Mutation of the Highly Conserved Ser-40 of the HIV-1 p6 Gag Protein to Phe Causes the Formation of a Hydrophobic Patch, Enhances Membrane Association, and Polyubiquitination of Gag

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
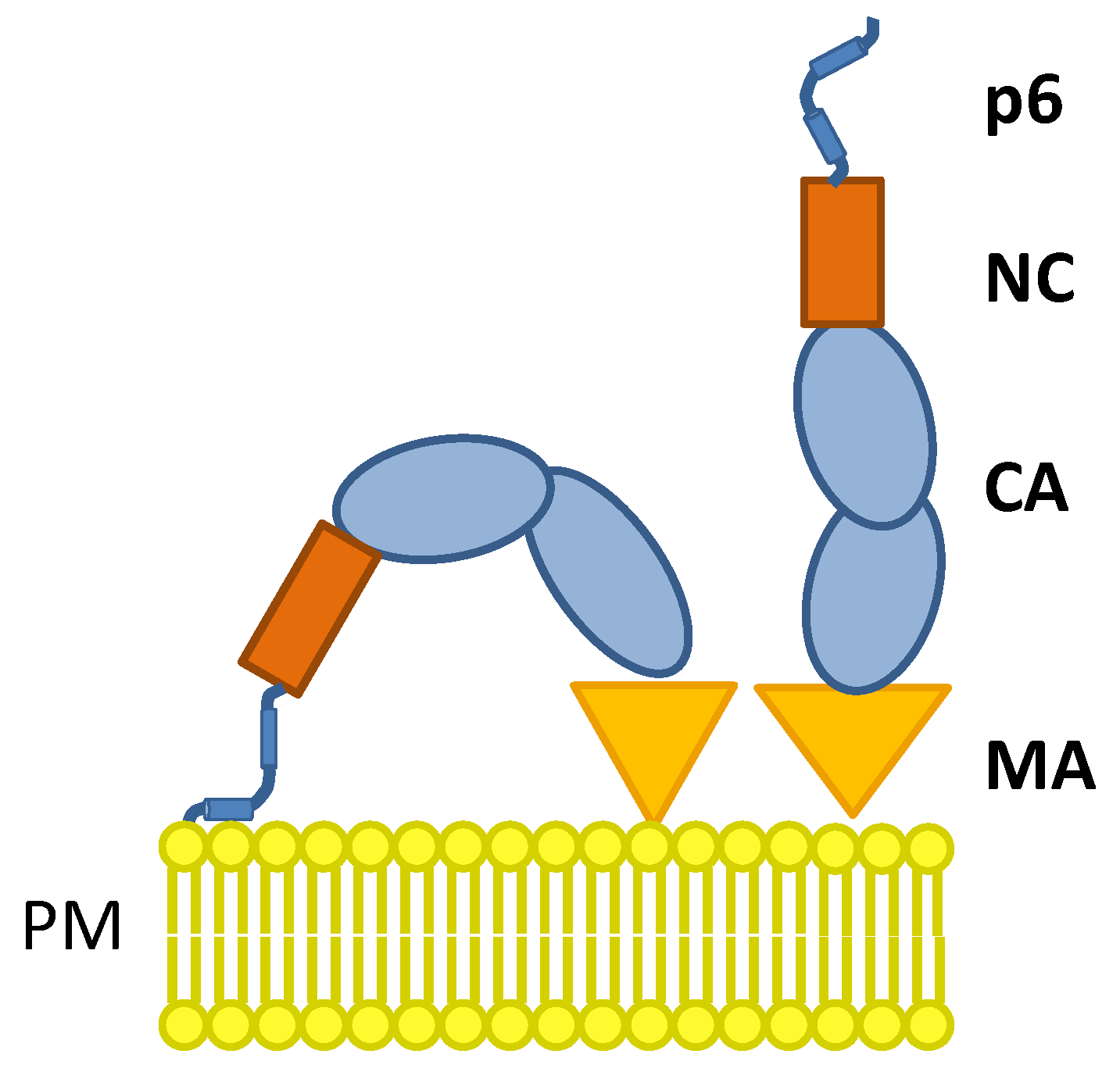
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Detection of Ubiquitinated Gag
2.3. SDS PAGE and Western Blotting
2.4. Expression Plasmids
2.5. Flow Cytometry
2.6. T-Cell Activation Assay
2.7. Membrane Flotation
2.8. Peptide Synthesis
2.9. Preparation of Samples for NMR Spectroscopy
2.10. NMR Spectroscopy
2.11. Structural Calculations
3. Results
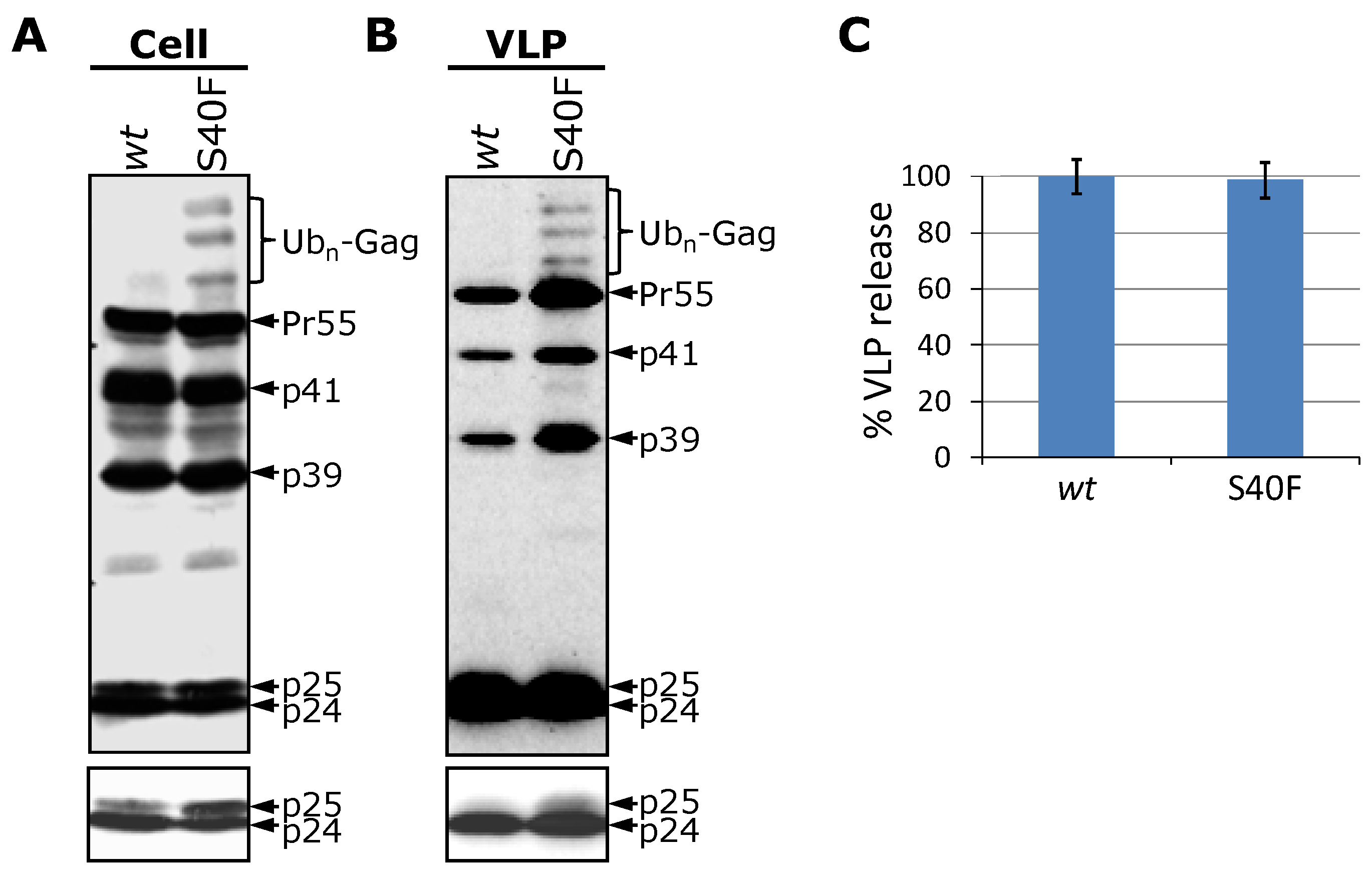
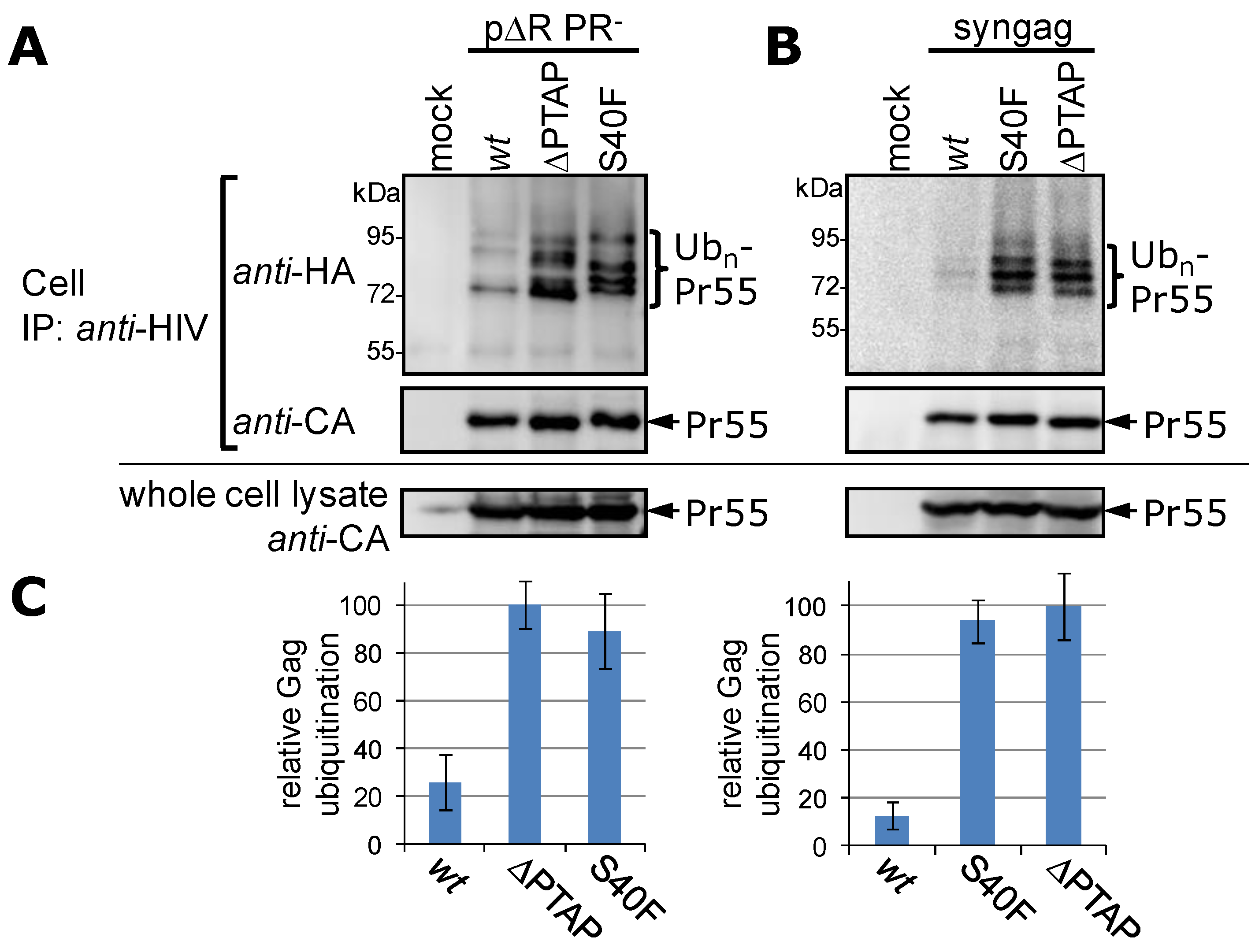
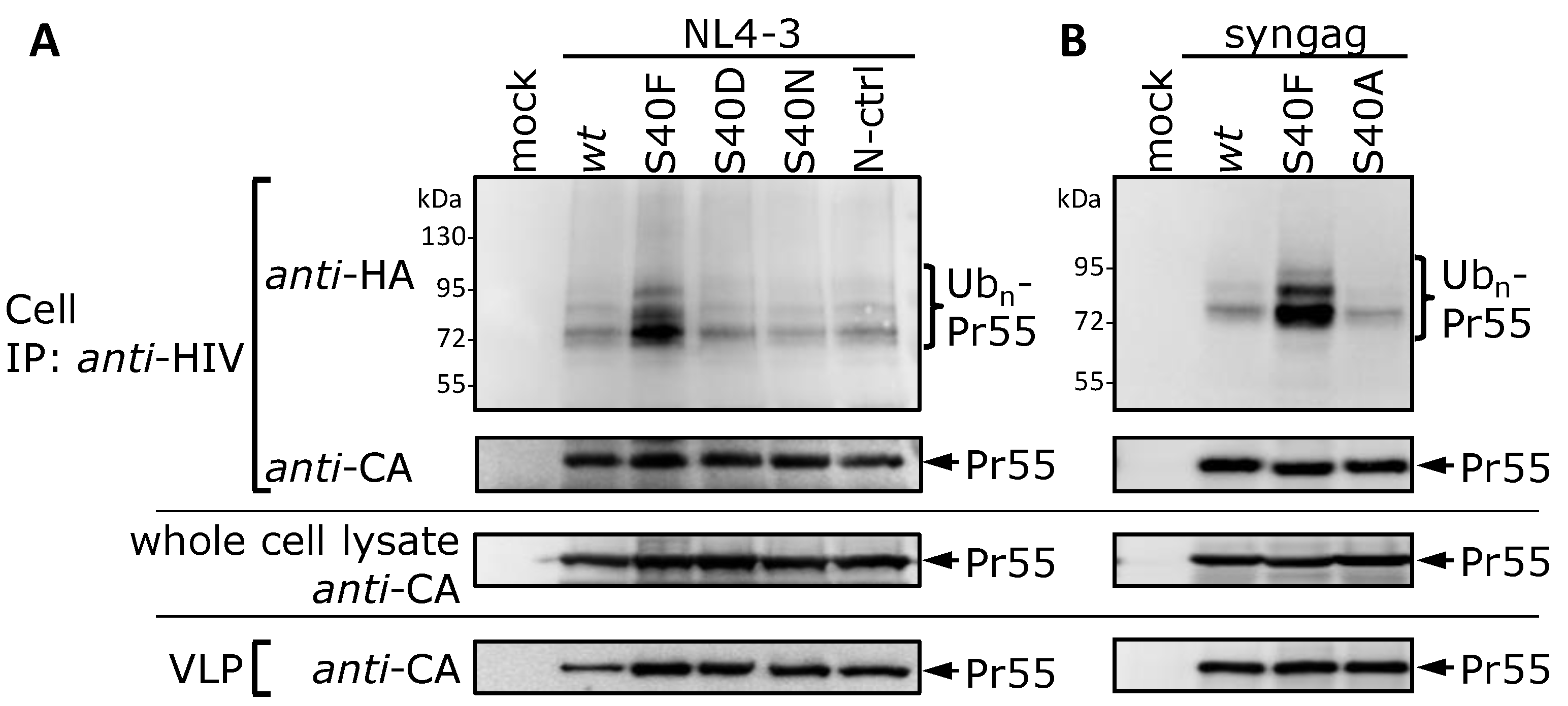
3.1. Mutation of Ser-40 Elevates Gag Ubiquitination
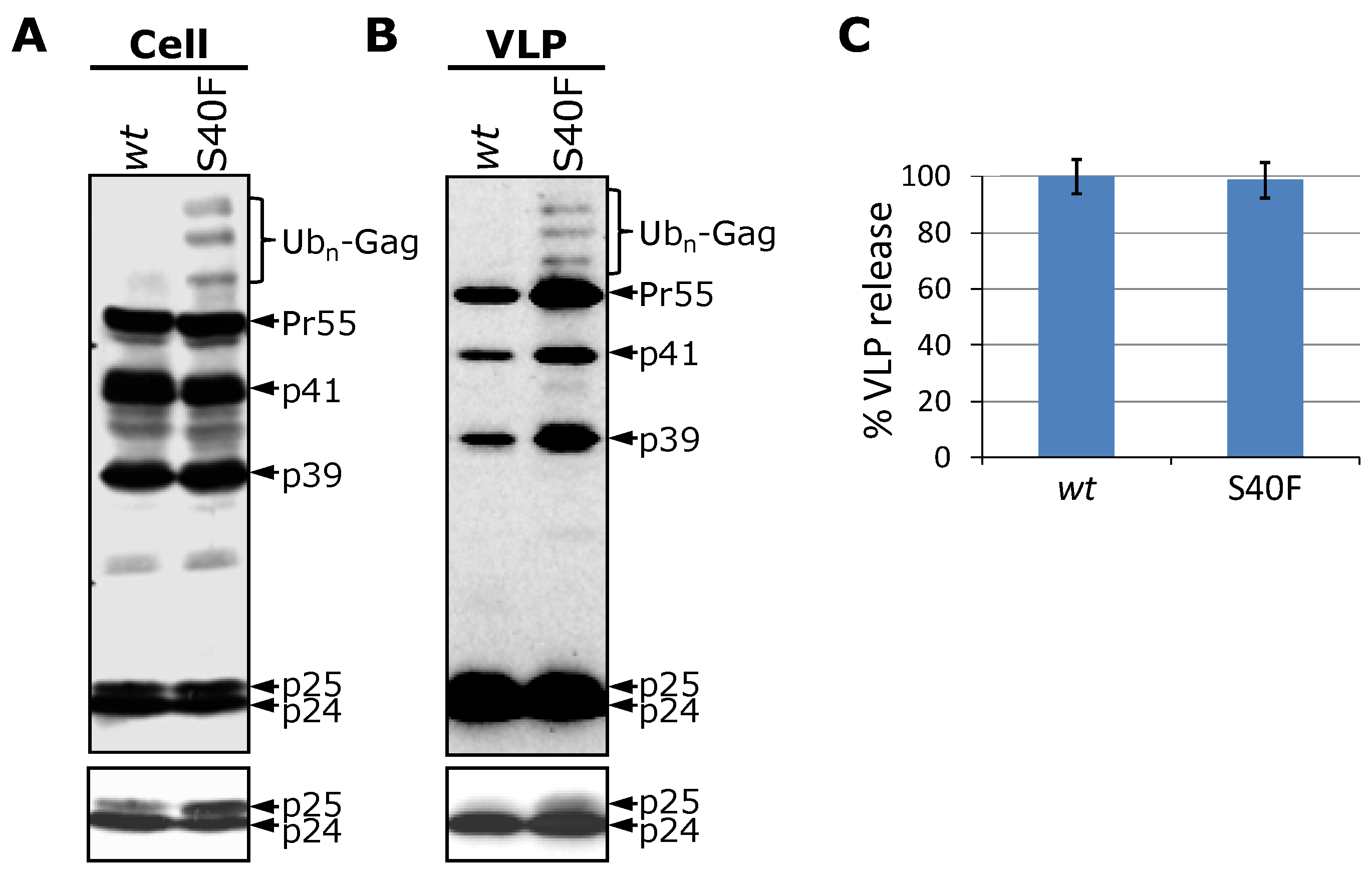
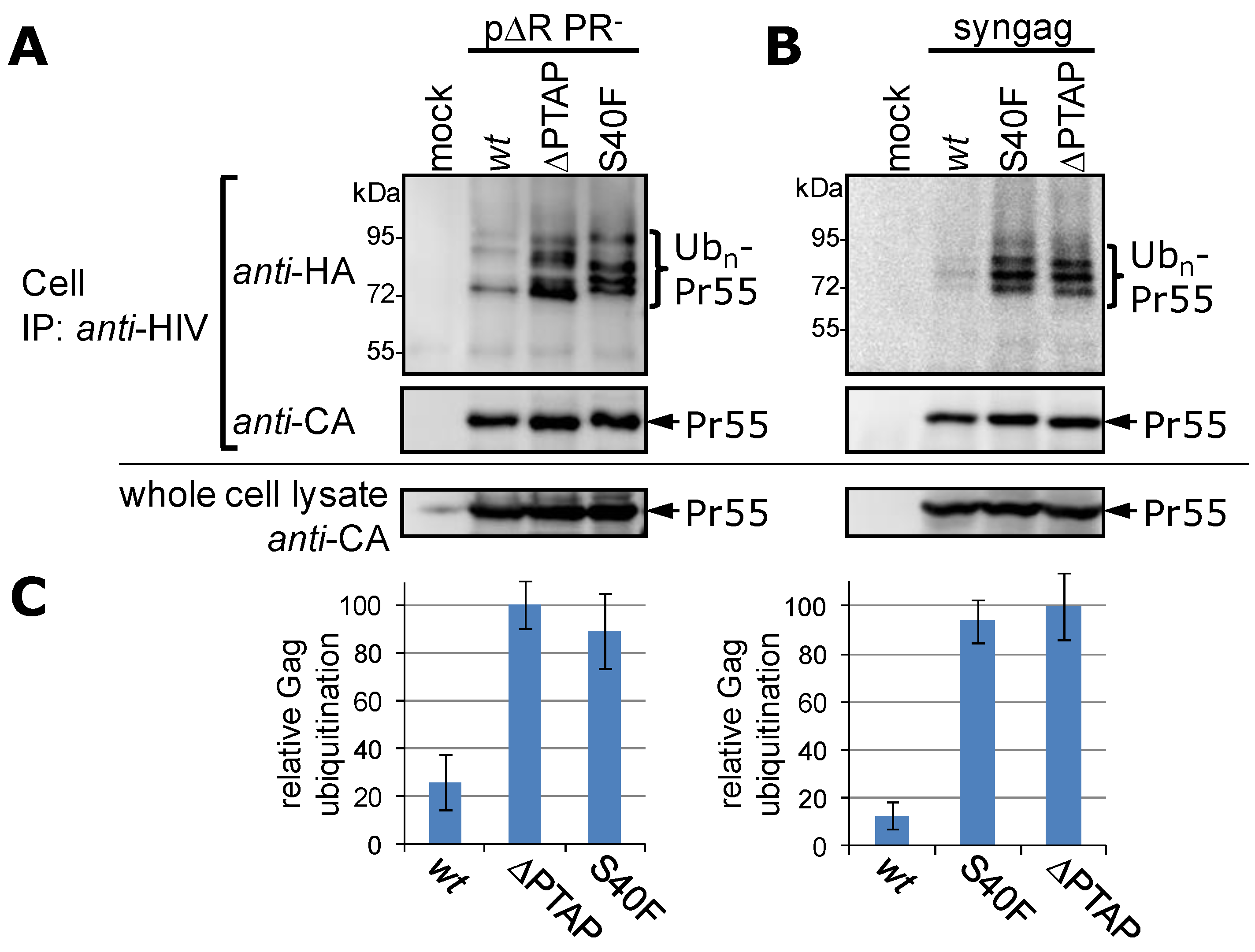
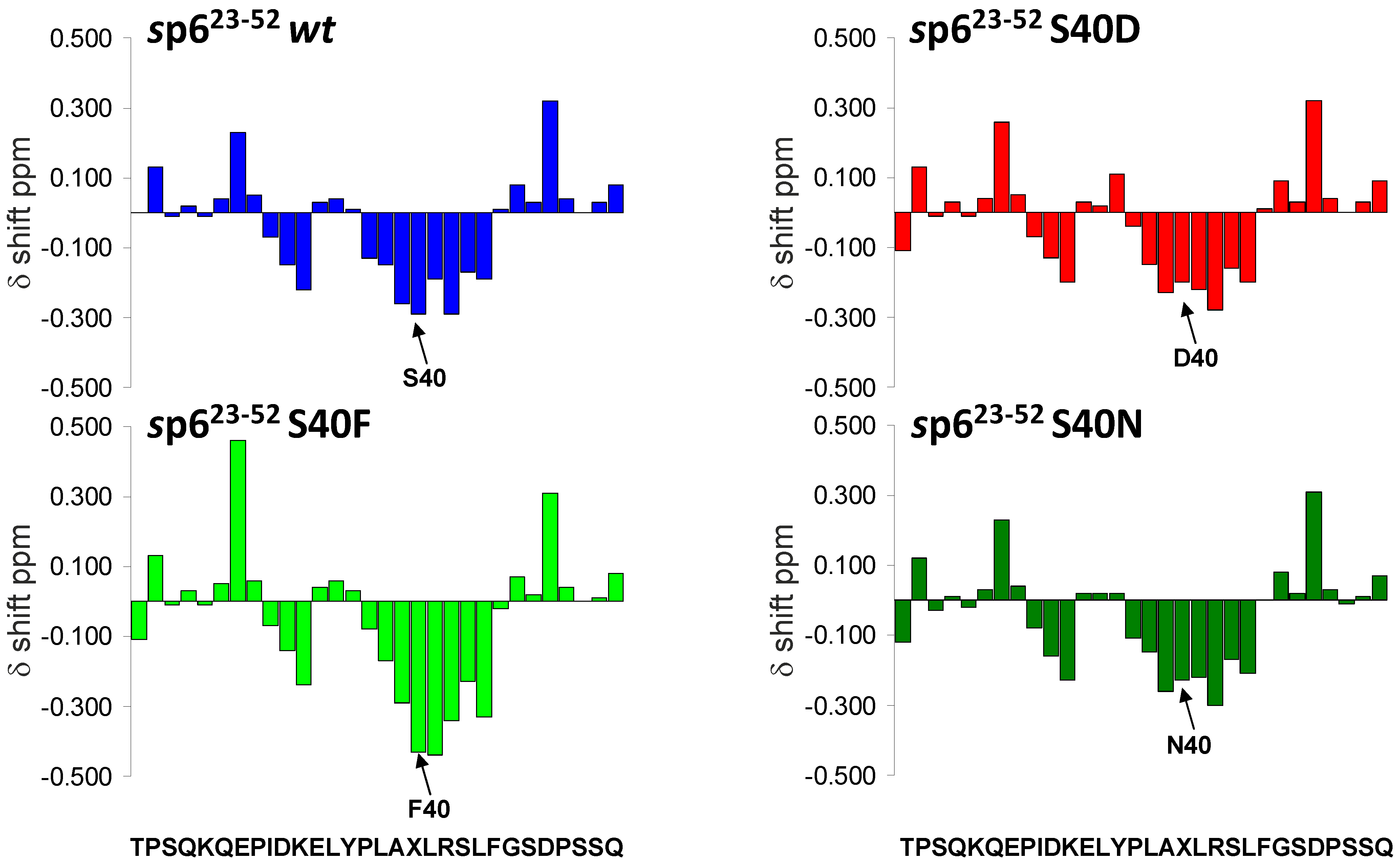
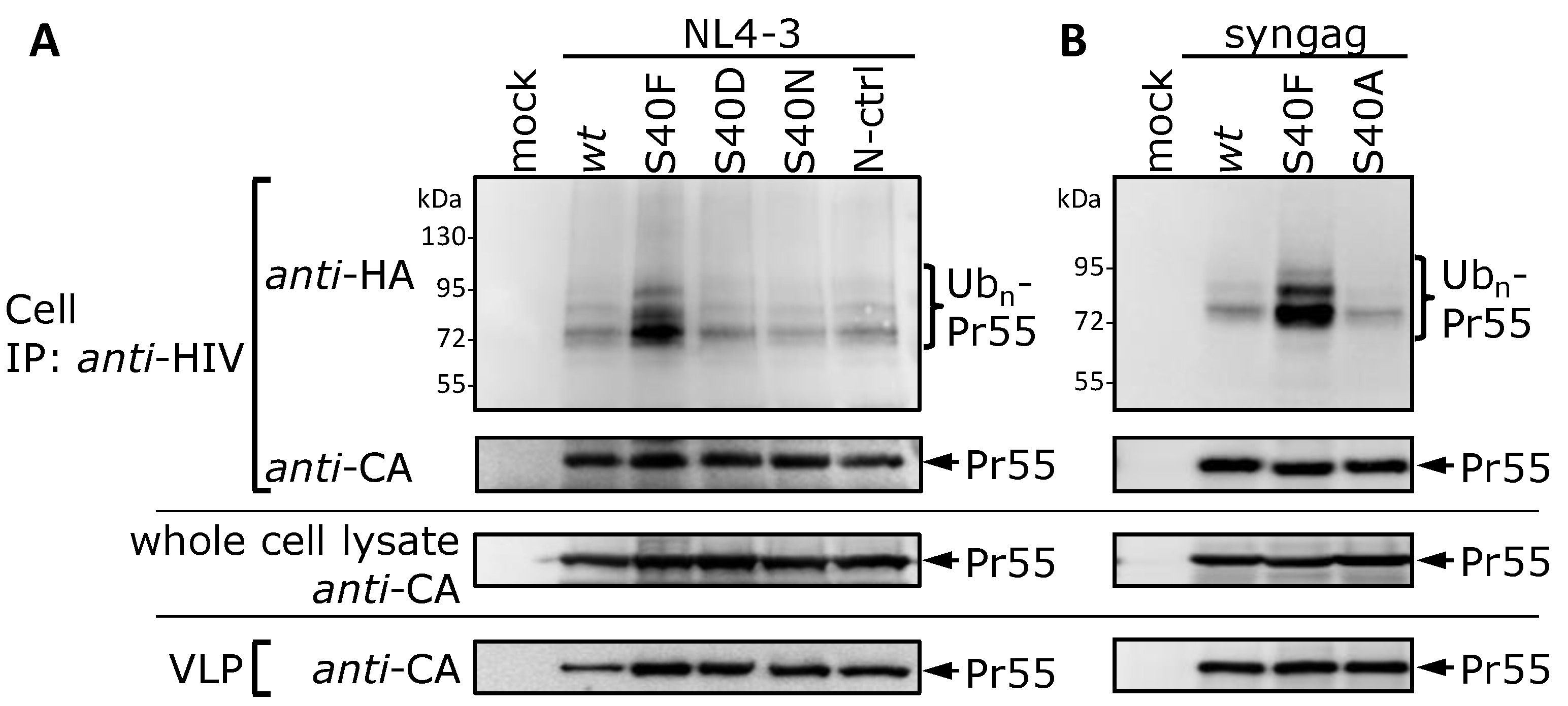
3.2. The Non-Conservative Amino Acid Exchange S40F, but Not the Conservative Substitutions S40D or S40N, Induces Elevated Gag Ubiquitination
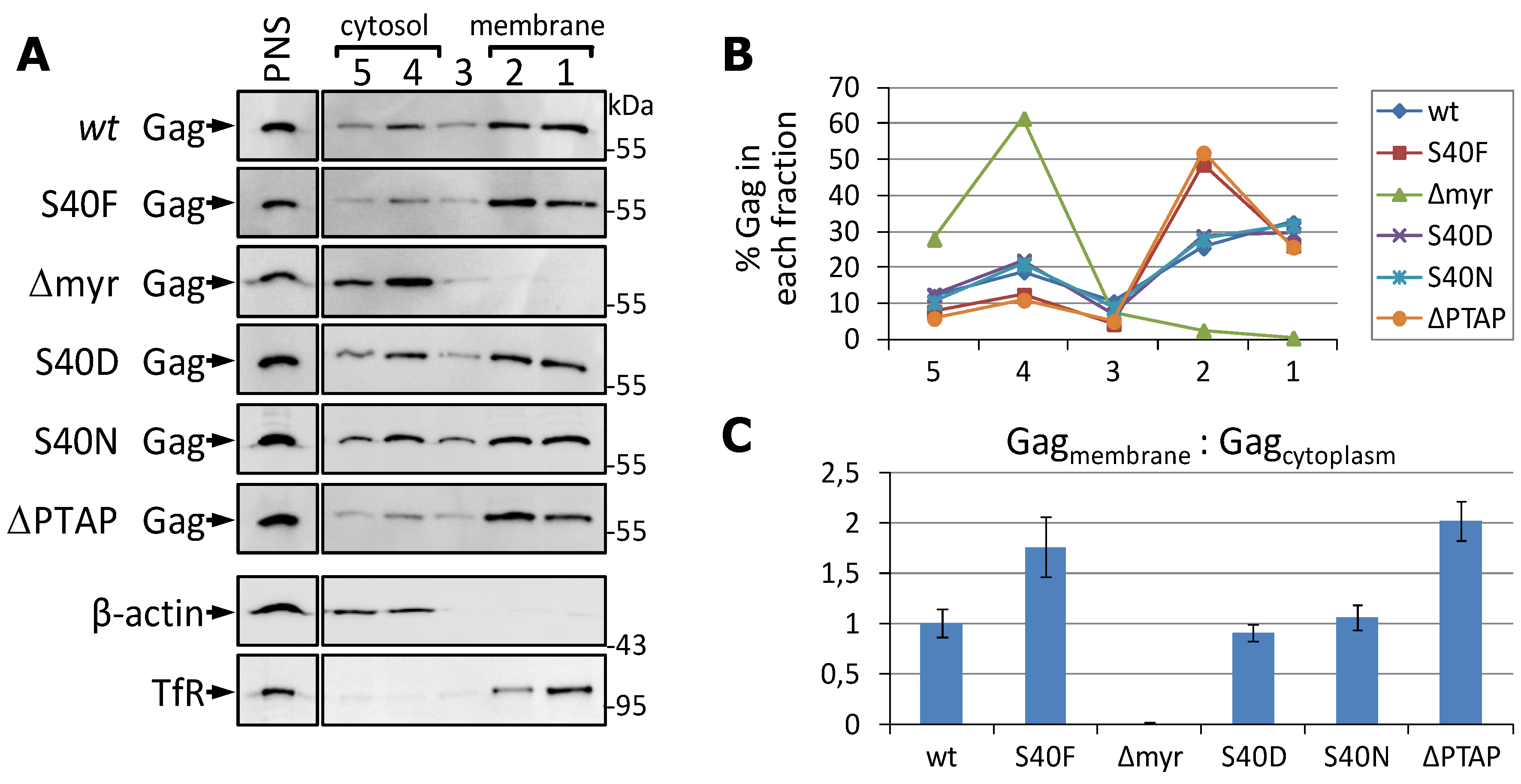
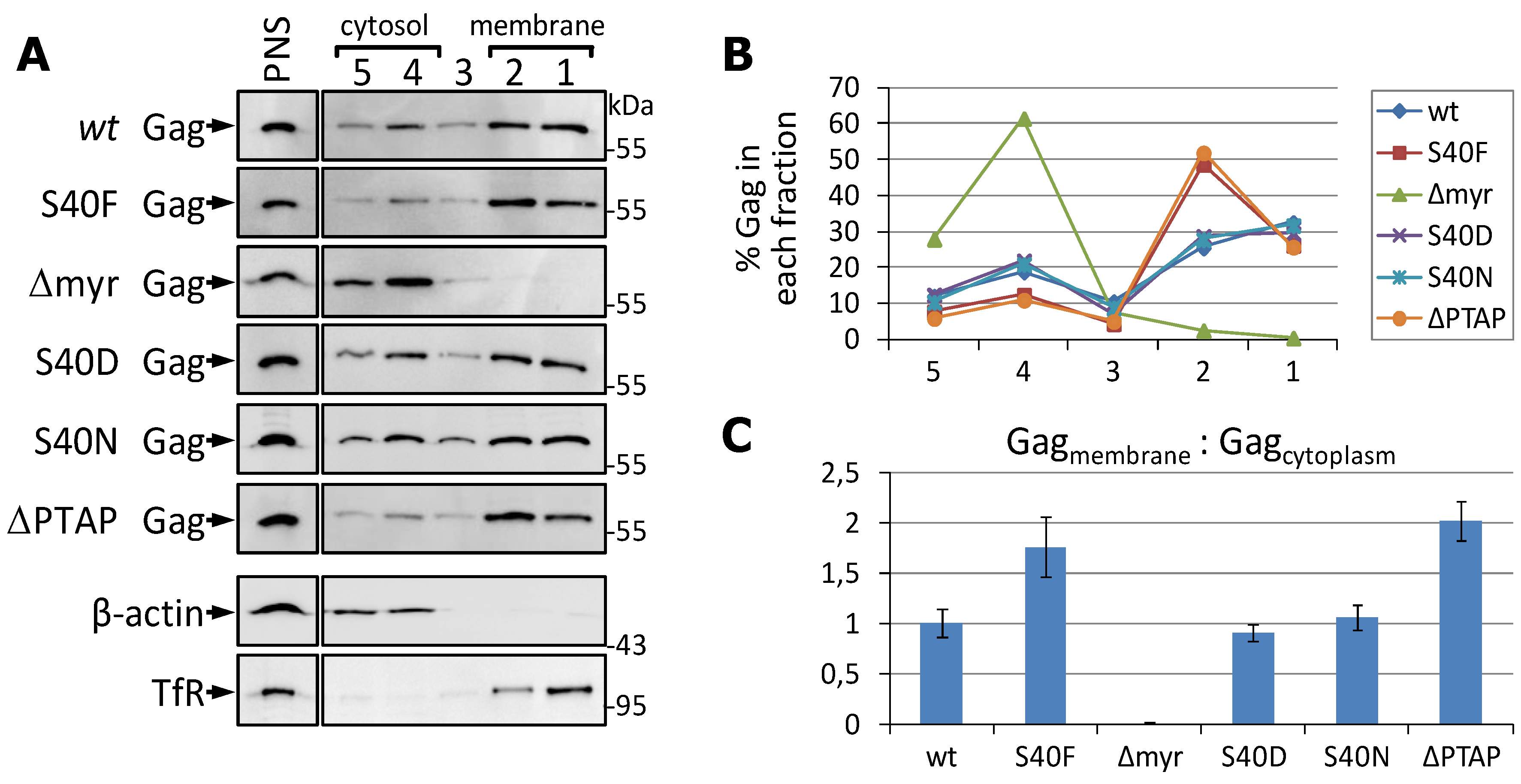
3.3. The S40F Mutation Increases the Amount of Membrane Associated Gag
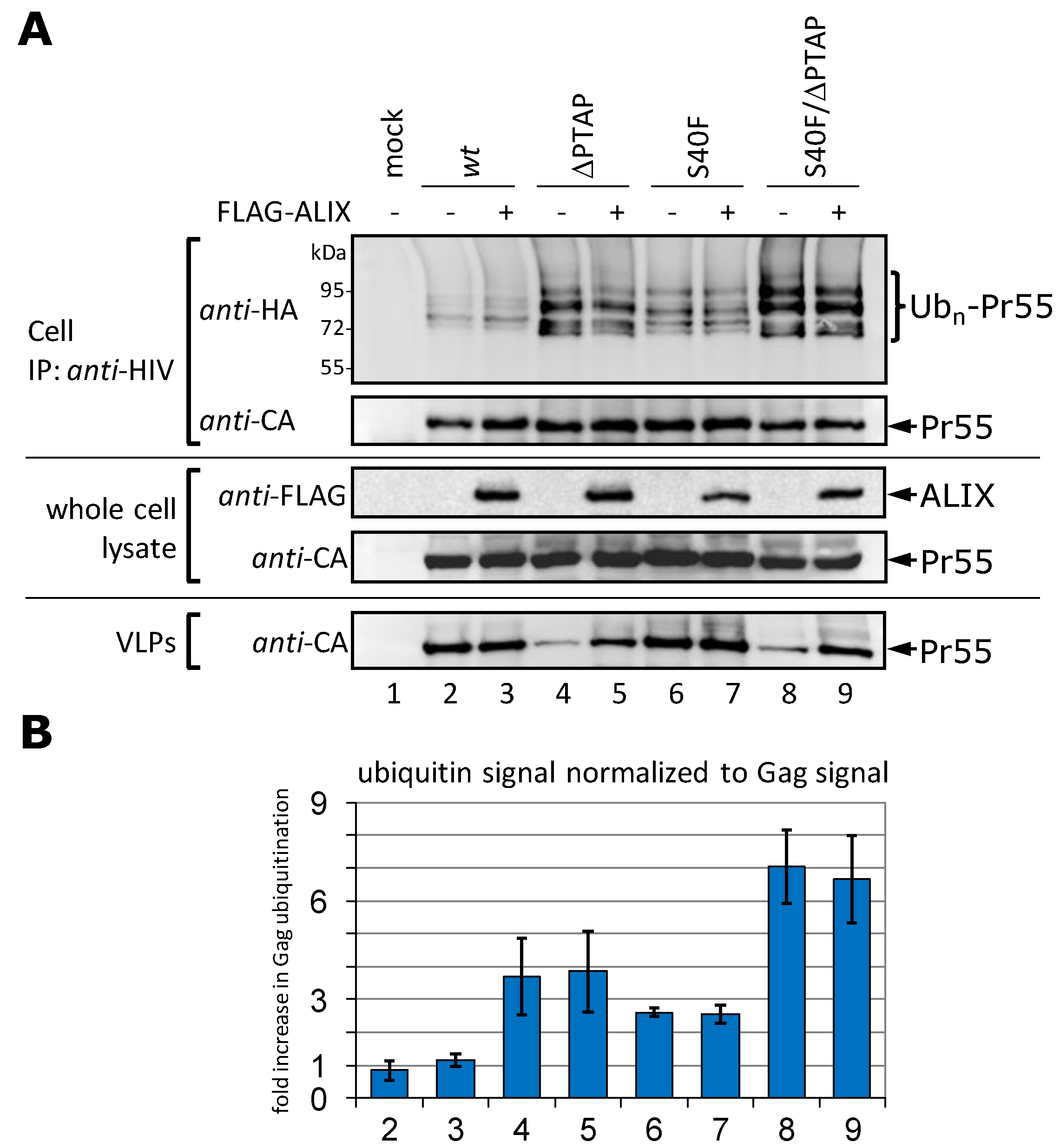
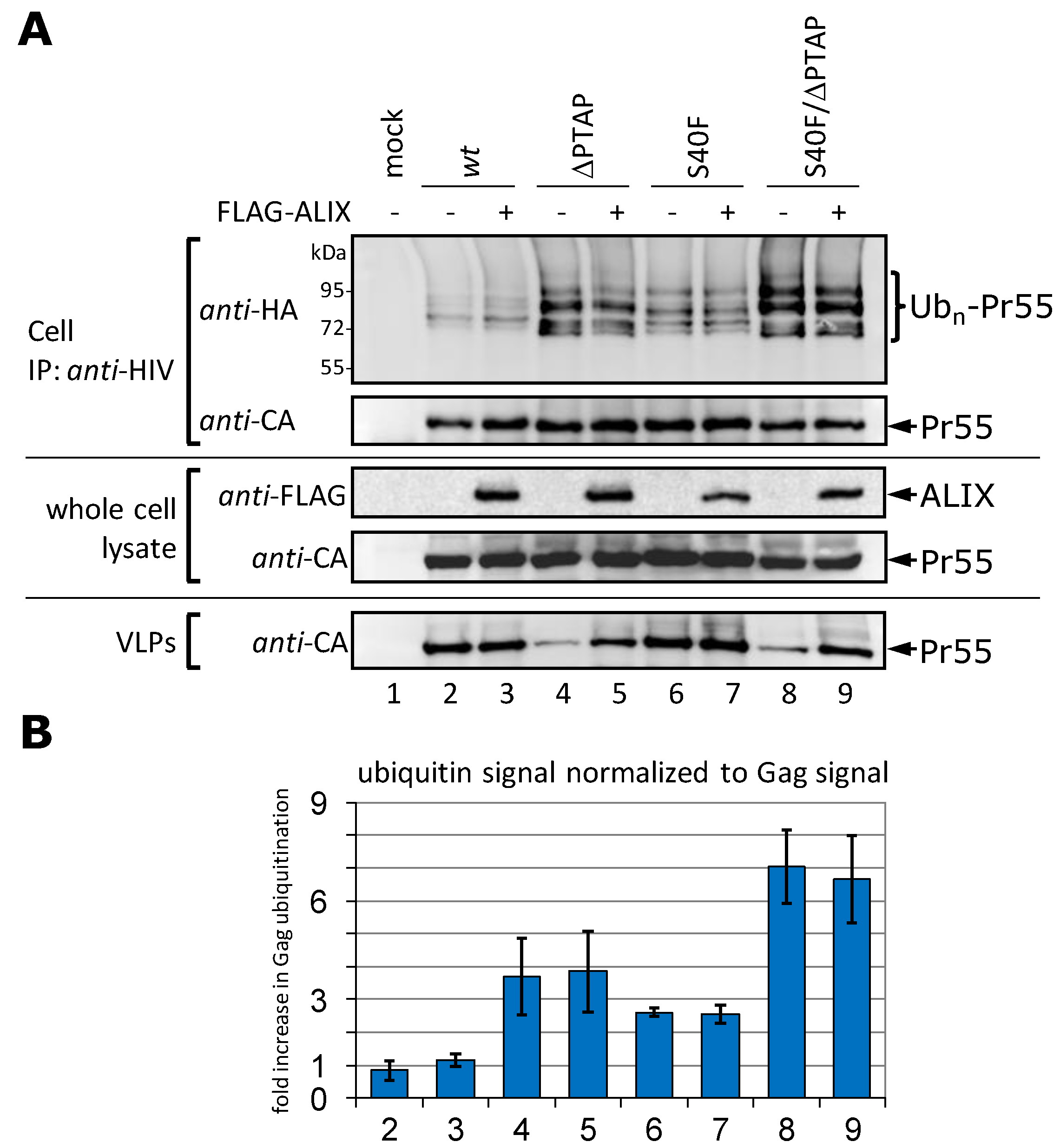
3.4. Augmentation of the VLP Release by ALIX Does Not Affect Gag Ubiquitination
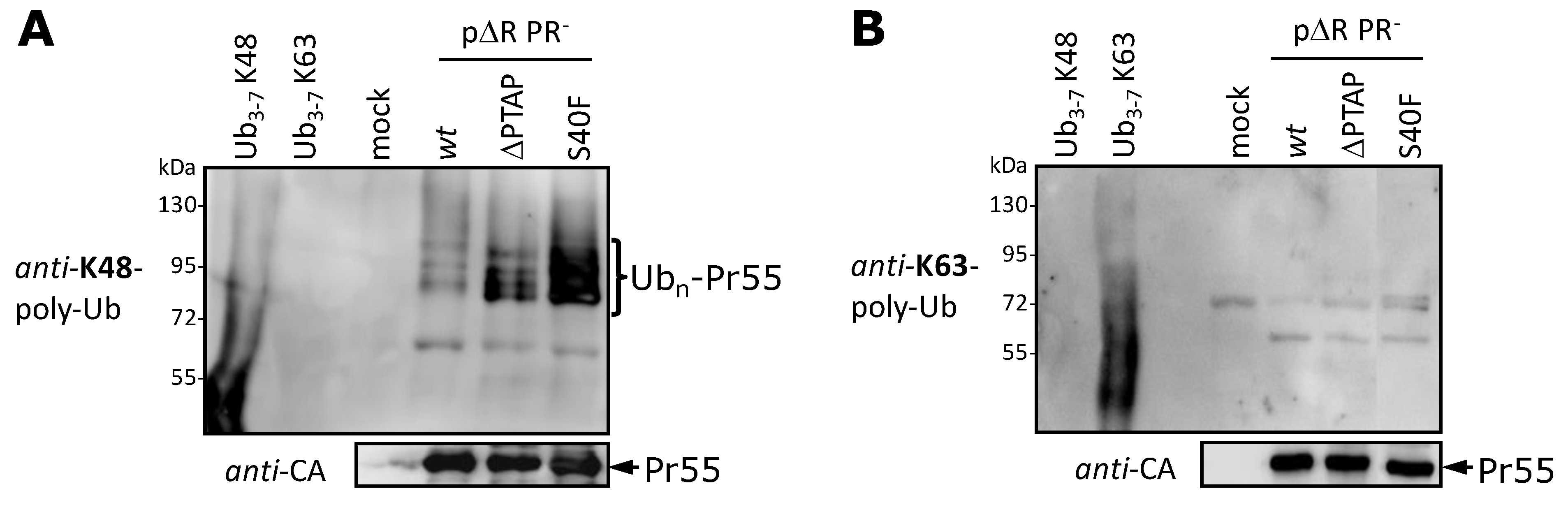
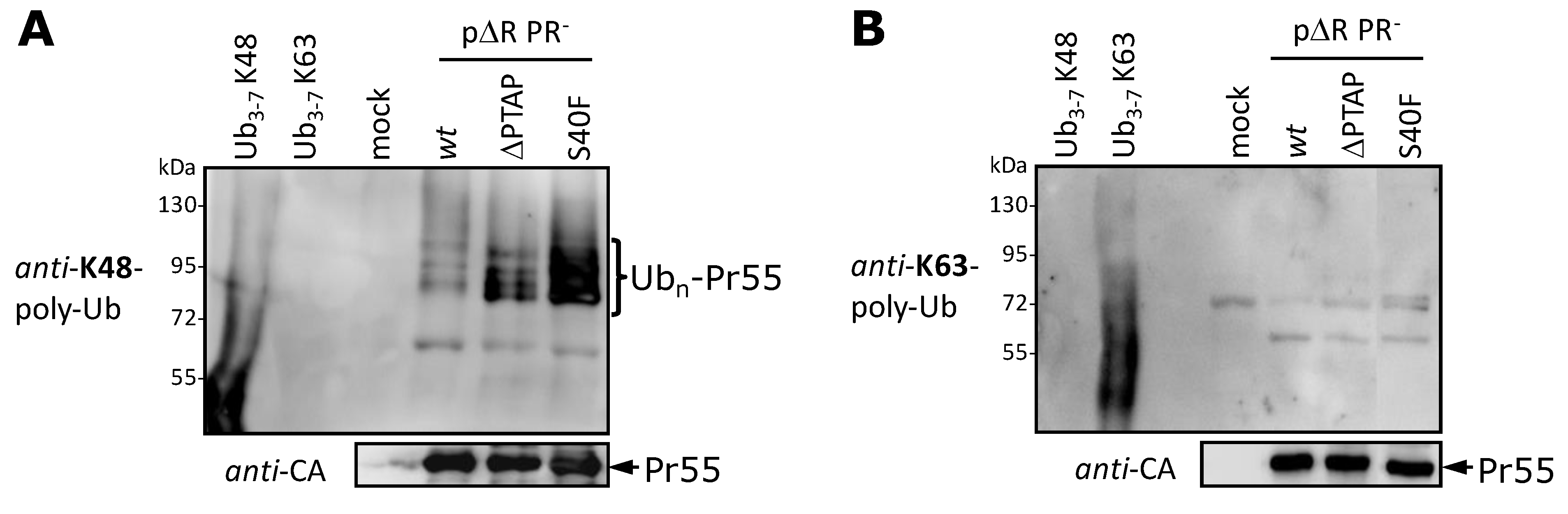
3.5. The S40F Mutation Increases the K48-Linked Polyubiquitination of Gag
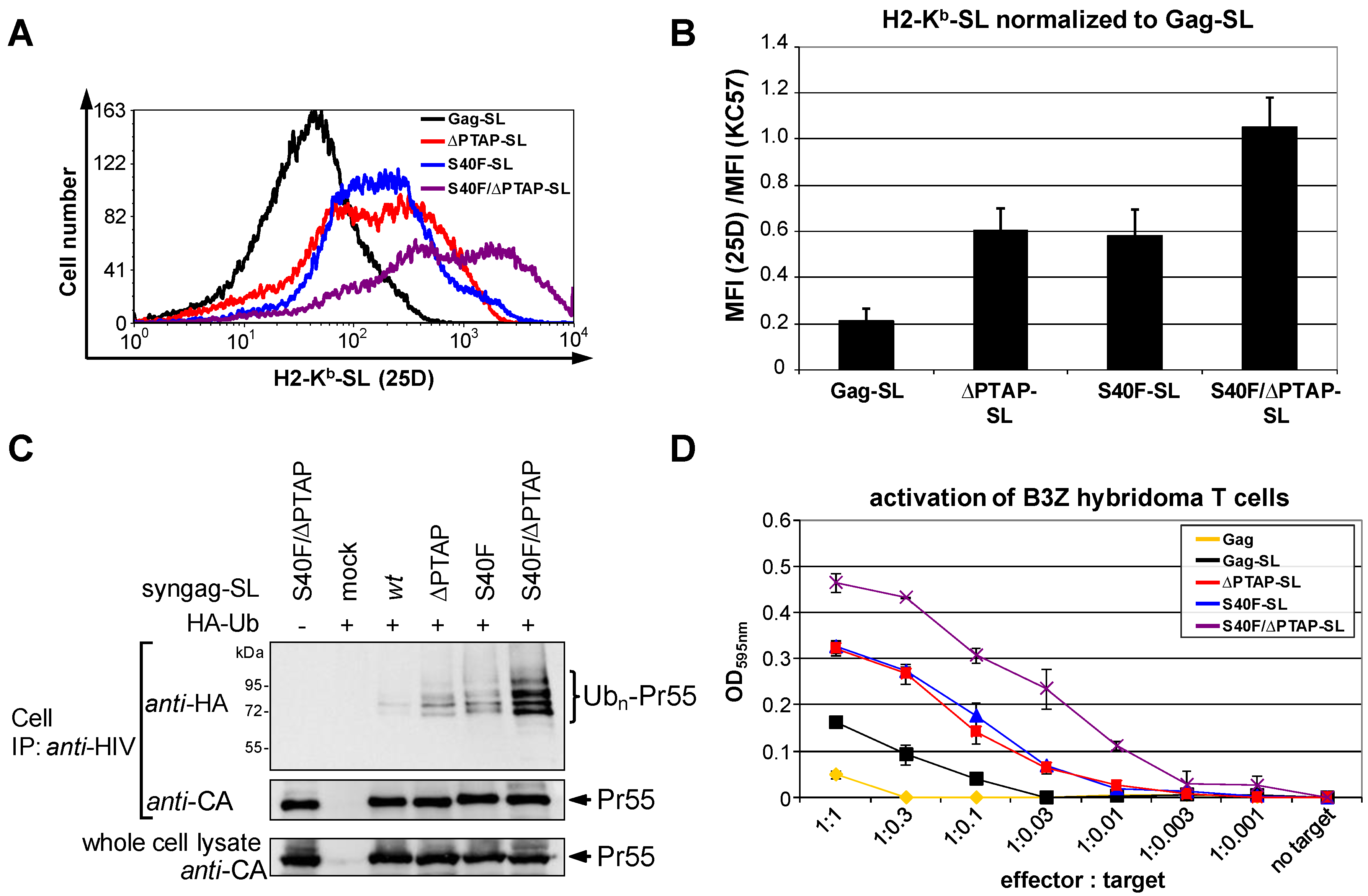
