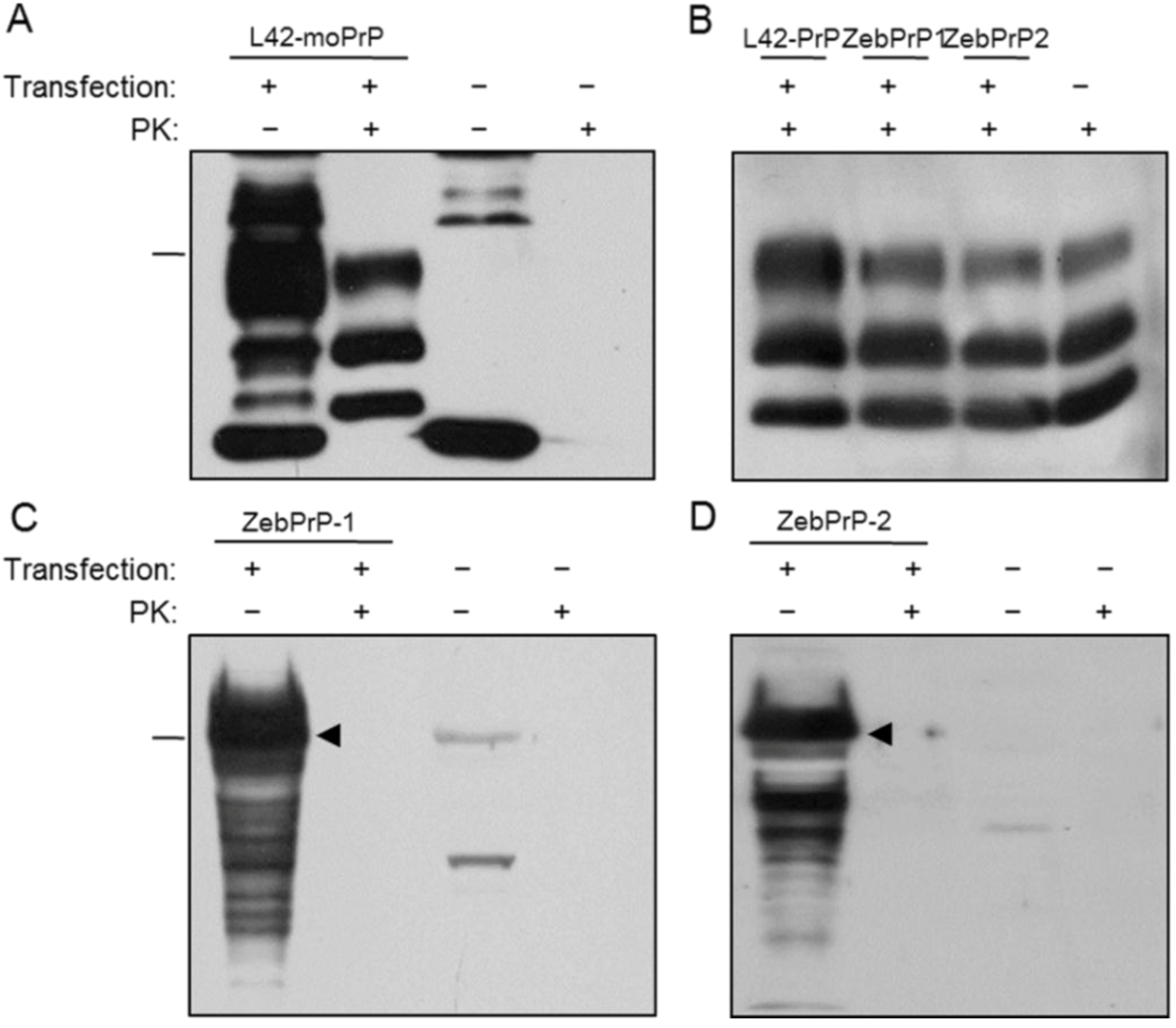
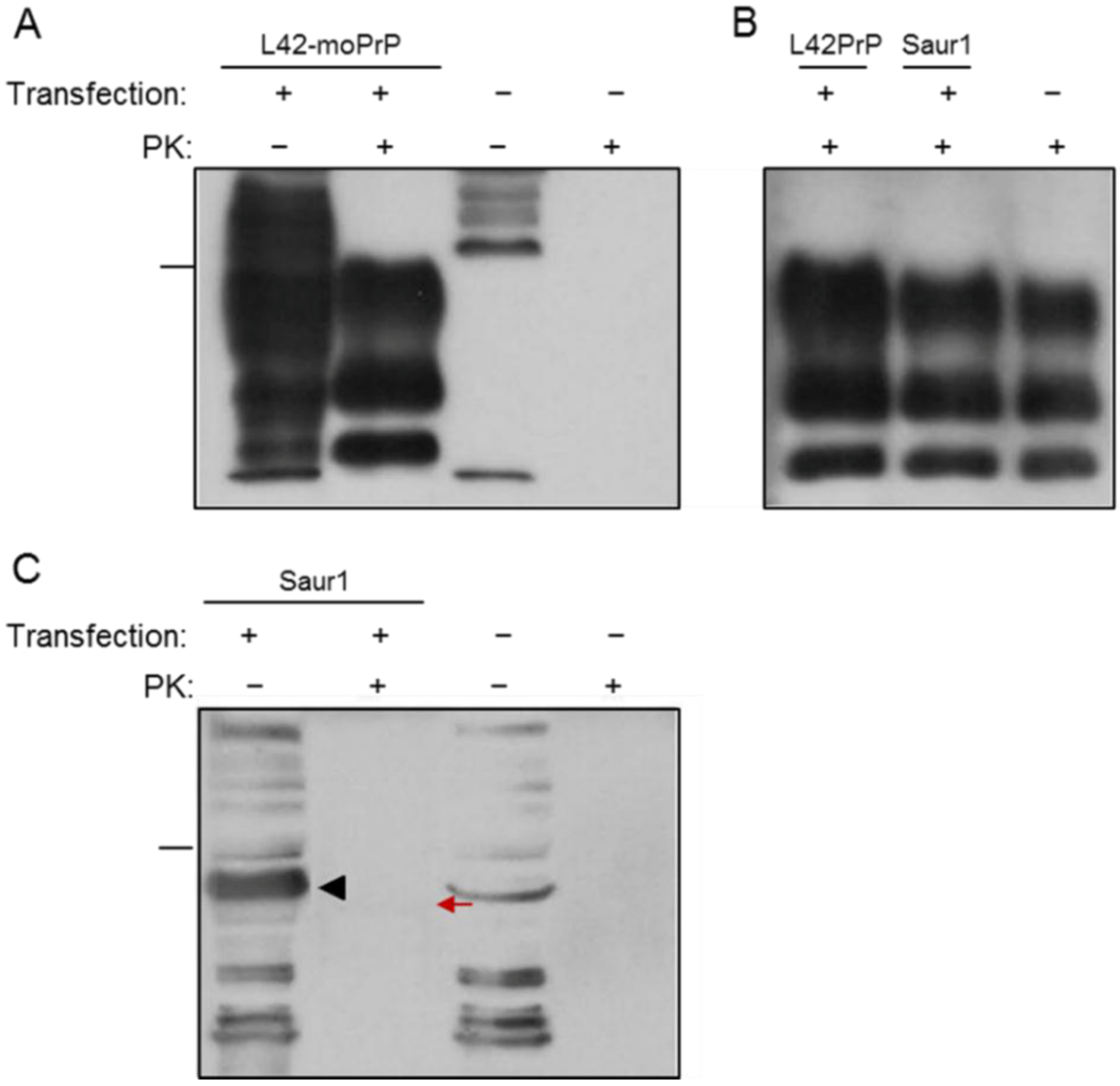
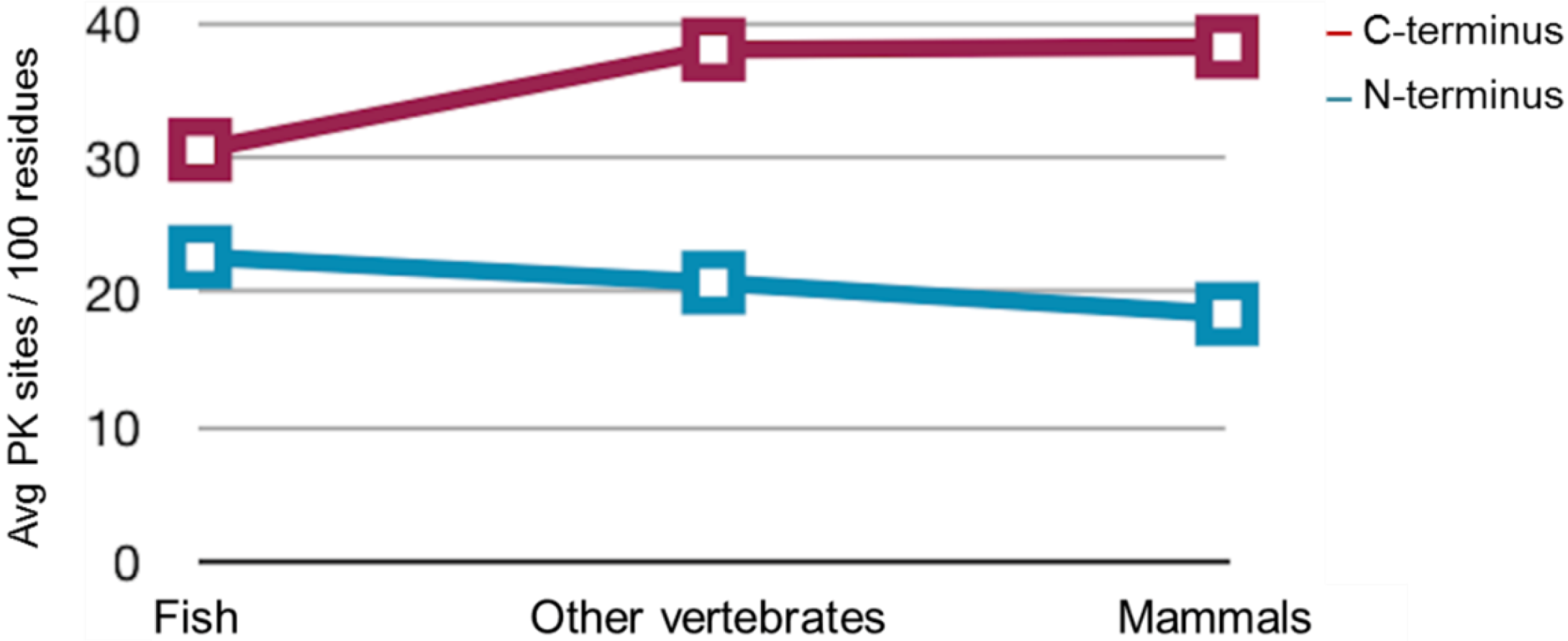
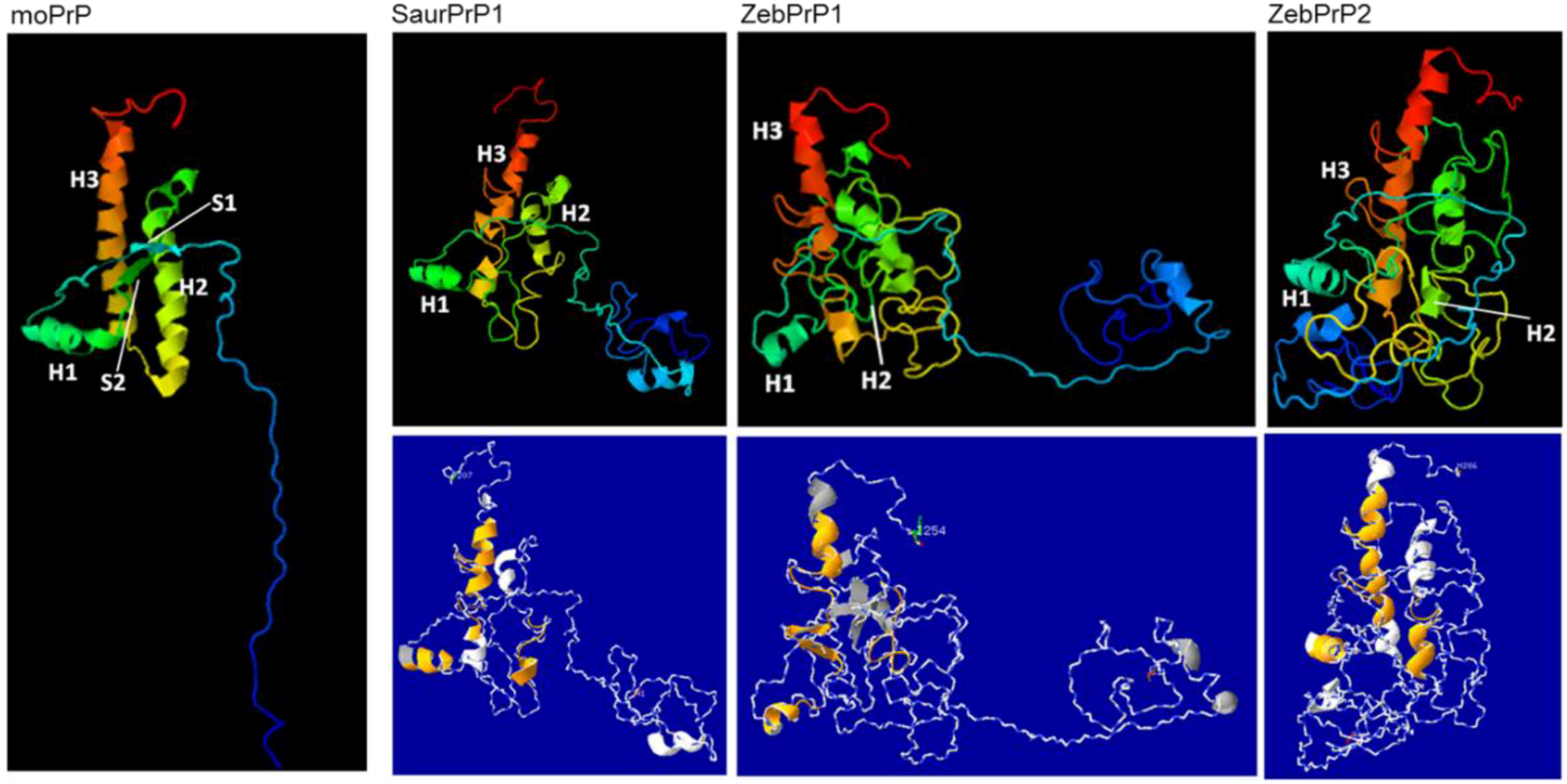
2.3. Sequence Analysis, Structural Predictions and PK Cleavage Site Simulation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | PrP length | PK sites | PK sites | PK sites | PK sites | PK sites |
|---|---|---|---|---|---|---|
| (Total) | N-terminus (total) | N-terminus (/100 residues) | C-terminus (total) | C-terminus (/100 residues) | ||
| Mo | 254 | 101 | 25 | 27.4 | 76 | 46.6 |
| Saur | 496 | 167 | 73 | 28.8 | 94 | 38.6 |
| ZebPrP1 | 606 | 182 | 80 | 24.7 | 102 | 36 |
| ZebPrP2 | 567 | 188 | 63 | 25.7 | 125 | 38.8 |

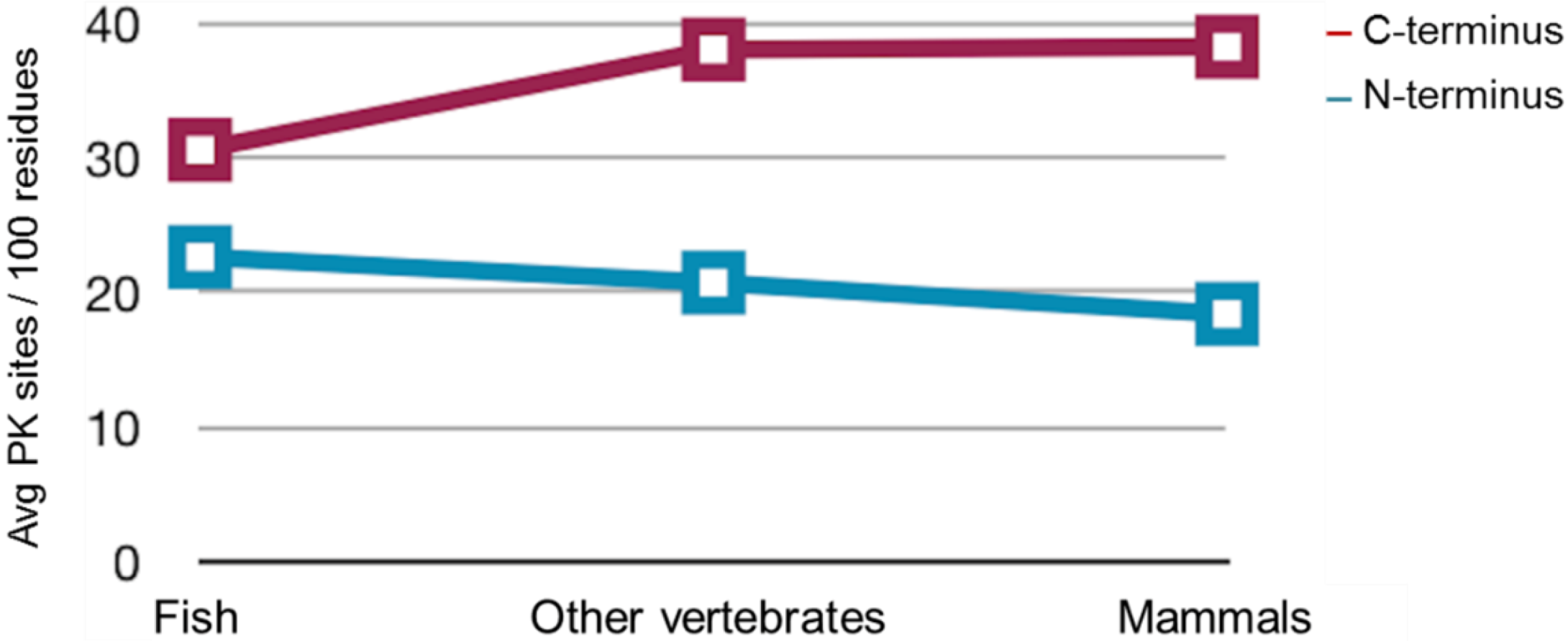

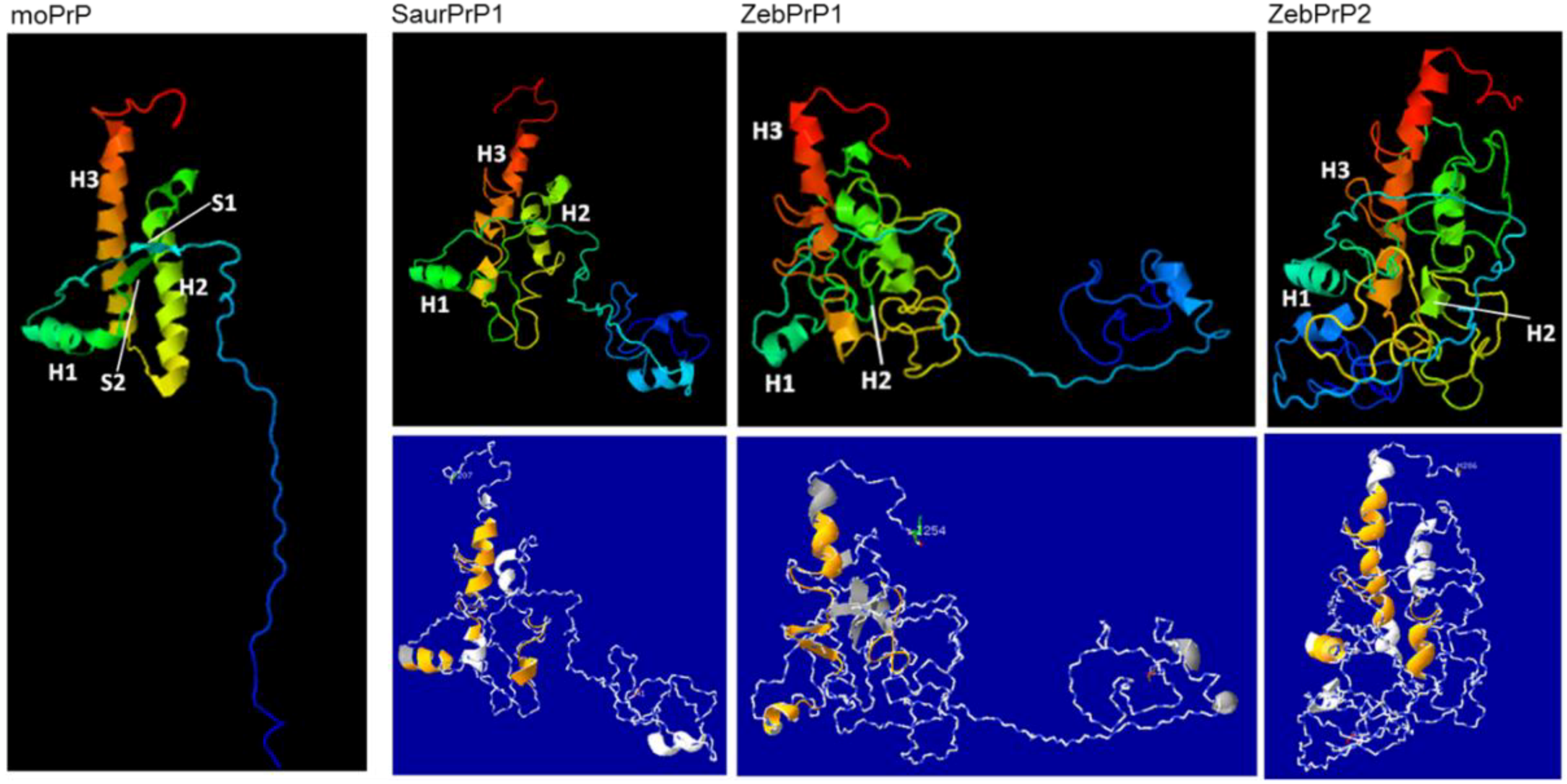
3. Discussion
4. Materials and Methods
4.1. Generation of DNA Constructs
4.2. Antibodies and Lectins
4.3. Cell Culture and Transfections
4.4. Trypsinization, Lysis and Proteinsase K Treatment
4.5. Western Blot Analysis
4.6. Lectin Staining
4.7. Indirect Immunofluorescence
4.8. PNGase Treatment
4.9. Immunoprecipitation
4.10. Sequence Analysis and Comparison
4.11. PK Cleavage Site Simulation
4.12. Structure Prediction and Modeling
Supplementary Files
Supplementary File 1Supplementary File 2Supplementary File 3Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Bueler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Beringue, V.; Vilotte, J.L.; Laude, H. Prion agent diversity and species barrier. Vet. Res. 2008, 39, 47. [Google Scholar] [CrossRef] [PubMed]
- Schatzl, H.M.; da Costa, M.; Taylor, L.; Cohen, F.E.; Prusiner, S.B. Prion protein gene variation among primates. J. Mol. Biol. 1995, 245, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J. Molecular neurology of prion disease. J. Neurol. Neurosurg. Psychiatry 2005, 76, 906–919. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, B.; Liu, Y.; Zhang, H.; Song, Y. New insight into the species barrier of mammalian prions obtained from structure-based conservation analysis. Intervirology 2008, 51, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Oidtmann, B.; Simon, D.; Holtkamp, N.; Hoffmann, R.; Baier, M. Identification of cDNAs from Japanese pufferfish (Fugu rubripes) and Atlantic salmon (Salmo salar) coding for homologues to tetrapod prion proteins. FEBS Lett. 2003, 538, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Maddison, B.C.; Patel, S.; James, R.F.; Conlon, H.E.; Oidtmann, B.; Baier, M.; Whitelam, G.C.; Gough, K.C. Generation and characterisation of monoclonal antibodies to Rainbow trout (Oncorhynchus mykiss) prion protein. J. Immunol. Methods 2005, 306, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Milla, E.; Oidtmann, B.; Panagiotidis, C.H.; Baier, M.; Sklaviadis, T.; Hoffmann, R.; Zhou, Y.; Solis, G.P.; Stuermer, C.A.; Malaga-Trillo, E. Disparate evolution of prion protein domains and the distinct origin of Doppel- and prion-related loci revealed by fish-to-mammal comparisons. FASEB J. 2006, 20, 317–319. [Google Scholar] [PubMed]
- Favre-Krey, L.; Theodoridou, M.; Boukouvala, E.; Panagiotidis, C.H.; Papadopoulos, A.I.; Sklaviadis, T.; Krey, G. Molecular characterization of a cDNA from the gilthead sea bream (Sparus aurata) encoding a fish prion protein. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2007, 147, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Cotto, E.; Andre, M.; Forgue, J.; Fleury, H.J.; Babin, P.J. Molecular characterization, phylogenetic relationships, and developmental expression patterns of prion genes in zebrafish (Danio rerio). FEBS J. 2005, 272, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Baumann, F.; Bremer, J. The prion’s elusive reason for being. Annu. Rev. Neurosci. 2008, 31, 439–477. [Google Scholar] [CrossRef] [PubMed]
