A Unique Evolution of the S2 Gene of Equine Infectious Anemia Virus in Hosts Correlated with Particular Infection Statuses


Abstract
:1. Introduction
2. Materials and Methods
2.1. EIAV Strains
2.2. Horses Experimentally Infected with EIAV
2.3. Analysis of S2 Gene Variation
2.4. Detection of Selection Pressures
3. Results
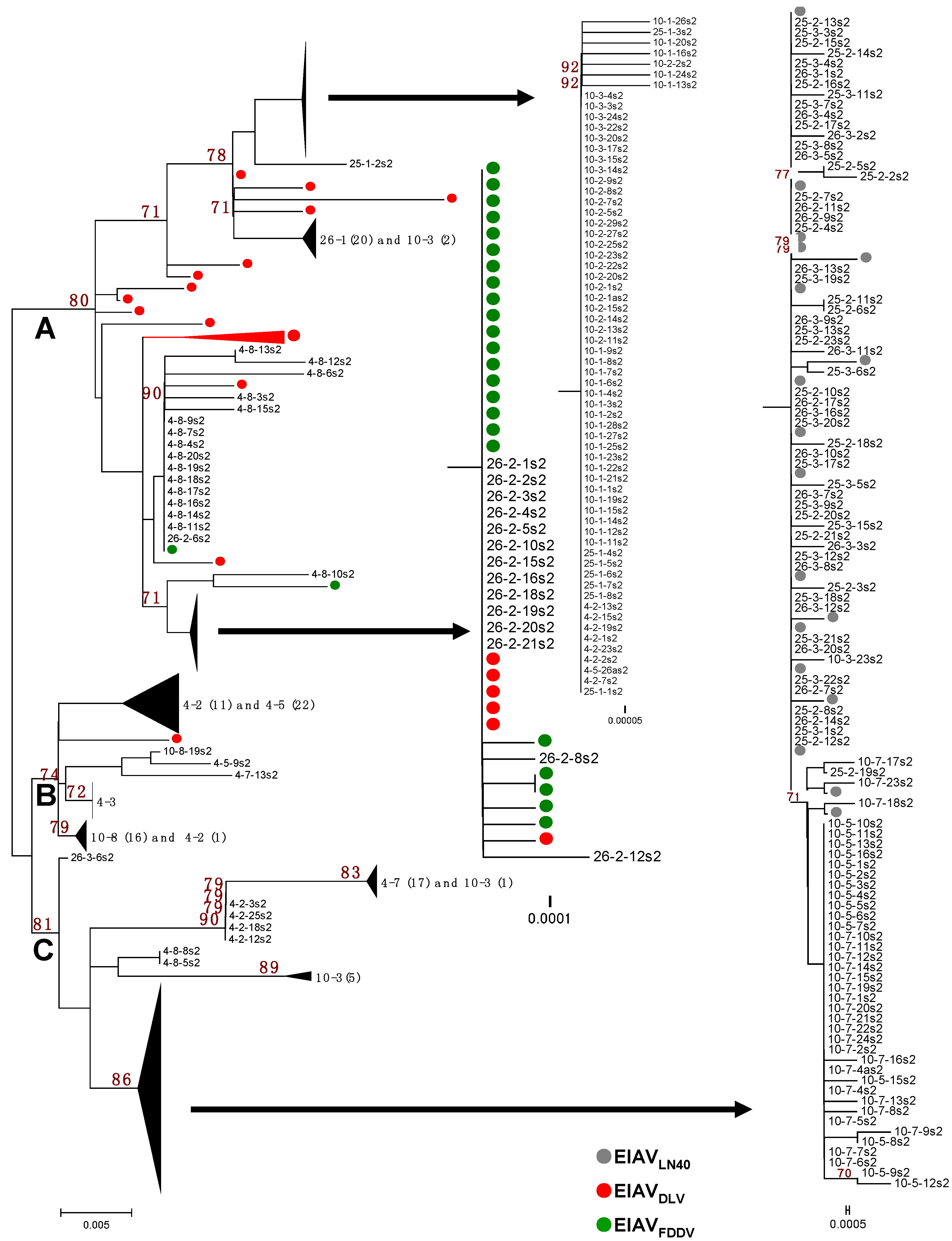
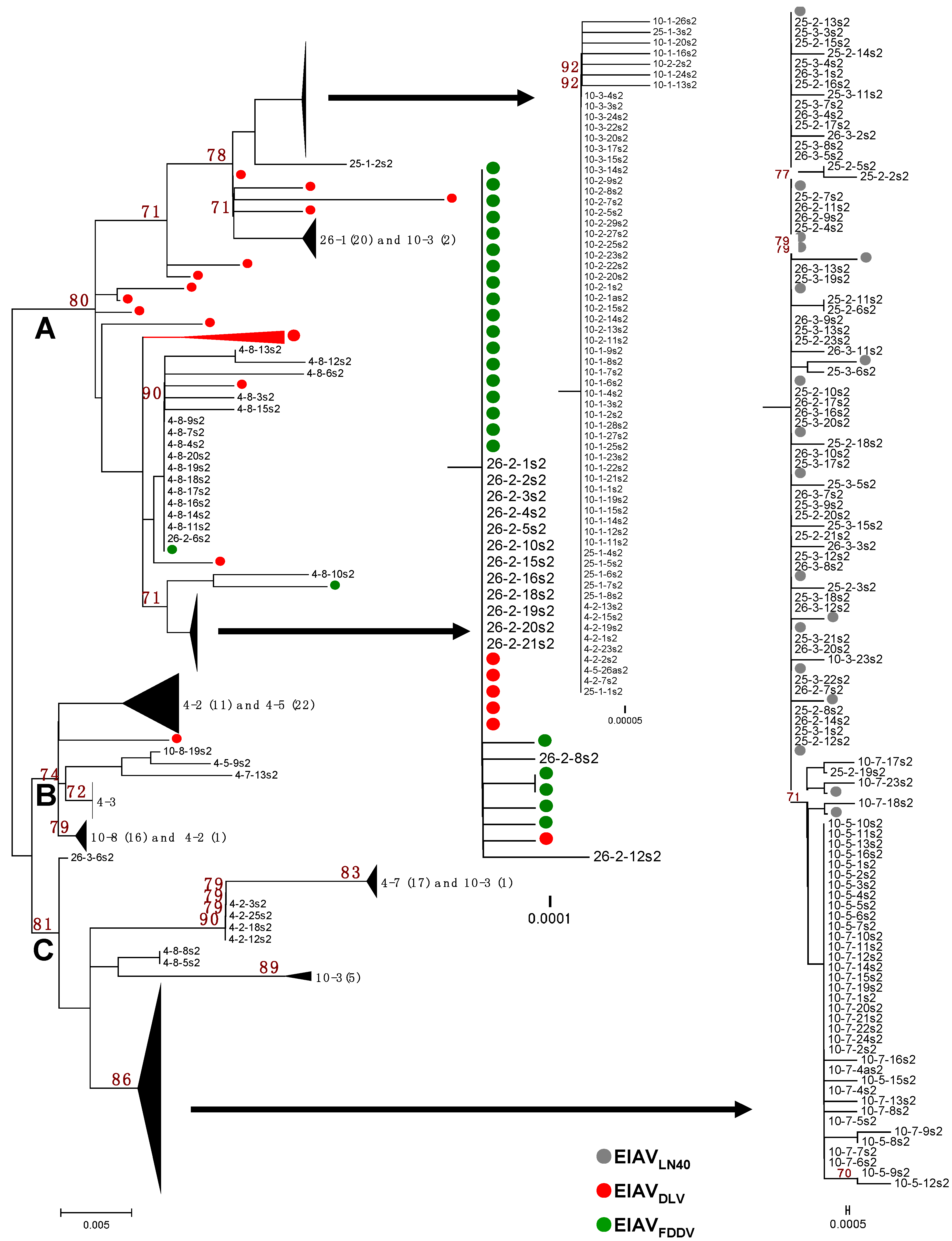
3.1. The S2 Gene Highly Varied among the Isolates of Experimentally Infected Horses and in Vitro Attenuated Strains

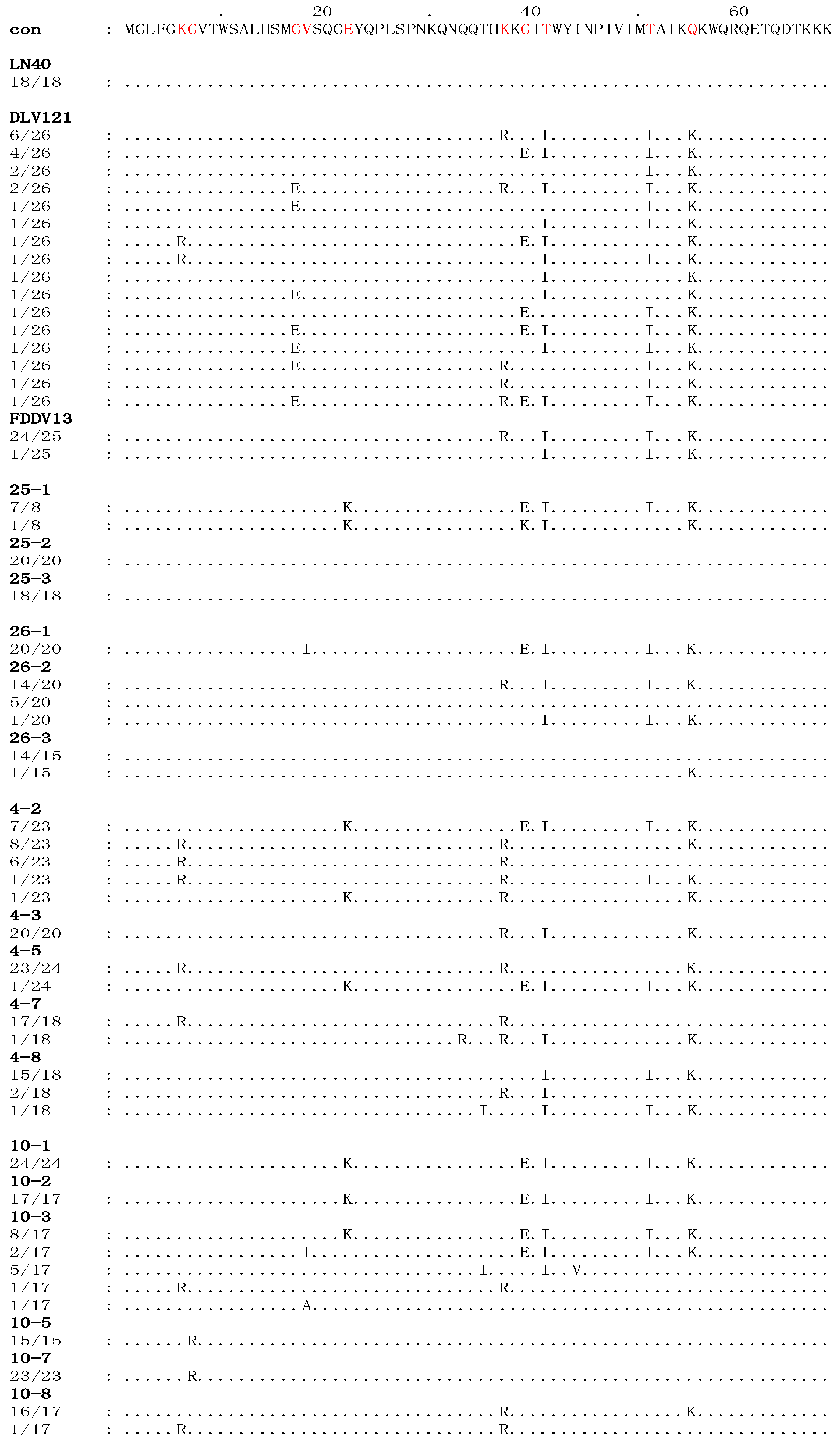{kind=link}
{kind=link}
| Sample | EIAVLN40 | EIAVDLV121 | EIAVFDDV13 | ||||
|---|---|---|---|---|---|---|---|
| name | dpi | nt | aa | nt | aa | nt | aa |
| DLV121 | V | 2.32(0.97–4.53) | 6.84(2.99–14.20) | ||||
| FDDV13 | V | 2.31(1.47–4.00) | 6.65(4.51–10.86) | 1.17(0–3.49) | 3.21(0–7.64) | ||
| 25-1 | 14 | 2.75(2.47–4.00) | 7.02(6.06–10.86) | 1.81(0.49–3.50) | 5.22(1.48–10.86) | 1.78(0.98–3.50) | 4.96(2.99–7.64) |
| 25-2 * | 18 | 0.44(0–1.96) | 1.03(0–4.51) | 2.32(0.97–4.51) | 6.78(2.99–12.52) | 2.31(1.47–3.99) | 6.60(4.51–9.24) |
| 25-3 * | 28 | 0.32(0–1.47) | 0.74(0–4.51) | 2.21(0.97–4.00) | 6.50(2.99–12.52) | 2.20(1.47–3.49) | 6.30(4.51–9.24) |
| 26-1 | 14 | 2.89(2.47–4.50) | 8.57(7.64–14.20) | 1.84(0.49–3.47) | 5.28(1.48–10.86) | 1.79(0.98–3.47) | 5.04(2.99–9.24) |
| 26-2 * | 18 | 1.74(0–4.00) | 5.17(0–10.86) | 1.37(0–3.49) | 3.97(0–10.86) | 0.73(0–2.97) | 1.88(0–7.64) |
| 26-3 * | 30 | 0.35(0–1.47) | 0.87(0–4.51) | 2.17(0.49–4.00) | 6.43(1.48–12.52) | 2.15(0.98–3.49) | 6.23(2.99–9.2) |
| 4-2 | 28 | 2.05(0.98–3.49) | 6.2(2.98–10.86) | 2.17(0.49–5.04) | 6.51(1.48–14.20) | 1.81(0.98–3.49) | 5.27(2.99–9.24) |