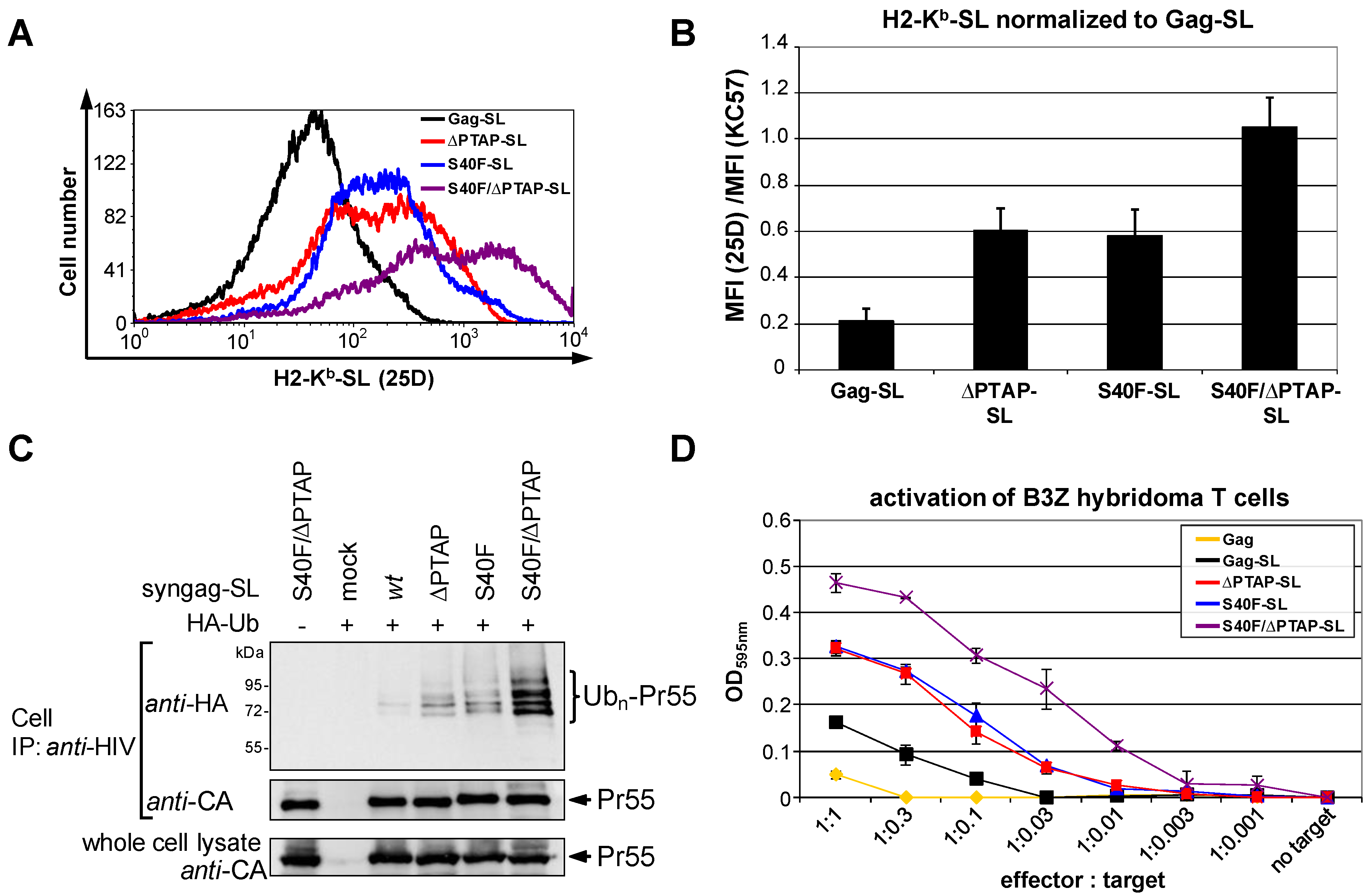
3.6. The S40F Mutation Enhances MHC-I Antigen Presentation of Gag
3.7. Mutations of Ser-40 Do Not Change the Extent or Localization of Secondary Structure of p6
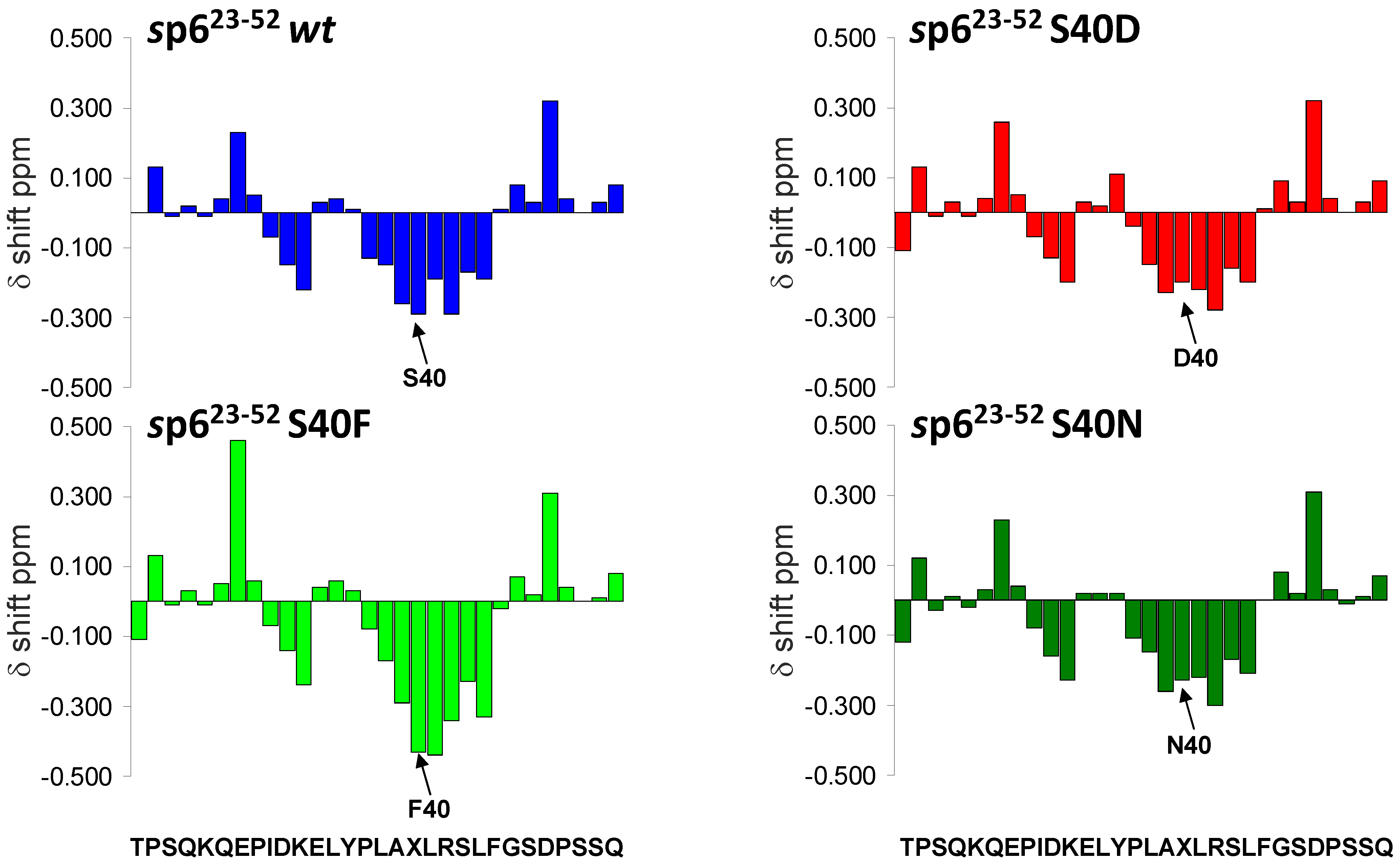
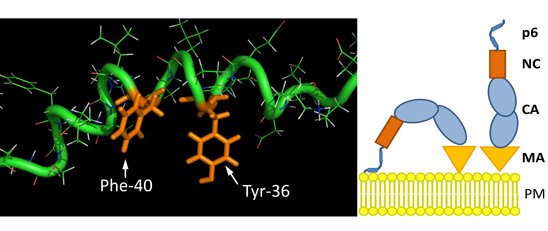
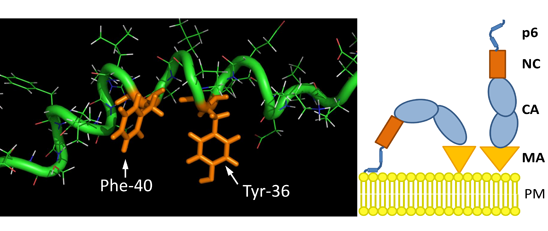
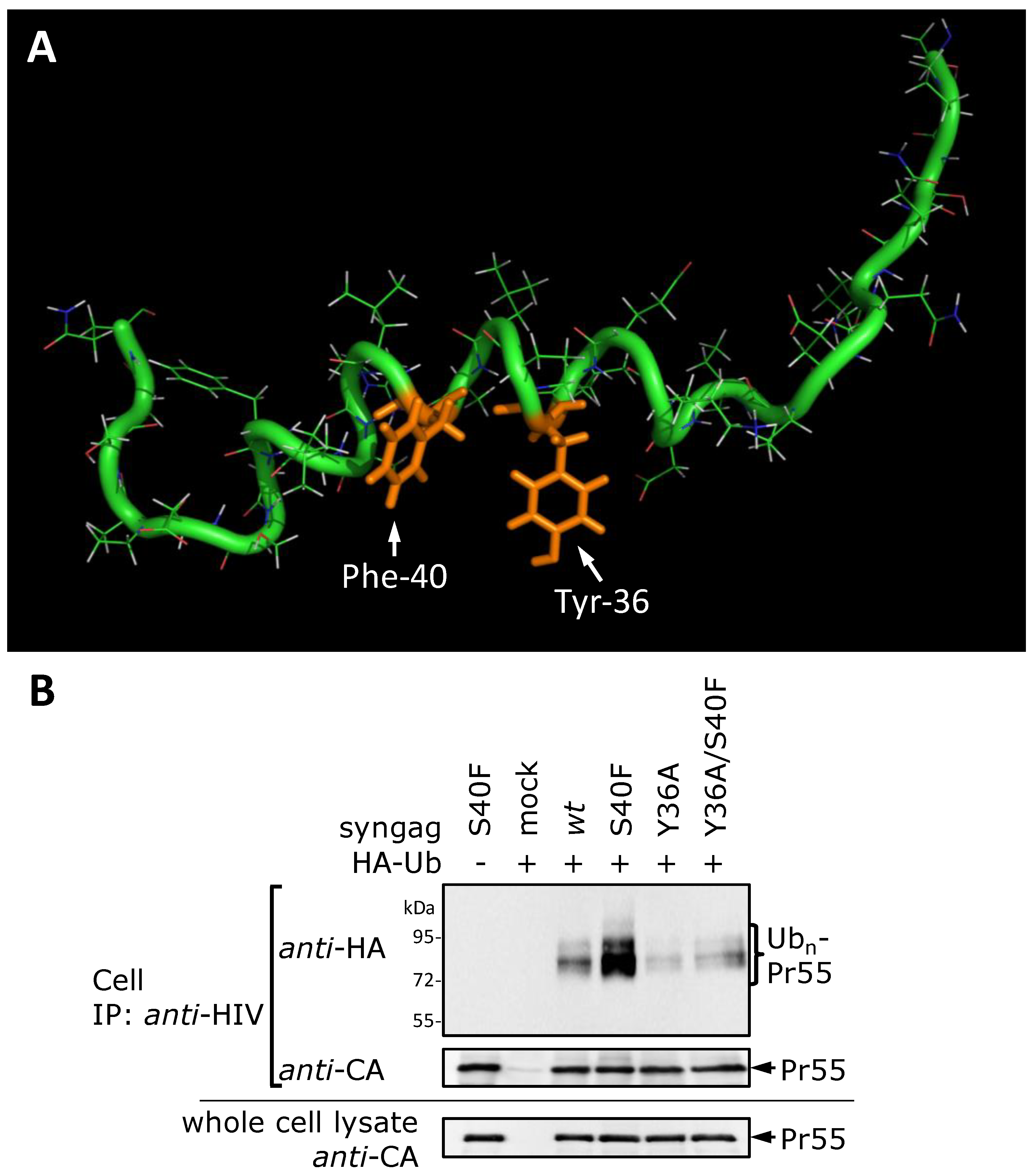
3.8. The S40F Mutation Creates a Hydrophobic Domain Involving Aromatic Residues Tyr-36 and Phe-40 at the Same Surface of the C-Terminal Helix of p6
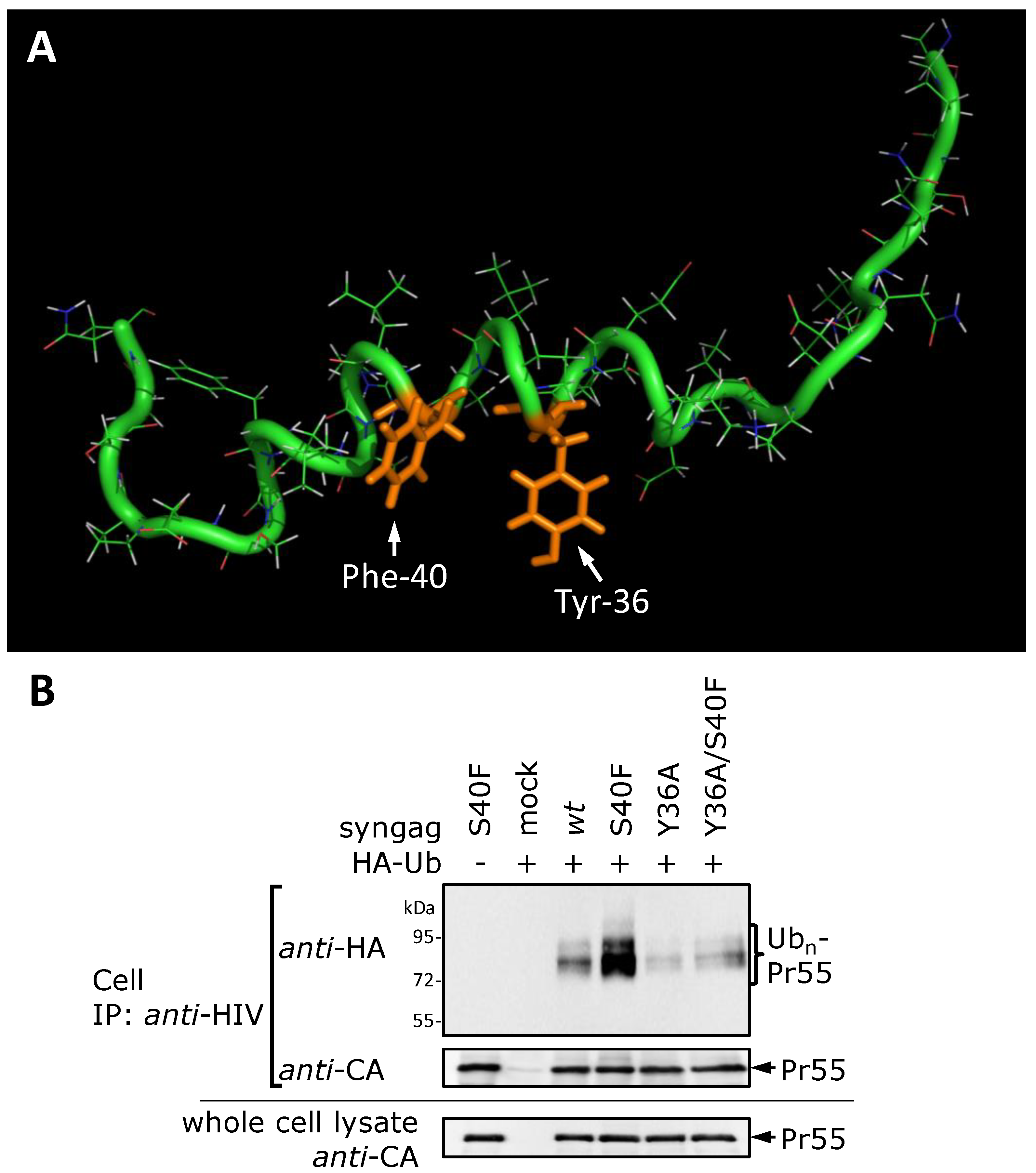
4. Discussion

Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Sundquist, W.I.; Krausslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, M.; Freed, E.O. New insights into HIV assembly and trafficking. Physiology Bethesda 2011, 26, 236–251. [Google Scholar] [CrossRef] [PubMed]
- Gottlinger, H.G.; Sodroski, J.G.; Haseltine, W.A. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 1989, 86, 5781–5785. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Parent, L.J.; Wills, J.W.; Resh, M.D. Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phospholipids. J. Virol. 1994, 68, 2556–2569. [Google Scholar] [PubMed]
- Darlix, J.L.; Godet, J.; Ivanyi-Nagy, R.; Fosse, P.; Mauffret, O.; Mely, Y. Flexible nature and specific functions of the HIV-1 nucleocapsid protein. J. Mol. Biol. 2011, 410, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Heng, X.; Summers, M.F. Structural determinants and mechanism of HIV-1 genome packaging. J. Mol. Biol. 2011, 410, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Cote, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef] [PubMed]