- Malaga-Trillo, E.; Solis, G.P.; Schrock, Y.; Geiss, C.; Luncz, L.; Thomanetz, V.; Stuermer, C.A. Regulation of embryonic cell adhesion by the prion protein. PLoS Biol. 2009, 7, e55. [Google Scholar] [CrossRef] [PubMed]
- Fleisch, V.C.; Leighton, P.L.; Wang, H.; Pillay, L.M.; Ritzel, R.G.; Bhinder, G.; Roy, B.; Tierney, K.B.; Ali, D.W.; Waskiewicz, A.J.; et al. Targeted mutation of the gene encoding prion protein in zebrafish reveals a conserved role in neuron excitability. Neurobiol. Dis. 2013, 55, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Ingrosso, L.; Novoa, B.; Valle, A.Z.; Cardone, F.; Aranguren, R.; Sbriccoli, M.; Bevivino, S.; Iriti, M.; Liu, Q.; Vetrugno, V.; et al. Scrapie infectivity is quickly cleared in tissues of orally-infected farmed fish. BMC Vet. Res. 2006, 2, 21. [Google Scholar] [CrossRef] [PubMed]
- Dalla Valle, A.Z.; Iriti, M.; Faoro, F.; Berti, C.; Ciappellano, S. In vivo prion protein intestinal uptake in fish. APMIS 2008, 116, 173–180. [Google Scholar]
- Malaga-Trillo, E.; Salta, E.; Figueras, A.; Panagiotidis, C.; Sklaviadis, T. Fish models in prion biology: Underwater issues. Biochim. Biophys. Acta 2011, 1812, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Salta, E.; Panagiotidis, C.; Teliousis, K.; Petrakis, S.; Eleftheriadis, E.; Arapoglou, F.; Grigoriadis, N.; Nicolaou, A.; Kaldrymidou, E.; Krey, G.; et al. Evaluation of the possible transmission of BSE and scrapie to gilthead sea bream (Sparus aurata). PLoS One 2009, 4, e6175. [Google Scholar] [CrossRef] [PubMed]
- Race, R.E.; Fadness, L.H.; Chesebro, B. Characterization of scrapie infection in mouse neuroblastoma cells. J. Gen. Virol. 1987, 68, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Race, R.E.; Ernst, D.; Buchmeier, M.J.; Chesebro, B. Prion protein biosynthesis in scrapie-infected and uninfected neuroblastoma cells. J. Virol. 1989, 63, 175–181. [Google Scholar] [PubMed]