| 4-3 | 42 | 1.69(1.47–2.47) | 5.03(4.51–7.64) | 1.42(0.49–2.97) | 4.18(1.48–7.64) | 0.64(0.49–1.47) | 1.66(1.48–2.9) |
| 4-5 | 70 | 2.04(1.47–4.00) | 5.72(4.51–10.86) | 2.41(0.49–5.59) | 6.86(1.48–14.20) | 1.94(0.98–4.02) | 5.27(2.99–9.24) |
| 4-7 | 112 | 2.36(1.97–3.50) | 3.92(2.99–9.24) | 3.67(1.47–6.11) | 7.96(2.99–14.20) | 3.2(1.47–4.53) | 6.26(2.99–9.2) |
| 4-8 | 162 | 1.88(0.98–3.49) | 5.47(2.99–10.86) | 1.31(0.49–3.47) | 3.68(1.48–9.24) | 0.90(0-2.48) | 2.30(0–6.06) |
| 10-1 | 14 | 2.8(2.47–4) | 8.44(7.64–12.52) | 1.75(0.49–2.99) | 5.08(1.48–9.24) | 1.70(0.98–2.99) | 4.83(2.99–7.64) |
| 10-2 | 28 | 2.72(2.47–4) | 8.27(7.64–12.52) | 1.67(0.49–2.99) | 4.90(1.48–9.24) | 1.62(0.98–2.99) | 4.67(2.99–7.64) |
| 10-3 | 42 | 2.34(0.49–3.49) | 6.92(1.48–10.86) | 2.26(0.49–5.57) | 6.59(1.48–14.20) | 2.12(0.98–4.00) | 6.06(2.99–10.81) |
| 10-5 * | 70 | 0.87(0.49–2.48) | 2.07(1.48–4.51) | 2.77(1.47–5.06) | 7.90(4,51–12.52) | 2.76(1.96–4.53) | 7.70(6.06–9.24) |
| 10-7 | 112 | 0.86(0.49–2.48) | 2.38(1.48–6.06) | 2.77(1.47–5.06) | 8.25(4.51–14.20) | 2.76(1.96–4.54) | 8.05(6.06–10.81) |
| 10-8 | 162 | 1.34(0.97–2.47) | 3.85(2.99–7.6) | 1.83(0.98–4.00) | 5.37(2.99–10.87) | 1.28(0.98–2.48) | 3.53(2.99–6.06) |
3.2. Mutations in the S2 Gene Were the Result of Positive Selection
| Model | dN/dS | Parameters b,c | 2ΔlnL | Positively Selected Codons d |
|---|---|---|---|---|
| M0 (ratio of one) 1 a | 2.2197 | ω = 2.2197 | 34.71 p < 0.01 | None |
| M3 (discrete) 5 a | 2.4099 | P0 = 0.54824 p1 = 0.34821 p2 = 0.10355 ω0 = 0.79215 ω1 = 3.59132 ω2 = 7.00158 | 17, 18, 22, 37, 39, 41 | |