- VerPlank, L.; Bouamr, F.; LaGrassa, T.J.; Agresta, B.; Kikonyogo, A.; Leis, J.; Carter, C.A. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc. Natl. Acad. Sci. USA 2001, 98, 7724–7729. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Demirov, D.G.; Orenstein, J.M.; Freed, E.O. The late domain of human immunodeficiency virus type 1 p6 promotes virus release in a cell type-dependent manner. J. Virol. 2002, 76, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Strack, B.; Calistri, A.; Craig, S.; Popova, E.; Gottlinger, H.G. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 2003, 114, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Popov, S.; Gottlinger, H.G. Potent rescue of human immunodeficiency virus type 1 late domain mutants by ALIX/AIP1 depends on its CHMP4 binding site. J. Virol. 2007, 81, 6614–6622. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.D.; Chung, H.Y.; Zhai, Q.; Robinson, H.; Sundquist, W.I.; Hill, C.P. Structural and biochemical studies of ALIX/AIP1 and its role in retrovirus budding. Cell 2007, 128, 841–852. [Google Scholar] [CrossRef] [PubMed]
- von Schwedler, U.K.; Stuchell, M.; Muller, B.; Ward, D.M.; Chung, H.Y.; Morita, E.; Wang, H.E.; Davis, T.; He, G.P.; Cimbora, D.M.; et al. The protein network of HIV budding. Cell 2003, 114, 701–713. [Google Scholar]
- McCullough, J.; Colf, L.A.; Sundquist, W.I. Membrane fission reactions of the mammalian ESCRT pathway. Annu. Rev. Biochem. 2013, 82, 663–692. [Google Scholar] [CrossRef] [PubMed]