- Bosque, P.J.; Prusiner, S.B. Cultured cell sublines highly susceptible to prion infection. J. Virol. 2000, 74, 4377–4386. [Google Scholar] [CrossRef] [PubMed]
- Bach, C.; Gilch, S.; Rost, R.; Greenwood, A.D.; Horsch, M.; Hajj, G.N.; Brodesser, S.; Facius, A.; Schadler, S.; Sandhoff, K.; et al. Prion-induced activation of cholesterogenic gene expression by Srebp2 in neuronal cells. J. Biol. Chem. 2009, 284, 31260–31269. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.A. Trafficking, turnover and membrane topology of PrP. Br. Med. Bull. 2003, 66, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Xanthopoulos, K.; Polymenidou, M.; Bellworthy, S.J.; Benestad, S.L.; Sklaviadis, T. Species and strain glycosylation patterns of PrPSc. PLoS One 2009, 4, e5633. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, K.; Nakamura, S.; Wang, H.; Iwasaki, H.; Maebara, K.; Cheng, L.; Hirabayashi, J.; Narimatsu, H. Elucidation of binding specificity of Jacalin toward O-glycosylated peptides: Quantitative analysis by frontal affinity chromatography. Glycobiology 2006, 16, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Vorberg, I.; Buschmann, A.; Harmeyer, S.; Saalmuller, A.; Pfaff, E.; Groschup, M.H. A novel epitope for the specific detection of exogenous prion proteins in transgenic mice and transfected murine cell lines. Virology 1999, 255, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Vilette, D. Cell models of prion infection. Vet. Res. 2008, 39, 10. [Google Scholar] [CrossRef] [PubMed]