| M1a (neutral) 1 a | 0.8434 | p0 = 0.17661 p1 = 0.82339 ω1 = 0.11319 ω1 = 1.00000 | 47.544 p < 0.01 | Not permitted |
| M2a (selection) 3 a | 2.4296 | p0 = 0.06756 p1 = 0.56448 p2 = 0.36796 ω0 = 0.32811 ω1=1.00000 ω2 = 5.00850 | 17, 18, 22, 37, 39, 41 | |
| M7 (β) 2 a | 1.0000 | P = 97.53765 q = 0.00500 | 51.384 p < 0.01 | Not permitted |
| M8 (β & ω) 4 a | 2.4337 | p0 = 0.63252 p1 = 0.36748 p = 0.19920 q = 0.01258 ω = 5.02072 | 17, 18, 22, 37, 39, 41 |
3.3. The Vaccine- and Pathogenic-Specific S2 Sequences Were Identified in Inoculated Horses, Depending on the Inoculation Dose and Infection Status
| S2 Variation a | No. (%) of Occurrences in: | p Value b | |
|---|---|---|---|
| A (n = 192) | C (n = 144) | ||
| 6K/R | 25(13) | 22(15) | > 0.05 |
| 7G/R | 0(0) | 38(26) | < 0.01 |
| 17G/E | 8(4) | 0(0) | < 0.05 |
| 18V/I | 22(11) | 0(0) | < 0.01 |
| 22E/K | 64(33) | 0(0) | < 0.01 |
| 37K/R | 72(38) | 24(17) | < 0.01 |
| 39G/E | 93(48) | 0(0) | < 0.01 |
| 41T/I | 162(84) | 7(5) | < 0.01 |
| 51T/I | 164(85) | 0(0) | < 0.01 |
| 55Q/K | 190(99) | 1(1) | < 0.01 |
4. Discussion
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Issel, C.J.; Adams, W.V., Jr.; Meek, L.; Ochoa, R. Transmission of equine infectious anemia virus from horses without clinical signs of disease. J. Am. Vet. Med. Assoc. 1982, 180, 272–275. [Google Scholar] [PubMed]
- Craigo, J.K.; Ezzelarab, C.; Cook, S.J.; Chong, L.; Horohov, D.; Issel, C.J.; Montelaro, R.C. Envelope determinants of equine lentiviral vaccine protection. PLoS One 2013, 8, e66093. [Google Scholar] [CrossRef] [PubMed]
- Leroux, C.; Cadore, J.L.; Montelaro, R.C. Equine Infectious Anemia Virus (EIAV): What has HIV’s country cousin got to tell us? Vet. Res. 2004, 35, 485–512. [Google Scholar]