- Caballe, A.; Martin-Serrano, J. ESCRT machinery and cytokinesis: The road to daughter cell separation. Traffic 2011, 12, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Carlton, J.G.; Caballe, A.; Agromayor, M.; Kloc, M.; Martin-Serrano, J. ESCRT-III governs the Aurora B—Mediated abscission checkpoint through CHMP4C. Science 2012, 336, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Hanson, P.I.; Cashikar, A. Multivesicular body morphogenesis. Annu. Rev. Cell Dev. Biol. 2012, 28, 337–362. [Google Scholar] [CrossRef] [PubMed]
- Raiborg, C.; Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Shields, S.B.; Piper, R.C. How ubiquitin functions with ESCRTs. Traffic 2011, 12, 1306–1317. [Google Scholar] [CrossRef] [PubMed]
- Ott, D.E.; Coren, L.V.; Chertova, E.N.; Gagliardi, T.D.; Schubert, U. Ubiquitination of HIV-1 and MuLV Gag. Virology 2000, 278, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Ott, D.E.; Coren, L.V.; Copeland, T.D.; Kane, B.P.; Johnson, D.G.; Sowder, R.C., 2nd; Yoshinaka, Y.; Oroszlan, S.; Arthur, L.O.; Henderson, L.E. Ubiquitin is covalently attached to the p6Gag proteins of human immunodeficiency virus type 1 and simian immunodeficiency virus and to the p12Gag protein of Moloney murine leukemia virus. J. Virol. 1998, 72, 2962–2968. [Google Scholar]
- Patnaik, A.; Chau, V.; Wills, J.W. Ubiquitin is part of the retrovirus budding machinery. Proc. Natl. Acad. Sci. USA 2000, 97, 13069–13074. [Google Scholar] [CrossRef] [PubMed]
