Replication Cycle and Molecular Biology of the West Nile Virus

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Virion Morphology, Attachment and Entry
3. Early Effects of Infection on Cells
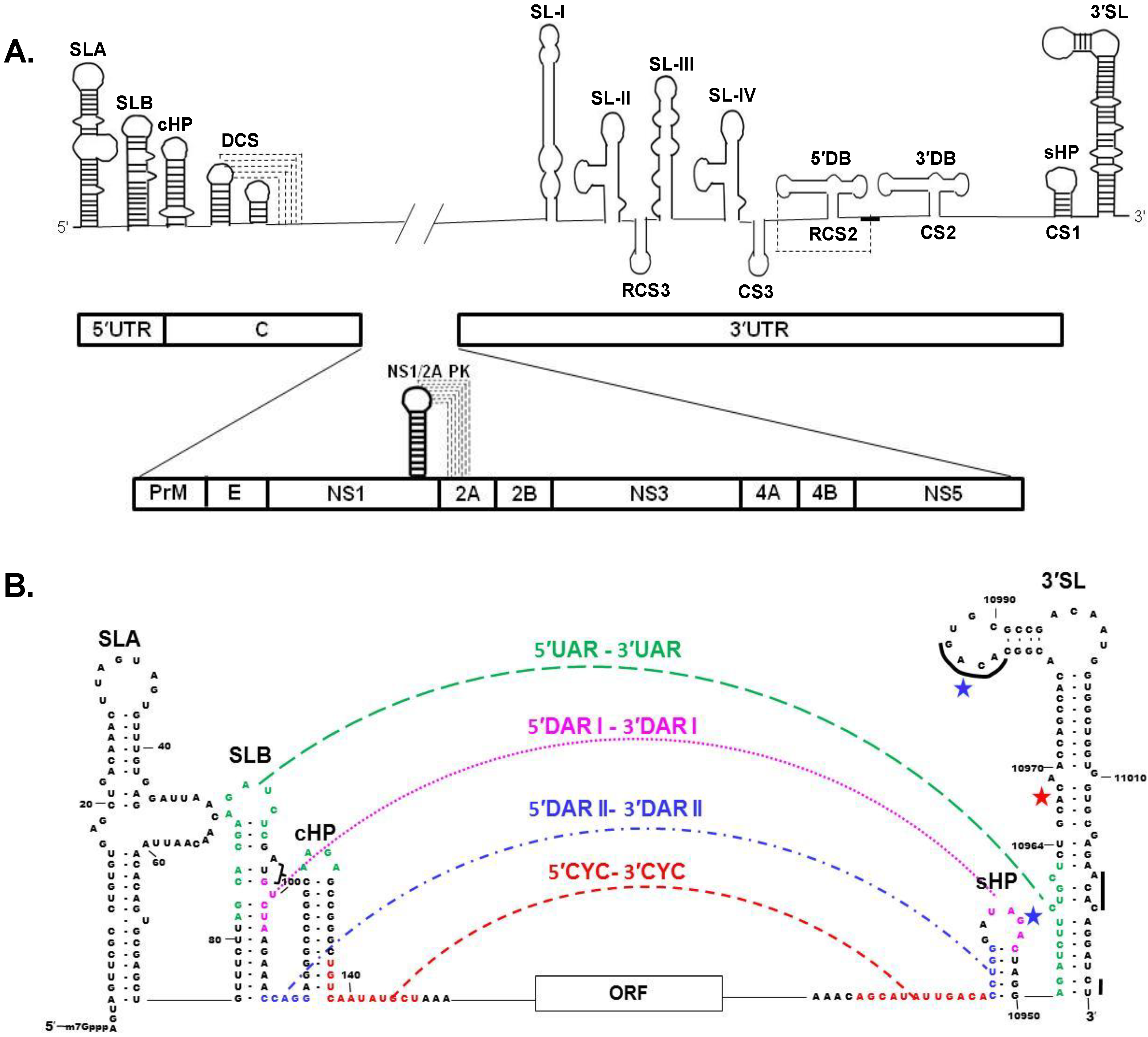
4. Viral Genome RNA
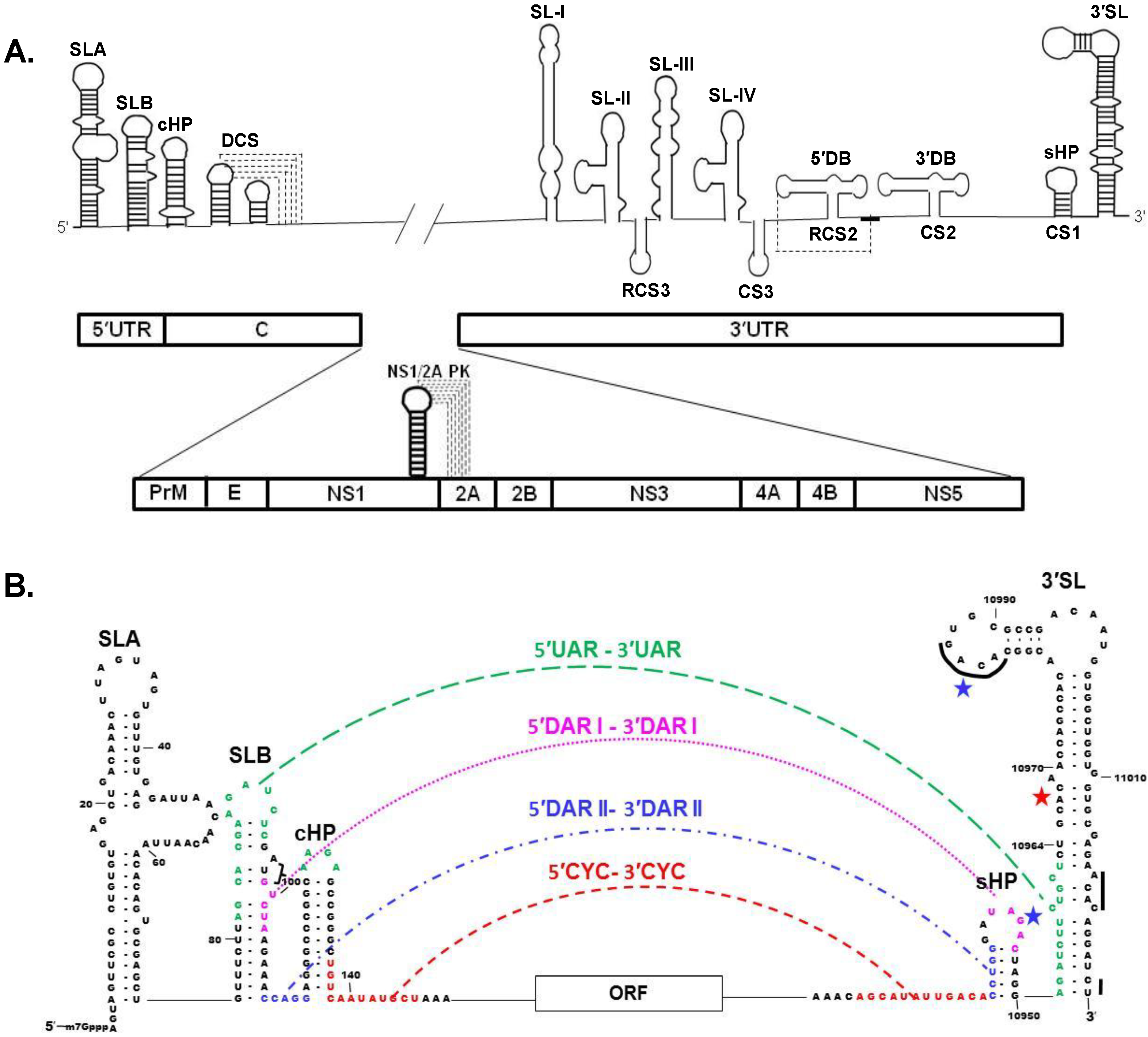
Long Distance 3′-5′ RNA-RNA Interactions
5. Viral Polyprotein
6. Viral Nonstructural Proteins
6.1. Four Nonstructural Membrane Proteins
6.2. Nonstructural Protein 1
6.3. Nonstructural Protein 3
6.4. Nonstructural Protein 5
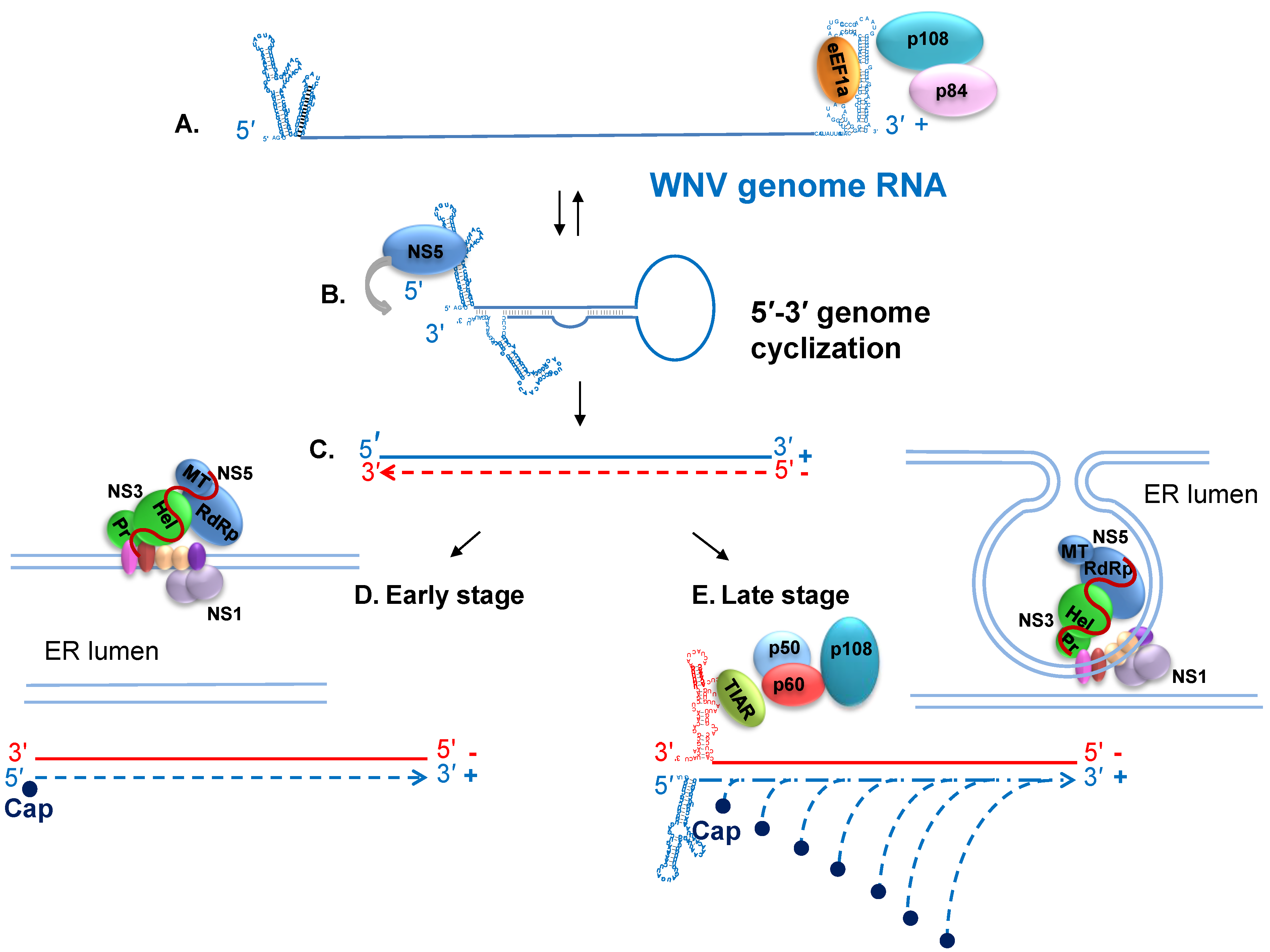
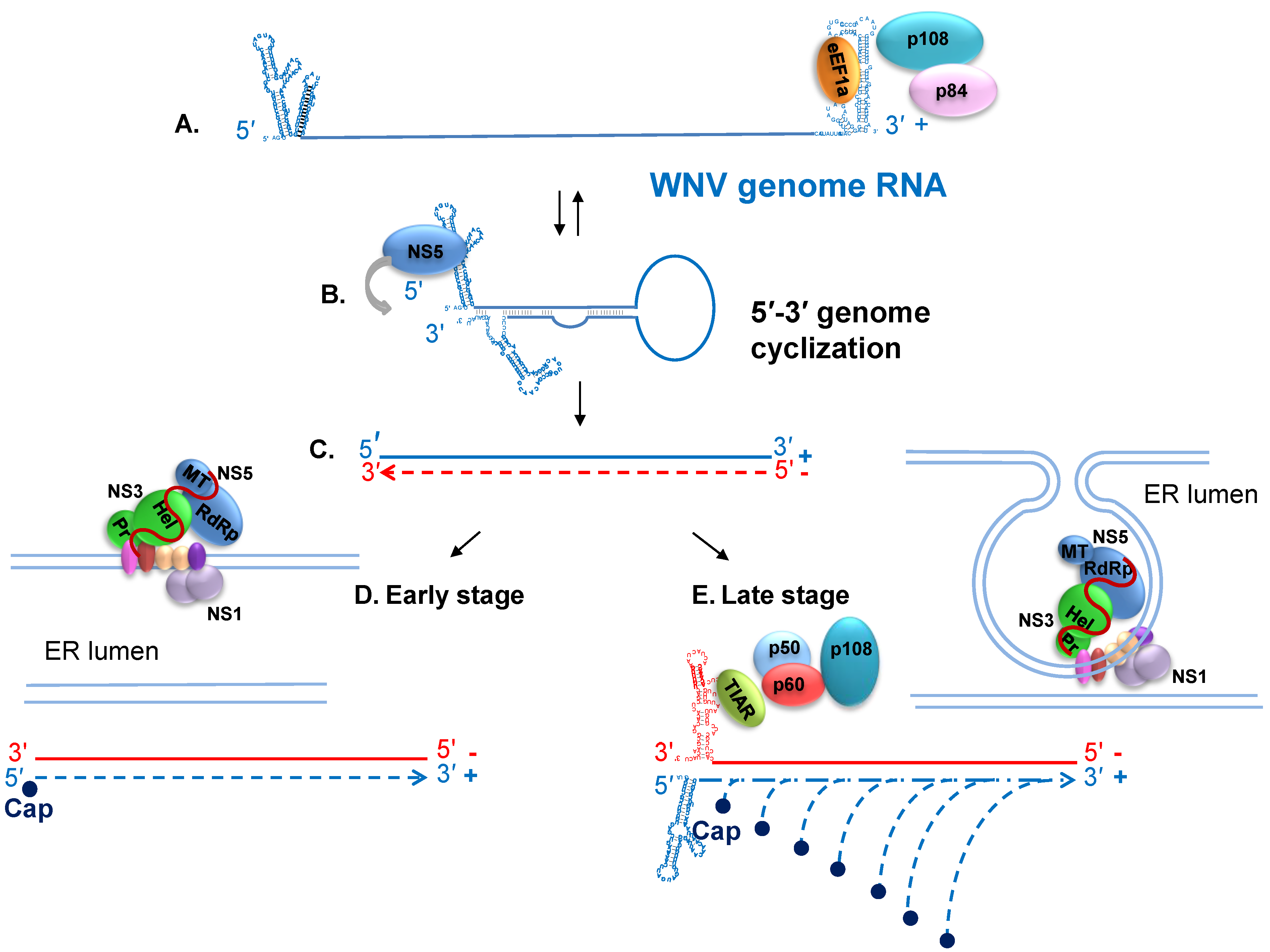
7. Viral RNA Translation and Replication
8. Host Cell Proteins that Interact with the WNV Genomic 3' Terminal SL RNAs and Facilitate RNA Synthesis
9. Nonstructural Protein Interactions and Regulation of Viral RNA Replication
10. Progeny Virus Assembly and Release
11. Conclusions
Acknowledgements
Conflicts of Interest
References and Notes
- Berthet, F.X.; Zeller, H.G.; Drouet, M.T.; Rauzier, J.; Digoutte, J.P.; Deubel, V. Extensive nucleotide changes and deletions within the envelope glycoprotein gene of Euro-African West Nile viruses. J. Gen. Virol. 1997, 78, 2293–2297. [Google Scholar]
- Jia, X.Y.; Briese, T.; Jordan, I.; Rambaut, A.; Chi, H.C.; Mackenzie, J.S.; Hall, R.A.; Scherret, J.; Lipkin, W.I. Genetic analysis of West Nile New York 1999 encephalitis virus. Lancet 1999, 354, 1971–1972. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Roehrig, J.T.; Deubel, V.; Smith, J.; Parker, M.; Steele, K.; Crise, B.; Volpe, K.E.; Crabtree, M.B.; Scherret, J.H.; et al. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science 1999, 286, 2333–2337. [Google Scholar] [CrossRef]
- Beasley, D.W.; Li, L.; Suderman, M.T.; Barrett, A.D. Mouse neuroinvasive phenotype of West Nile virus strains varies depending upon virus genotype. Virology 2002, 296, 17–23. [Google Scholar] [CrossRef]
- Heinz, F.X.; Purcell, M.S.; Gould, E.A.; Howard, C.R.; Houghton, M.; Moormann, R.J.M.; Rice, C.M.; Thiel, H.-J. Family Flaviviridae. In Virus Taxonomy; Regenmortel, C.F., Bishop, D.H.L., Carstens, E.B., Estes, M.K., Eds.; Academic Press: San Diego, CA, USA, 2000; pp. 860–878. [Google Scholar]
- Murray, J.M.; Aaskov, J.G.; Wright, P.J. Processing of the dengue virus type 2 proteins prM and C-prM. J. Gen. Virol. 1993, 74, 175–182. [Google Scholar] [CrossRef]
- Adams, S.C.; Broom, A.K.; Sammels, L.M.; Hartnett, A.C.; Howard, M.J.; Coelen, R.J.; Mackenzie, J.S.; Hall, R.A. Glycosylation and antigenic variation among Kunjin virus isolates. Virology 1995, 206, 49–56. [Google Scholar] [CrossRef]
- Zhang, W.; Chipman, P.R.; Corver, J.; Johnson, P.R.; Zhang, Y.; Mukhopadhyay, S.; Baker, T.S.; Strauss, J.H.; Rossmann, M.G.; Kuhn, R.J. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat. Struct. Biol. 2003, 10, 907–912. [Google Scholar] [CrossRef]
- Kiermayr, S.; Kofler, R.M.; Mandl, C.W.; Messner, P.; Heinz, F.X. Isolation of capsid protein dimers from the tick-borne encephalitis flavivirus and in vitro assembly of capsid-like particles. J. Virol. 2004, 78, 8078–8084. [Google Scholar] [CrossRef]