- Fagerness, A.J.; Flaherty, M.T.; Perry, S.T.; Jia, B.; Payne, S.L.; Fuller, F.J. The S2 accessory gene of equine infectious anemia virus is essential for expression of disease in ponies. Virology 2006, 349, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, S.; Lin, Y.; Jiang, C.; Ma, J.; Zhao, L.; Lv, X.; Wang, F.; Shen, R.; Kong, X.; Zhou, J. Genomic comparison between attenuated Chinese equine infectious anemia virus vaccine strains and their parental virulent strains. Arch. Virol. 2010, 156, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Covaleda, L.; Gno, B.T.; Fuller, F.J.; Payne, S.L. Identification of cellular proteins interacting with equine infectious anemia virus S2 protein. Virus Res. 2010, 151, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.L.; Fuller, F.J. Virulence determinants of equine infectious anemia virus. Curr. HIV Res. 2010, 8, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Kirchhoff, F.; Easterbrook, P.J.; Douglas, N.; Troop, M.; Greenough, T.C.; Weber, J.; Carl, S.; Sullivan, J.L.; Daniels, R.S. Sequence variations in human immunodeficiency virus type 1 Nef are associated with different stages of disease. J. Virol. 1999, 73, 5497–5508. [Google Scholar] [PubMed]
- Walker, P.R.; Ketunuti, M.; Choge, I.A.; Meyers, T.; Gray, G.; Holmes, E.C.; Morris, L. Polymorphisms in Nef associated with different clinical outcomes in HIV type 1 subtype C-infected children. AIDS Res. Hum. Retrovir. 2007, 23, 204–215. [Google Scholar] [PubMed]