- Strack, B.; Calistri, A.; Accola, M.A.; Palu, G.; Gottlinger, H.G. A role for ubiquitin ligase recruitment in retrovirus release. Proc. Natl. Acad. Sci. USA 2000, 97, 13063–13068. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Jager, S.; Habermann, A.; Krausslich, H.G. Cumulative mutations of ubiquitin acceptor sites in human immunodeficiency virus type 1 gag cause a late budding defect. J. Virol. 2006, 80, 6267–6275. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Gottwein, E.; Krausslich, H.G. Ubiquitination of human immunodeficiency virus type 1 Gag is highly dependent on Gag membrane association. J. Virol. 2007, 81, 9193–9201. [Google Scholar] [CrossRef] [PubMed]
- Muller, B.; Patschinsky, T.; Krausslich, H.G. The late-domain-containing protein p6 is the predominant phosphoprotein of human immunodeficiency virus type 1 particles. J. Virol. 2002, 76, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Kudoh, A.; Takahama, S.; Sawasaki, T.; Ode, H.; Yokoyama, M.; Okayama, A.; Ishikawa, A.; Miyakawa, K.; Matsunaga, S.; Kimura, H.; et al. The phosphorylation of HIV-1 Gag by atypical protein kinase C facilitates viral infectivity by promoting Vpr incorporation into virions. Retrovirology 2014, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Hemonnot, B.; Cartier, C.; Gay, B.; Rebuffat, S.; Bardy, M.; Devaux, C.; Boyer, V.; Briant, L. The host cell MAP kinase ERK-2 regulates viral assembly and release by phosphorylating the p6gag protein of HIV-1. J. Biol. Chem. 2004, 279, 32426–32434. [Google Scholar] [CrossRef] [PubMed]
- Votteler, J.; Neumann, L.; Hahn, S.; Hahn, F.; Rauch, P.; Schmidt, K.; Studtrucker, N.; Solbak, S.M.; Fossen, T.; Henklein, P.; et al. Highly conserved serine residue 40 in HIV-1 p6 regulates capsid processing and virus core assembly. Retrovirology 2011, 8, 11. [Google Scholar] [CrossRef]