- Ma, L.; Jones, C.T.; Groesch, T.D.; Kuhn, R.J.; Post, C.B. Solution structure of dengue virus capsid protein reveals another fold. Proc. Natl. Acad. Sci. USA 2004, 101, 3414–3419. [Google Scholar]
- Pierson, T.C.; Diamond, M.S. Flaviviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer/Lippencott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 747–794. [Google Scholar]
- Mukhopadhyay, S.; Kuhn, R.J.; Rossmann, M.G. A structural perspective of the flavivirus life cycle. Nat. Rev. 2005, 3, 13–22. [Google Scholar]
- Lee, E.; Hall, R.A.; Lobigs, M. Common E protein determinants for attenuation of glycosaminoglycan-binding variants of Japanese encephalitis and West Nile viruses. J. Virol. 2004, 78, 8271–8280. [Google Scholar] [CrossRef]
- Davis, C.W.; Nguyen, H.Y.; Hanna, S.L.; Sanchez, M.D.; Doms, R.W.; Pierson, T.C. West Nile virus discriminates between DC-SIGN and DC-SIGNR for cellular attachment and infection. J. Virol. 2006, 80, 1290–1301. [Google Scholar] [CrossRef]
- Rios, M.; Daniel, S.; Chancey, C.; Hewlett, I.K.; Stramer, S.L. West Nile virus adheres to human red blood cells in whole blood. Clin. Infect. Dis. 2007, 45, 181–186. [Google Scholar] [CrossRef]
- Jemielity, S.; Wang, J.J.; Chan, Y.K.; Ahmed, A.A.; Li, W.; Monahan, S.; Bu, X.; Farzan, M.; Freeman, G.J.; Umetsu, D.T.; et al. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS pathog. 2013, 9, e1003232. [Google Scholar] [CrossRef]
- Chu, J.J.; Ng, M.L. Infectious entry of West Nile virus occurs through a clathrin-mediated endocytic pathway. J. Virol. 2004, 78, 10543–10555. [Google Scholar] [CrossRef]
- Bogachek, M.V.; Zaitsev, B.N.; Sekatskii, S.K.; Protopopova, E.V.; Ternovoi, V.A.; Ivanova, A.V.; Kachko, A.V.; Ivanisenko, V.A.; Dietler, G.; Loktev, V.B. Characterization of glycoprotein E C-end of West Nile virus and evaluation of its interaction force with alphaVbeta3 integrin as putative cellular receptor. Biochemistry 2010, 75, 472–480. [Google Scholar]
- Lee, J.W.; Chu, J.J.; Ng, M.L. Quantifying the specific binding between West Nile virus envelope domain III protein and the cellular receptor αvβ3 integrin. J. Biol. Chem. 2006, 281, 1352–1360. [Google Scholar]
- Schmidt, K.; Keller, M.; Bader, B.L.; Korytar, T.; Finke, S.; Ziegler, U.; Groschup, M.H. Integrins modulate the infection efficiency of West Nile virus into cells. J. Gen. Virol. 2013, 94, 1723–1733. [Google Scholar] [CrossRef]
- Wan, S.W.; Lin, C.F.; Lu, Y.T.; Lei, H.Y.; Anderson, R.; Lin, Y.S. Endothelial cell surface expression of protein disulfide isomerase activates β1 and β3 integrins and facilitates dengue virus infection. J. Cell. Biochem. 2012, 113, 1681–1691. [Google Scholar]
- Medigeshi, G.R.; Hirsch, A.J.; Streblow, D.N.; Nikolich-Zugich, J.; Nelson, J.A. West Nile virus entry requires cholesterol-rich membrane microdomains and is independent of αvβ3 integrin. J. Virol. 2008, 82, 5212–5219. [Google Scholar] [CrossRef]