- Gilch, S.; Winklhofer, K.F.; Groschup, M.H.; Nunziante, M.; Lucassen, R.; Spielhaupter, C.; Muranyi, W.; Riesner, D.; Tatzelt, J.; Schatzl, H.M. Intracellular re-routing of prion protein prevents propagation of PrP(Sc) and delays onset of prion disease. EMBO J. 2001, 20, 3957–3966. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Veith, N.M.; Plattner, H.; Stuermer, C.A.; Schulz-Schaeffer, W.J.; Burkle, A. Immunolocalisation of PrPSc in scrapie-infected N2a mouse neuroblastoma cells by light and electron microscopy. Eur. J. Cell Biol. 2009, 88, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, A.C.; Baron, G.S.; Lee, K.S.; Steele-Mortimer, O.; Dorward, D.; Prado, M.A.; Caughey, B. Uptake and neuritic transport of scrapie prion protein coincident with infection of neuronal cells. J. Neurosci. 2005, 25, 5207–5216. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Preusser, M.; Strohschneider, M.; Budka, H. Subcellular localization of disease-associated prion protein in the human brain. Am. J. Pathol. 2005, 166, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Mange, A.; Crozet, C.; Lehmann, S.; Beranger, F. Scrapie-like prion protein is translocated to the nuclei of infected cells independently of proteasome inhibition and interacts with chromatin. J. Cell Sci. 2004, 117, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Malaga-Trillo, E.; Sempou, E. PrPs: Proteins with a purpose: Lessons from the zebrafish. Prion 2009, 3, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Solis, G.P.; Radon, Y.; Sempou, E.; Jechow, K.; Stuermer, C.A.; Malaga-Trillo, E. Conserved roles of the prion protein domains on subcellular localization and cell-cell adhesion. PLoS One 2013, 8, e70327. [Google Scholar] [CrossRef] [PubMed]
- Miesbauer, M.; Bamme, T.; Riemer, C.; Oidtmann, B.; Winklhofer, K.F.; Baier, M.; Tatzelt, J. Prion protein-related proteins from zebrafish are complex glycosylated and contain a glycosylphosphatidylinositol anchor. Biochem. Biophys. Res. Commun. 2006, 341, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Priola, S.A.; Lawson, V.A. Glycosylation influences cross-species formation of protease-resistant prion protein. EMBO J. 2001, 20, 6692–6699. [Google Scholar] [CrossRef] [PubMed]