- Watanabe, S.M.; Chen, M.H.; Khan, M.; Ehrlich, L.; Kemal, K.S.; Weiser, B.; Shi, B.; Chen, C.; Powell, M.; Anastos, K.; et al. The S40 residue in HIV-1 Gag p6 impacts local and distal budding determinants, revealing additional late domain activities. Retrovirology 2013, 10, 143. [Google Scholar] [CrossRef] [PubMed]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef]
- Okumura, F.; Yoshida, K.; Liang, F.; Hatakeyama, S. MDA-9/syntenin interacts with ubiquitin via a novel ubiquitin-binding motif. Mol. Cell. Biochem. 2011, 352, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, S.; Bago, R.; Odintsova, E.; Muratov, G.; Baldwin, G.; Sridhar, P.; Overduin, M.; Berditchevski, F. Binding to syntenin-1 protein defines a new mode of ubiquitin-based interactions regulated by phosphorylation. J. Biol. Chem. 2011, 286, 39606–39614. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T. The age of crosstalk: Phosphorylation, ubiquitination, and beyond. Mol. Cell 2007, 28, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Salgado, G.F.; Vogel, A.; Marquant, R.; Feller, S.E.; Bouaziz, S.; Alves, I.D. The role of membranes in the organization of HIV-1 Gag p6 and Vpr: p6 shows high affinity for membrane bilayers which substantially increases the interaction between p6 and Vpr. J. Med. Chem. 2009, 52, 7157–7162. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Krausslich, H.G. Analysis of human immunodeficiency virus type 1 Gag ubiquitination. J. Virol. 2005, 79, 9134–9144. [Google Scholar] [CrossRef] [PubMed]