- Barrows, N.J.; Le Sommer, C.; Garcia-Blanco, M.A.; Pearson, J.L. Factors affecting reproducibility between genome-scale siRNA-based screens. J. Biomol. Screen. 2010, 15, 735–747. [Google Scholar] [CrossRef]
- Krishnan, M.N.; Ng, A.; Sukumaran, B.; Gilfoy, F.D.; Uchil, P.D.; Sultana, H.; Brass, A.L.; Adametz, R.; Tsui, M.; Qian, F.; et al. RNA interference screen for human genes associated with West Nile virus infection. Nature 2008, 455, 242–245. [Google Scholar] [CrossRef]
- Sessions, O.M.; Barrows, N.J.; Souza-Neto, J.A.; Robinson, T.J.; Hershey, C.L.; Rodgers, M.A.; Ramirez, J.L.; Dimopoulos, G.; Yang, P.L.; Pearson, J.L.; et al. Discovery of insect and human dengue virus host factors. Nature 2009, 458, 1047–1050. [Google Scholar] [CrossRef]
- Le Sommer, C.; Barrows, N.J.; Bradrick, S.S.; Pearson, J.L.; Garcia-Blanco, M.A. G protein-coupled receptor kinase 2 promotes flaviviridae entry and replication. PLoS Negl. Trop. Dis. 2012, 6, e1820. [Google Scholar] [CrossRef]
- Fernandez-Garcia, M.D.; Meertens, L.; Bonazzi, M.; Cossart, P.; Arenzana-Seisdedos, F.; Amara, A. Appraising the roles of CBLL1 and the ubiquitin/proteasome system for flavivirus entry and replication. J. Virol. 2011, 85, 2980–2989. [Google Scholar] [CrossRef]
- Cheng, G.; Cox, J.; Wang, P.; Krishnan, M.N.; Dai, J.; Qian, F.; Anderson, J.F.; Fikrig, E. A C-type lectin collaborates with a CD45 phosphatase homolog to facilitate West Nile virus infection of mosquitoes. Cell 2010, 142, 714–725. [Google Scholar] [CrossRef]
- Chu, J.J.; Leong, P.W.; Ng, M.L. Analysis of the endocytic pathway mediating the infectious entry of mosquito-borne flavivirus West Nile into Aedes albopictus mosquito (C6/36) cells. Virology 2006, 349, 463–475. [Google Scholar] [CrossRef]
- Krishnan, M.N.; Sukumaran, B.; Pal, U.; Agaisse, H.; Murray, J.L.; Hodge, T.W.; Fikrig, E. Rab 5 is required for the cellular entry of dengue and West Nile viruses. J. Virol. 2007, 81, 4881–4885. [Google Scholar] [CrossRef]
- Van der Schaar, H.M.; Rust, M.J.; Chen, C.; van der Ende-Metselaar, H.; Wilschut, J.; Zhuang, X.; Smit, J.M. Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog. 2008, 4, e1000244. [Google Scholar] [CrossRef]
- Allison, S.L.; Schalich, J.; Stiasny, K.; Mandl, C.W.; Kunz, C.; Heinz, F.X. Oligomeric rearrangementof tick-borne encephalitis virus envelope proteins induced by an acidic pH. J. Virol. 1995, 69, 695–700. [Google Scholar]
- Heinz, F.X.; Allison, S.L. Structures and mechanisms in flavivirus fusion. Adv. Virus Res. 2000, 55, 231–269. [Google Scholar]
- Ivanyi-Nagy, R.; Lavergne, J.P.; Gabus, C.; Ficheux, D.; Darlix, J.L. RNA chaperoning and intrinsic disorder in the core proteins of Flaviviridae. Nucleic Acids Res. 2007, 36, 712–725. [Google Scholar] [CrossRef]
- Pong, W.L.; Huang, Z.S.; Teoh, P.G.; Wang, C.C.; Wu, H.N. RNA binding property and RNA chaperone activity of dengue virus core protein and other viral RNA-interacting proteins. FEBS Lett. 2011, 585, 2575–2581. [Google Scholar] [CrossRef]