- Priola, S.A.; Caughey, B.; Race, R.E.; Chesebro, B. Heterologous PrP molecules interfere with accumulation of protease-resistant PrP in scrapie-infected murine neuroblastoma cells. J. Virol. 1994, 68, 4873–4878. [Google Scholar] [PubMed]
- Horiuchi, M.; Priola, S.A.; Chabry, J.; Caughey, B. Interactions between heterologous forms of prion protein: Binding, inhibition of conversion, and species barriers. Proc. Natl. Acad. Sci. USA 2000, 97, 5836–5841. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, K.; Hara, H.; Hanada, K. Species-barrier phenomenon in prion transmissibility from a viewpoint of protein science. J. Biochem. 2013, 153, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Norstrom, E.M.; Mastrianni, J.A. The AGAAAAGA palindrome in PrP is required to generate a productive PrPSc-PrPC complex that leads to prion propagation. J. Biol. Chem. 2005, 280, 27236–27243. [Google Scholar] [CrossRef] [PubMed]
- Solforosi, L.; Bellon, A.; Schaller, M.; Cruite, J.T.; Abalos, G.C.; Williamson, R.A. Toward molecular dissection of PrPC-PrPSc interactions. J. Biol. Chem. 2007, 282, 7465–7471. [Google Scholar] [CrossRef] [PubMed]
- Abalos, G.C.; Cruite, J.T.; Bellon, A.; Hemmers, S.; Akagi, J.; Mastrianni, J.A.; Williamson, R.A.; Solforosi, L. Identifying key components of the PrPC-PrPSc replicative interface. J. Biol. Chem. 2008, 283, 34021–34028. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Okemoto-Nakamura, Y.; Shinkai-Ouchi, F.; Hanada, K.; Yamakawa, Y.; Hagiwara, K. Mouse prion protein (PrP) segment 100 to 104 regulates conversion of PrP(C) to PrP(Sc) in prion-infected neuroblastoma cells. J. Virol. 2012, 86, 5626–5636. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Clarke, A.R. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Tzaban, S.; Friedlander, G.; Schonberger, O.; Horonchik, L.; Yedidia, Y.; Shaked, G.; Gabizon, R.; Taraboulos, A. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry 2002, 41, 12868–12875. [Google Scholar] [CrossRef] [PubMed]
- Gambetti, P.; Dong, Z.; Yuan, J.; Xiao, X.; Zheng, M.; Alshekhlee, A.; Castellani, R.; Cohen, M.; Barria, M.A.; Gonzalez-Romero, D.; et al. A novel human disease with abnormal prion protein sensitive to protease. Ann. Neurol. 2008, 63, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Harris, D.A.; Vilette, D.; Laude, H.; Frobert, Y.; Grassi, J.; Casanova, D.; Milhavet, O.; Lehmann, S. Successful transmission of three mouse-adapted scrapie strains to murine neuroblastoma cell lines overexpressing wild-type mouse prion protein. J. Virol. 2000, 74, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bolton, E.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucl. Acids Res. 2011, 39, D38–D51. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Promponas, V.J.; Enright, A.J.; Tsoka, S.; Kreil, D.P.; Leroy, C.; Hamodrakas, S.; Sander, C.; Ouzounis, C.A. CAST: An iterative algorithm for the complexity analysis of sequence tracts. Complexity analysis of sequence tracts. Bioinformatics 2000, 16, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- ExPASy Bioinformatics Resourse Portal, PeptideCutter tool. Available online: http://web.expasy.org/peptide_cutter/ (accessed on 11 October 2014).
- Gasteiger, E.H.C.; Gattiger, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server; Humana Press: New York, NY, USA, 2008. [Google Scholar]