- Votteler, J.; Iavnilovitch, E.; Fingrut, O.; Shemesh, V.; Taglicht, D.; Erez, O.; Sorgel, S.; Walther, T.; Bannert, N.; Schubert, U.; et al. Exploring the functional interaction between POSH and ALIX and the relevance to HIV-1 release. BMC Biochem. 2009, 10, 12. [Google Scholar]
- Adachi, A.; Gendelman, H.E.; Koenig, S.; Folks, T.; Willey, R.; Rabson, A.; Martin, M.A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986, 59, 284–291. [Google Scholar] [PubMed]
- Schubert, U.; Clouse, K.A.; Strebel, K. Augmentation of virus secretion by the human immunodeficiency virus type 1 Vpu protein is cell type independent and occurs in cultured human primary macrophages and lymphocytes. J. Virol. 1995, 69, 7699–7711. [Google Scholar]
- Hahn, S.; Setz, C.; Wild, J.; Schubert, U. The PTAP sequence within the p6 domain of human immunodeficiency virus type 1 Gag regulates its ubiquitination and MHC class I antigen presentation. J. Immunol. 2011, 186, 5706–5718. [Google Scholar] [CrossRef] [PubMed]
- Goldwich, A.; Hahn, S.S.; Schreiber, S.; Meier, S.; Kampgen, E.; Wagner, R.; Lutz, M.B.; Schubert, U. Targeting HIV-1 Gag into the defective ribosomal product pathway enhances MHC class I antigen presentation and CD8+ T cell activation. J. Immunol. 2008, 180, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Köhler, G.; Fisher-Lindahl, K.; Heusser, C. Characterization of a Monoclonal Anti-H-2Kd Antibody; Karger AG: Basel, Switzerland, 1981; Volume 2, pp. 202–208. [Google Scholar]
- FCS Express Version 3; Version number 3.00.0819, Professional Network; De Novo software: Los Angeles, CA, USA, 2001.