- Scherbik, S.V.; Brinton, M.A. Virus-induced Ca2+ influx extends survival of west nile virus-infected cells. J. Virol. 2010, 84, 8721–8731. [Google Scholar] [CrossRef]
- Urbanowski, M.D.; Hobman, T.C. The West Nile virus capsid protein blocks apoptosis through a phosphatidylinositol 3-kinase-dependent mechanism. J. Virol. 2013, 87, 872–881. [Google Scholar] [CrossRef]
- Beasley, D.W.; Li, L.; Suderman, M.T.; Barrett, A.D. West Nile virus strains differ in mouse neurovirulence and binding to mouse or human brain membrane receptor preparations. Ann. NY Acad. Sci. 2001, 951, 332–335. [Google Scholar]
- Rice, C.M.; Lenches, E.M.; Eddy, S.R.; Shin, S.J.; Sheets, R.L.; Strauss, J.H. Nucleotide sequence of yellow fever virus: Implications for flavivirus gene expression and evolution. Science 1985, 229, 726–733. [Google Scholar]
- Brinton, M.A.; Fernandez, A.V.; Dispoto, J.H. The 3'-nucleotides of flavivirus genomic RNA form a conserved secondary structure. Virology 1986, 153, 113–121. [Google Scholar] [CrossRef]
- Wengler, G.; Wengler, G. The carboxy-terminal part of the NS 3 protein of the West Nile flavivirus can be isolated as a soluble protein after proteolytic cleavage and represents an RNA-stimulated NTPase. Virology 1991, 184, 707–715. [Google Scholar] [CrossRef]
- Brinton, M.A.; Dispoto, J.H. Sequence and secondary structure analysis of the 5'-terminal region of flavivirus genome RNA. Virology 1988, 162, 290–299. [Google Scholar] [CrossRef]
- Rauscher, S.; Flamm, C.; Mandl, C.W.; Heinz, F.X.; Stadler, P.F. Secondary structure of the 3'-noncoding region of flavivirus genomes: Comparative analysis of base pairing probabilities. RNA 1997, 3, 779–791. [Google Scholar]
- Lai, C.J.; Men, R.; Pethel, M.; Bray, M. Infectious RNA transcribed from stably cloned full-length cDNA: Construction of growth-restricted dengue mutants. In Vaccines 92; Brown, F., Chanock, R.M., Ginsberg, H.S., Lerner, R.A., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1992; pp. 265–270. [Google Scholar]
- Cahour, A.; Pletnev, A.; Vazielle-Falcoz, M.; Rosen, L.; Lai, C.J. Growth-restricted dengue virus mutants containing deletions in the 5' noncoding region of the RNA genome. Virology 1995, 207, 68–76. [Google Scholar] [CrossRef]
- Yu, L.; Markoff, L. The topology of bulges in the long stem of the flavivirus 3' stem-loop is a major determinant of RNA replication competence. J. Virol. 2005, 79, 2309–2324. [Google Scholar] [CrossRef]
- Davis, W.G.; Basu, M.; Elrod, E.J.; Germann, M.W.; Brinton, M.A. Identification of cis-acting nucleotides and a structural feature in West Nile virus 3'-terminus RNA that facilitate viral minus strand RNA synthesis. J. Virol. 2013, 87, 7622–7636. [Google Scholar] [CrossRef]
- Sztuba-Solinska, J.; Teramoto, T.; Rausch, J.W.; Shapiro, B.A.; Padmanabhan, R.; Le Grice, S.F.J. Structural complexity of Dengue virus untranslated regions: cis-Acting RNA motifs and pseudoknot interactions modulating functionality of the viral genome. Nucleic Acids Res. 2013, 41, 5075–5089. [Google Scholar] [CrossRef]