- Li, F.; Puffer, B.A.; Montelaro, R.C. The S2 gene of equine infectious anemia virus is dispensable for viral replication in vitro. J. Virol. 1998, 72, 8344–8348. [Google Scholar] [PubMed]
- Li, F.; Leroux, C.; Craigo, J.K.; Cook, S.J.; Issel, C.J.; Montelaro, R.C. The S2 gene of equine infectious anemia virus is a highly conserved determinant of viral replication and virulence properties in experimentally infected ponies. J. Virol. 2000, 74, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Jiang, C.G.; Wang, X.F.; Lin, Y.Z.; Ma, J.; Han, X.E.; Zhao, L.P.; Shen, R.X.; Xiang, W.H.; Zhou, J.H. Reverse mutation of the virulence-associated S2 gene does not cause an attenuated equine infectious anemia virus strain to revert to pathogenicity. Virology 2013, 443, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, S.; Lin, Y.; Jiang, C.; Ma, J.; Zhao, L.; Lv, X.; Wang, F.; Shen, R.; Zhou, J. Unique evolution characteristics of the envelope protein of EIAVLN40, a virulent strain of equine infectious anemia virus. Virus Genes 2011, 42, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wong, W.S.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [PubMed]
- Wong, W.S.; Yang, Z.; Goldman, N.; Nielsen, R. Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics 2004, 168, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.E.; Soares, M.A.; Schrago, C.G. Positive selection on HIV accessory proteins and the analysis of molecular adaptation after interspecies transmission. J. Mol. Evol. 2008, 66, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Craigo, J.K.; Montelaro, R.C. EIAV envelope diversity: Shaping viral persistence and encumbering vaccine efficacy. Curr. HIV Res. 2010, 8, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Mo, H.; Wang, N.; Nam, D.S.; Cao, Y.; Koup, R.A.; Ho, D.D. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science 1993, 261, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.Z.; Shen, R.X.; Zhu, Z.Y.; Deng, X.L.; Cao, X.Z.; Wang, X.F.; Ma, J.; Jiang, C.G.; Zhao, L.P.; Lv, X.L.; et al. An attenuated EIAV vaccine strain induces significantly different immune responses from its pathogenic parental strain although with similar in vivo replication pattern. Antivir. Res. 2011, 92, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, S.S.; Lin, Y.Z.; Liu, H.F.; Wei, H.M.; Du, C.; Wang, X.F.; Zhou, J.H. An attenuated EIAV strain and its molecular clone strain differentially induce the expression of Toll-like receptors and type-I interferons in equine monocyte-derived macrophages. Vet. Microbiol. 2013, 166, 263–269. [Google Scholar] [PubMed]
- Covaleda, L.; Fuller, F.J.; Payne, S.L. EIAV S2 enhances pro-inflammatory cytokine and chemokine response in infected macrophages. Virology 2010, 397, 217–223. [Google Scholar] [CrossRef]
- Cook, R.F.; Leroux, C.; Issel, C.J. Equine infectious anemia and equine infectious anemia virus in 2013: A review. Vet. Microbiol. 2013, 167, 181–204. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Liddament, M.T. Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 2004, 4, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Sakurai, M.; Gupta, R.; Valente, L.; Wulff, B.E.; Ariyoshi, K.; Iizasa, H.; Davuluri, R.V.; Nishikura, K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 2013, 153, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.M.; Wang, X.F.; Wang, S.S.; Du, C.; Liu, H.F.; Liu, Q.; Zhou, J.H. The application of single-genome amplification and sequencing in genomic analysis of an attenuated EIAV vaccine. Bing Du Xue Bao 2012, 28, 431–438. (In Chinese) [Google Scholar] [PubMed]