- Bordoli, L.; Kiefer, F.; Arnold, K.; Benkert, P.; Battey, J.; Schwede, T. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 2009, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Shatsky, M.; Nussinov, R.; Wolfson, H.J. A method for simultaneous alignment of multiple protein structures. Proteins 2004, 56, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Rosenstrom, P. Dali server: Conservation mapping in 3D. Nucl. Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Tang, M.; Grishin, N.V. PROMALS3D web server for accurate multiple protein sequence and structure alignments. Nucl. Acids Res. 2008, 36, W30–W34. [Google Scholar] [CrossRef] [PubMed]
- Jmol: An open-source Java viewer for chemical structures in 3D. Available online: http://www.jmol.org/ (accessed on 26 November 2011).
- Kelley, L.A.; Sternberg, M.J. Protein structure prediction on the Web: A case study using the Phyre server. Nat. Protoc. 2009, 4, 363–371. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salta, E.; Kanata, E.; Ouzounis, C.A.; Gilch, S.; Schätzl, H.; Sklaviadis, T. Assessing Proteinase K Resistance of Fish Prion Proteins in a Scrapie-Infected Mouse Neuroblastoma Cell Line. Viruses 2014, 6, 4398-4421. https://doi.org/10.3390/v6114398
Salta E, Kanata E, Ouzounis CA, Gilch S, Schätzl H, Sklaviadis T. Assessing Proteinase K Resistance of Fish Prion Proteins in a Scrapie-Infected Mouse Neuroblastoma Cell Line. Viruses. 2014; 6(11):4398-4421. https://doi.org/10.3390/v6114398
Chicago/Turabian StyleSalta, Evgenia, Eirini Kanata, Christos A. Ouzounis, Sabine Gilch, Hermann Schätzl, and Theodoros Sklaviadis. 2014. "Assessing Proteinase K Resistance of Fish Prion Proteins in a Scrapie-Infected Mouse Neuroblastoma Cell Line" Viruses 6, no. 11: 4398-4421. https://doi.org/10.3390/v6114398
APA StyleSalta, E., Kanata, E., Ouzounis, C. A., Gilch, S., Schätzl, H., & Sklaviadis, T. (2014). Assessing Proteinase K Resistance of Fish Prion Proteins in a Scrapie-Infected Mouse Neuroblastoma Cell Line. Viruses, 6(11), 4398-4421. https://doi.org/10.3390/v6114398