- Elghonemy, S.; Davis, W.G.; Brinton, M.A. The majority of the nucleotides in the top loop of the genomic 3' terminal stem loop structure are cis-acting in a West Nile virus infectious clone. Virology 2005, 331, 238–246. [Google Scholar] [CrossRef]
- Tilgner, M.; Deas, T.S.; Shi, P.Y. The flavivirus-conserved penta-nucleotide in the 3' stem-loop of the West Nile virus genome requires a specific sequence and structure for RNA synthesis, but not for viral translation. Virology 2005, 331, 375–386. [Google Scholar] [CrossRef]
- Villordo, S.M.; Gamarnik, A.V. Differential RNA sequence requirement for dengue virus replication in mosquito and mammalian cells. J. Virol. 2013, 87, 9365–9372. [Google Scholar] [CrossRef]
- Shi, P.Y.; Brinton, M.A.; Veal, J.M.; Zhong, Y.Y.; Wilson, W.D. Evidence for the existence of a pseudoknot structure at the 3' terminus of the flavivirus genomic RNA. Biochemistry 1996, 35, 4222–4230. [Google Scholar] [CrossRef]
- Gritsun, T.S.; Gould, E.A. Direct repeats in the flavivirus 3' untranslated region; a strategy for survival in the environment? Virology 2007, 358, 258–265. [Google Scholar] [CrossRef]
- Olsthoorn, R.C.; Bol, J.F. Sequence comparison and secondary structure analysis of the 3' noncoding region of flavivirus genomes reveals multiple pseudoknots. RNA 2001, 7, 1370–1377. [Google Scholar]
- Manzano, M.; Reichert, E.D.; Polo, S.; Falgout, B.; Kasprzak, W.; Shapiro, B.A.; Padmanabhan, R. Identification of cis-acting elements in the 3'-untranslated region of the dengue virus type 2 RNA that modulate translation and replication. J. Biol. Chem. 2011, 286, 22521–22534. [Google Scholar]
- Funk, A.; Truong, K.; Nagasaki, T.; Torres, S.; Floden, N.; Balmori Melian, E.; Edmonds, J.; Dong, H.; Shi, P.Y.; Khromykh, A.A. RNA structures required for production of subgenomic flavivirus RNA. J. Virol. 2010, 84, 11407–11417. [Google Scholar] [CrossRef]
- Clyde, K.; Harris, E. RNA secondary structure in the coding region of dengue virus type 2 directs translation start codon selection and is required for viral replication. J. Virol. 2006, 80, 2170–2182. [Google Scholar] [CrossRef]
- Shi, P.Y.; Li, W.; Brinton, M.A. Cell proteins bind specifically to West Nile virus minus-strand 3' stem-loop RNA. J. Virol. 1996, 70, 6278–6287. [Google Scholar]
- Ray, D.; Shah, A.; Tilgner, M.; Guo, Y.; Zhao, Y.; Dong, H.; Deas, T.S.; Zhou, Y.; Li, H.; Shi, P.Y. West Nile virus 5'-cap structure is formed by sequential guanine N-7 and ribose 2'-O methylations by nonstructural protein 5. J. Virol. 2006, 80, 8362–8370. [Google Scholar] [CrossRef]
- Zhou, Y.; Ray, D.; Zhao, Y.; Dong, H.; Ren, S.; Li, Z.; Guo, Y.; Bernard, K.A.; Shi, P.Y.; Li, H. Structure and function of flavivirus NS5 methyltransferase. J. Virol. 2007, 81, 3891–3903. [Google Scholar]
- Issur, M.; Geiss, B.J.; Bougie, I.; Picard-Jean, F.; Despins, S.; Mayette, J.; Hobdey, S.E.; Bisaillon, M. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two-step reaction to form the RNA cap structure. RNA 2009, 15, 2340–2350. [Google Scholar] [CrossRef]