- Keele, B.F.; Giorgi, E.E.; Salazar-Gonzalez, J.F.; Decker, J.M.; Pham, K.T.; Salazar, M.G.; Sun, C.; Grayson, T.; Wang, S.; Li, H.; et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 2008, 105, 7552–7557. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.-F.; Wang, S.; Liu, Q.; Lin, Y.-Z.; Du, C.; Tang, Y.-D.; Na, L.; Wang, X.; Zhou, J.-H. A Unique Evolution of the S2 Gene of Equine Infectious Anemia Virus in Hosts Correlated with Particular Infection Statuses. Viruses 2014, 6, 4265-4279. https://doi.org/10.3390/v6114265
Wang X-F, Wang S, Liu Q, Lin Y-Z, Du C, Tang Y-D, Na L, Wang X, Zhou J-H. A Unique Evolution of the S2 Gene of Equine Infectious Anemia Virus in Hosts Correlated with Particular Infection Statuses. Viruses. 2014; 6(11):4265-4279. https://doi.org/10.3390/v6114265
Chicago/Turabian StyleWang, Xue-Feng, Shuai Wang, Qiang Liu, Yue-Zhi Lin, Cheng Du, Yan-Dong Tang, Lei Na, Xiaojun Wang, and Jian-Hua Zhou. 2014. "A Unique Evolution of the S2 Gene of Equine Infectious Anemia Virus in Hosts Correlated with Particular Infection Statuses" Viruses 6, no. 11: 4265-4279. https://doi.org/10.3390/v6114265
APA StyleWang, X.-F., Wang, S., Liu, Q., Lin, Y.-Z., Du, C., Tang, Y.-D., Na, L., Wang, X., & Zhou, J.-H. (2014). A Unique Evolution of the S2 Gene of Equine Infectious Anemia Virus in Hosts Correlated with Particular Infection Statuses. Viruses, 6(11), 4265-4279. https://doi.org/10.3390/v6114265