- Sanderson, S.; Shastri, N. LacZ inducible, antigen/MHC-specific T cell hybrids. Int. Immunol. 1994, 6, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Henklein, P.; Bruns, K.; Sherman, M.P.; Tessmer, U.; Licha, K.; Kopp, J.; de Noronha, C.M.; Greene, W.C.; Wray, V.; Schubert, U. Functional and structural characterization of synthetic HIV-1 Vpr that transduces cells, localizes to the nucleus, and induces G2 cell cycle arrest. J. Biol. Chem. 2000, 275, 32016–32026. [Google Scholar] [CrossRef] [PubMed]
- Fossen, T.; Wray, V.; Bruns, K.; Rachmat, J.; Henklein, P.; Tessmer, U.; Maczurek, A.; Klinger, P.; Schubert, U. Solution structure of the human immunodeficiency virus type 1 p6 protein. J. Biol. Chem. 2005, 280, 42515–42527. [Google Scholar] [CrossRef] [PubMed]
- Bruker Topspin 2.1 Software; Version 2.1; Bruker Biospin AG: Fällanden, Switzerland.
- Wüthrich, K. NMR of Proteins and Nucleic Acids; John Wiley and Sons, Inc.: New York, NY, USA, 1986. [Google Scholar]
- Sparky 3, Version 3.1.1.4. Goddard, T.D.; Kneller, D.G (Eds.) University of California: San Francisco, USA, 2007.
- Guntert, P.; Mumenthaler, C.; Wuthrich, K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 1997, 273, 283–298. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, version 1.3r1; Schrodinger, LLC: New York, NY, USA, 2010.
- Radestock, B.; Morales, I.; Rahman, S.A.; Radau, S.; Glass, B.; Zahedi, R.P.; Muller, B.; Krausslich, H.G. Comprehensive mutational analysis reveals p6Gag phosphorylation to be dispensable for HIV-1 morphogenesis and replication. J. Virol. 2013, 87, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, J.; Perez-Caballero, D.; Bieniasz, P.D. Context-dependent effects of L domains and ubiquitination on viral budding. J. Virol. 2004, 78, 5554–5563. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Orenstein, J.M.; Martin, M.A.; Freed, E.O. p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J. Virol. 1995, 69, 6810–6818. [Google Scholar] [PubMed]
- Deml, L.; Bojak, A.; Steck, S.; Graf, M.; Wild, J.; Schirmbeck, R.; Wolf, H.; Wagner, R. Multiple effects of codon usage optimization on expression and immunogenicity of DNA candidate vaccines encoding the human immunodeficiency virus type 1 Gag protein. J. Virol. 2001, 75, 10991–11001. [Google Scholar] [CrossRef] [PubMed]
- Solbak, S.M.; Reksten, T.R.; Hahn, F.; Wray, V.; Henklein, P.; Halskau, O.; Schubert, U.; Fossen, T. HIV-1 p6—A structured to flexible multifunctional membrane-interacting protein. Biochim. Biophys. Acta 2013, 1828, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Waheed, A.A.; Joshi, A.; Freed, E.O. Association of human immunodeficiency virus type 1 gag with membrane does not require highly basic sequences in the nucleocapsid: Use of a novel Gag multimerization assay. J. Virol. 2005, 79, 14131–14140. [Google Scholar] [CrossRef]
- Spearman, P.; Wang, J.J.; Vander Heyden, N.; Ratner, L. Identification of human immunodeficiency virus type 1 Gag protein domains essential to membrane binding and particle assembly. J. Virol. 1994, 68, 3232–3242. [Google Scholar] [PubMed]
- Ciechanover, A. Proteolysis: From the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 2005, 6, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Porgador, A.; Yewdell, J.W.; Deng, Y.; Bennink, J.R.; Germain, R.N. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity 1997, 6, 715–726. [Google Scholar] [CrossRef] [PubMed]
- York, I.A.; Chang, S.C.; Saric, T.; Keys, J.A.; Favreau, J.M.; Goldberg, A.L.; Rock, K.L. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues. Nat. Immunol. 2002, 3, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Puorro, K.A.; Porgador, A.; Eisenlohr, L.C. The induction of virus-specific CTL as a function of increasing epitope expression: Responses rise steadily until excessively high levels of epitope are attained. J. Immunol. 1999, 163, 3735–3745. [Google Scholar] [PubMed]
- Wishart, D.S.; Sykes, B.D.; Richards, F.M. The chemical shift index: A fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 1992, 31, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Putterman, D.; Pepinsky, R.B.; Vogt, V.M. Ubiquitin in avian leukosis virus particles. Virology 1990, 176, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Munshi, U.; Ablan, S.D.; Nagashima, K.; Freed, E.O. Functional Replacement of a Retroviral Late Domain by Ubiquitin Fusion. Traffic 2008, 9, 1972–1983. [Google Scholar] [CrossRef] [PubMed]
- Zhadina, M.; Bieniasz, P.D. Functional interchangeability of late domains, late domain cofactors and ubiquitin in viral budding. PLoS Pathog. 2010, 6, e1001153. [Google Scholar] [CrossRef] [PubMed]
- Sette, P.; Nagashima, K.; Piper, R.C.; Bouamr, F. Ubiquitin conjugation to Gag is essential for ESCRT-mediated HIV-1 budding. Retrovirology 2013, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- Keren-Kaplan, T.; Attali, I.; Estrin, M.; Kuo, L.S.; Farkash, E.; Jerabek-Willemsen, M.; Blutraich, N.; Artzi, S.; Peri, A.; Freed, E.O.; et al. Structure-based in silico identification of ubiquitin-binding domains provides insights into the ALIX-V: Ubiquitin complex and retrovirus budding. Embo J. 2013, 32, 538–551. [Google Scholar] [CrossRef] [PubMed]
- Dowlatshahi, D.P.; Sandrin, V.; Vivona, S.; Shaler, T.A.; Kaiser, S.E.; Melandri, F.; Sundquist, W.I.; Kopito, R.R. ALIX is a Lys63-specific polyubiquitin binding protein that functions in retrovirus budding. Dev. Cell 2012, 23, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Stringer, D.K.; Piper, R.C. A single ubiquitin is sufficient for cargo protein entry into MVBs in the absence of ESCRT ubiquitination. J. Cell Biol. 2011, 192, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Piper, R.C.; Katzmann, D.J. Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol. 2007, 23, 519–547. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Kirkpatrick, D.; Jiang, X.; Gygi, S.; Sorkin, A. Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol. Cell 2006, 21, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Lauwers, E.; Erpapazoglou, Z.; Haguenauer-Tsapis, R.; Andre, B. The ubiquitin code of yeast permease trafficking. Trends Cell Biol. 2010, 20, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Weiss, E.R.; Popova, E.; Yamanaka, H.; Kim, H.C.; Huibregtse, J.M.; Gottlinger, H. Rescue of HIV-1 release by targeting widely divergent NEDD4-type ubiquitin ligases and isolated catalytic HECT domains to Gag. PLoS Pathog. 2010, 6, e1001107. [Google Scholar] [CrossRef] [PubMed]
- Strack, B.; Calistri, A.; Gottlinger, H.G. Late assembly domain function can exhibit context dependence and involves ubiquitin residues implicated in endocytosis. J. Virol. 2002, 76, 5472–5479. [Google Scholar] [CrossRef] [PubMed]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Munro, J.B.; Nath, A.; Farber, M.; Datta, S.A.; Rein, A.; Rhoades, E.; Mothes, W. A conformational transition observed in single HIV-1 Gag molecules during in vitro assembly of virus-like particles. J. Virol. 2014, 88, 3577–3585. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.A.; Curtis, J.E.; Ratcliff, W.; Clark, P.K.; Crist, R.M.; Lebowitz, J.; Krueger, S.; Rein, A. Conformation of the HIV-1 Gag protein in solution. J. Mol. Biol. 2007, 365, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.A.; Heinrich, F.; Raghunandan, S.; Krueger, S.; Curtis, J.E.; Rein, A.; Nanda, H. HIV-1 Gag extension: Conformational changes require simultaneous interaction with membrane and nucleic acid. J. Mol. Biol. 2011, 406, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L.; Corey, R.B.; Branson, H.R. The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA 1951, 37, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Dathe, M.; Schumann, M.; Wieprecht, T.; Winkler, A.; Beyermann, M.; Krause, E.; Matsuzaki, K.; Murase, O.; Bienert, M. Peptide helicity and membrane surface charge modulate the balance of electrostatic and hydrophobic interactions with lipid bilayers and biological membranes. Biochemistry 1996, 35, 12612–12622. [Google Scholar] [CrossRef] [PubMed]
- McCullough, J.; Clague, M.J.; Urbe, S. AMSH is an endosome-associated ubiquitin isopeptidase. J. Cell Biol. 2004, 166, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.; Anton, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hahn, F.; Setz, C.; Friedrich, M.; Rauch, P.; Solbak, S.M.; Frøystein, N.Å.; Henklein, P.; Votteler, J.; Fossen, T.; Schubert, U. Mutation of the Highly Conserved Ser-40 of the HIV-1 p6 Gag Protein to Phe Causes the Formation of a Hydrophobic Patch, Enhances Membrane Association, and Polyubiquitination of Gag. Viruses 2014, 6, 3738-3765. https://doi.org/10.3390/v6103738
Hahn F, Setz C, Friedrich M, Rauch P, Solbak SM, Frøystein NÅ, Henklein P, Votteler J, Fossen T, Schubert U. Mutation of the Highly Conserved Ser-40 of the HIV-1 p6 Gag Protein to Phe Causes the Formation of a Hydrophobic Patch, Enhances Membrane Association, and Polyubiquitination of Gag. Viruses. 2014; 6(10):3738-3765. https://doi.org/10.3390/v6103738
Chicago/Turabian StyleHahn, Friedrich, Christian Setz, Melanie Friedrich, Pia Rauch, Sara Marie Solbak, Nils Åge Frøystein, Petra Henklein, Jörg Votteler, Torgils Fossen, and Ulrich Schubert. 2014. "Mutation of the Highly Conserved Ser-40 of the HIV-1 p6 Gag Protein to Phe Causes the Formation of a Hydrophobic Patch, Enhances Membrane Association, and Polyubiquitination of Gag" Viruses 6, no. 10: 3738-3765. https://doi.org/10.3390/v6103738
APA StyleHahn, F., Setz, C., Friedrich, M., Rauch, P., Solbak, S. M., Frøystein, N. Å., Henklein, P., Votteler, J., Fossen, T., & Schubert, U. (2014). Mutation of the Highly Conserved Ser-40 of the HIV-1 p6 Gag Protein to Phe Causes the Formation of a Hydrophobic Patch, Enhances Membrane Association, and Polyubiquitination of Gag. Viruses, 6(10), 3738-3765. https://doi.org/10.3390/v6103738