- Egloff, M.P.; Benarroch, D.; Selisko, B.; Romette, J.L.; Canard, B. An RNA cap (nucleoside-2'-O-)-methyltransferase in the flavivirus RNA polymerase NS5: Crystal structure and functional characterization. EMBO J. 2002, 21, 2757–2768. [Google Scholar] [CrossRef]
- Egloff, M.P.; Decroly, E.; Malet, H.; Selisko, B.; Benarroch, D.; Ferron, F.; Canard, B. Structural and functional analysis of methylation and 5'-RNA sequence requirements of short capped RNAs by the methyltransferase domain of dengue virus NS5. J. Mol. Biol. 2007, 372, 723–736. [Google Scholar] [CrossRef]
- Dong, H.; Ray, D.; Ren, S.; Zhang, B.; Puig-Basagoiti, F.; Takagi, Y.; Ho, C.K.; Li, H.; Shi, P.Y. Distinct RNA elements confer specificity to flavivirus RNA cap methylation events. J. Virol. 2007, 81, 4412–4421. [Google Scholar] [CrossRef]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2'-O-Methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef]
- Hahn, C.S.; Hahn, Y.S.; Rice, C.M.; Lee, E.; Dalgarno, L.; Strauss, E.G.; Strauss, J.H. Conserved elements in the 3' untranslated region of flavivirus RNAs and potential cyclization sequences. J. Mol. Biol. 1987, 198, 33–41. [Google Scholar] [CrossRef]
- Alvarez, D.E.; de Lella Ezcurra, A.L.; Fucito, S.; Gamarnik, A.V. Role of RNA structures present at the 3'UTR of dengue virus on translation, RNA synthesis, and viral replication. Virology 2005, 339, 200–212. [Google Scholar] [CrossRef]
- Corver, J.; Lenches, E.; Smith, K.; Robison, R.A.; Sando, T.; Strauss, E.G.; Strauss, J.H. Fine mapping of a cis-acting sequence element in yellow fever virus RNA that is required for RNA replication and cyclization. J. Virol. 2003, 77, 2265–2270. [Google Scholar] [CrossRef]
- Khromykh, A.A.; Meka, H.; Guyatt, K.J.; Westaway, E.G. Essential role of cyclization sequences in flavivirus RNA replication. J. Virol. 2001, 75, 6719–6728. [Google Scholar] [CrossRef]
- Lo, M.K.; Tilgner, M.; Bernard, K.A.; Shi, P.Y. Functional analysis of mosquito-borne flavivirus conserved sequence elements within 3' untranslated region of West Nile virus by use of a reporting replicon that differentiates between viral translation and RNA replication. J. Virol. 2003, 77, 10004–10014. [Google Scholar] [CrossRef]
- Basu, M.; Brinton, M.A. West Nile virus (WNV) genome RNAs with up to three adjacent mutations that disrupt long distance 5'–3' cyclization sequence basepairs are viable. Virology 2011, 412, 220–232. [Google Scholar] [CrossRef]
- Alvarez, D.E.; Lodeiro, M.F.; Luduena, S.J.; Pietrasanta, L.I.; Gamarnik, A.V. Long-range RNA-RNA interactions circularize the dengue virus genome. J. Virol. 2005, 79, 6631–6643. [Google Scholar] [CrossRef]
- Zhang, B.; Dong, H.; Stein, D.A.; Iversen, P.L.; Shi, P.Y. West Nile virus genome cyclization and RNA replication require two pairs of long-distance RNA interactions. Virology 2008, 373, 1–13. [Google Scholar] [CrossRef]
- Alvarez, D.E.; Filomatori, C.V.; Gamarnik, A.V. Functional analysis of dengue virus cyclization sequences located at the 5' and 3' UTRs. Virology 2008, 375, 223–235. [Google Scholar] [CrossRef]
- Dong, H.; Zhang, B.; Shi, P.Y. Terminal structures of West Nile virus genomic RNA and their interactions with viral NS5 protein. Virology 2008, 381, 123–135. [Google Scholar] [CrossRef]
- Friebe, P.; Harris, E. Interplay of RNA elements in the dengue virus 5' and 3' ends required for viral RNA replication. J. Virol. 2010, 84, 6103–6018. [Google Scholar] [CrossRef]
- Zhang, B.; Dong, H.; Ye, H.; Tilgner, M.; Shi, P.Y. Genetic analysis of West Nile virus containing a complete 3'CSI RNA deletion. Virology 2010, 408, 138–145. [Google Scholar] [CrossRef]
- Polacek, C.; Foley, J.E.; Harris, E. Conformational changes in the solution structure of the dengue virus 5' end in the presence and absence of the 3' untranslated region. J. Virol. 2009, 83, 1161–1166. [Google Scholar] [CrossRef]
- Friebe, P.; Shi, P.Y.; Harris, E. The 5' and 3' downstream AUG region elements are required for mosquito-borne flavivirus RNA replication. J. Virol. 2011, 85, 1900–1905. [Google Scholar] [CrossRef]
- Friebe, P.; Pena, J.; Pohl, M.O.; Harris, E. Composition of the sequence downstream of the dengue virus 5' cyclization sequence (dCS) affects viral RNA replication. Virology 2012, 422, 346–356. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Li, X.F.; Jiang, T.; Deng, Y.Q.; Zhao, H.; Wang, H.J.; Ye, Q.; Zhu, S.Y.; Qiu, Y.; Zhou, X.; et al. Novel cis-acting element within the capsid-coding region enhances flavivirus viral-RNA replication by regulating genome cyclization. J. Virol. 2013, 87, 6804–6818. [Google Scholar] [CrossRef]
- Vashist, S.; Bhullar, D.; Vrati, S. La protein can simultaneously bind to both 3'- and 5'-noncoding regions of Japanese encephalitis virus genome. DNA Cell Biol. 2011, 30, 339–346. [Google Scholar] [CrossRef]
- Ivanyi-Nagy, R.; Darlix, J.L. Core protein-mediated 5'–3' annealing of the West Nile virus genomic RNA in vitro. Virus Res. 2012, 167, 226–235. [Google Scholar] [CrossRef]
- Gebhard, L.G.; Kaufman, S.B.; Gamarnik, A.V. Novel ATP-independent RNA annealing activity of the dengue virus NS3 helicase. PLoS One 2012, 7, e36244. [Google Scholar]
- Villordo, S.M.; Alvarez, D.E.; Gamarnik, A.V. A balance between circular and linear forms of the dengue virus genome is crucial for viral replication. RNA 2010, 16, 2325–2335. [Google Scholar] [CrossRef]
- Clyde, K.; Barrera, J.; Harris, E. The capsid-coding region hairpin element (cHP) is a critical determinant of dengue virus and West Nile virus RNA synthesis. Virology 2008, 379, 314–323. [Google Scholar] [CrossRef]
- Iglesias, N.G.; Gamarnik, A.V. Dynamic RNA structures in the dengue virus genome. RNA Biol. 2011, 8, 249–257. [Google Scholar] [CrossRef]
- Lodeiro, M.F.; Filomatori, C.V.; Gamarnik, A.V. Structural and functional studies of the promoter element for dengue virus RNA replication. J. Virol. 2009, 83, 993–1008. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Thiel, H.J.; Rice, C.M. Flaviviridae: The viruses and their replication. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2007; pp. 1101–1152. [Google Scholar]
- Nowak, T.; Farber, P.M.; Wengler, G.; Wengler, G. Analyses of the terminal sequences of West Nile virus structural proteins and of the in vitro translation of these proteins allow the proposal of a complete scheme of the proteolytic cleavages involved in their synthesis. Virology 1989, 169, 365–376. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Murray, C.L.; Thiel, H.-J.; Rice, C.M. Flaviviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer/Lippencott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 712–746. [Google Scholar]
- Mackenzie, J.M.; Khromykh, A.A.; Jones, M.K.; Westaway, E.G. Subcellular localization and some biochemical properties of the flavivirus Kunjin nonstructural proteins NS2A and NS4A. Virology 1998, 245, 203–215. [Google Scholar] [CrossRef]
- Westaway, E.G.; Mackenzie, J.M.; Khromykh, A.A. Replication and gene function in Kunjin virus. Curr. Top. Microbiol. Immunol. 2002, 267, 323–351. [Google Scholar]
- Miller, S.; Sparacio, S.; Bartenschlager, R. Subcellular localization and membrane topology of the Dengue virus type 2 Non-structural protein 4B. J. Biol. Chem. 2006, 281, 8854–8863. [Google Scholar] [CrossRef]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Buhler, S.; Bartenschlager, R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar] [CrossRef]
- Khromykh, A.A.; Sedlak, P.L.; Westaway, E.G. cis- And trans-acting elements in flavivirus RNA replication. J. Virol. 2000, 74, 3253–3263. [Google Scholar] [CrossRef]
- Xie, X.; Wang, Q.Y.; Xu, H.Y.; Qing, M.; Kramer, L.; Yuan, Z.; Shi, P.Y. Inhibition of dengue virus by targeting viral NS4B protein. J. Virol. 2011, 85, 11183–11195. [Google Scholar] [CrossRef]
- Kummerer, B.M.; Rice, C.M. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J. Virol. 2002, 76, 4773–4784. [Google Scholar] [CrossRef]
- Liu, W.J.; Chen, H.B.; Khromykh, A.A. Molecular and functional analyses of Kunjin virus infectious cDNA clones demonstrate the essential roles for NS2A in virus assembly and for a nonconservative residue in NS3 in RNA replication. J. Virol. 2003, 77, 7804–7813. [Google Scholar] [CrossRef]
- Melian, E.B.; Edmonds, J.H.; Nagasaki, T.K.; Hinzman, E.; Floden, N.; Khromykh, A.A. West Nile virus NS2A protein facilitates virus-induced apoptosis independently of interferon response. J. Gen. Virol. 2013, 94, 308–313. [Google Scholar] [CrossRef]
- Xie, X.; Gayen, S.; Kang, C.; Yuan, Z.; Shi, P.Y. Membrane topology and function of dengue virus NS2A protein. J. Virol. 2013, 87, 4609–4622. [Google Scholar] [CrossRef]
- Erbel, P.; Schiering, N.; D’Arcy, A.; Renatus, M.; Kroemer, M.; Lim, S.P.; Yin, Z.; Keller, T.H.; Vasudevan, S.G.; Hommel, U. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat. Struct. Mol. Biol. 2006, 13, 372–373. [Google Scholar] [CrossRef]
- Roosendaal, J.; Westaway, E.G.; Khromykh, A.; Mackenzie, J.M. Regulated cleavages at the West Nile virus NS4A-2K-NS4B junctions play a major role in rearranging cytoplasmic membranes and Golgi trafficking of the NS4A protein. J. Virol. 2006, 80, 4623–4632. [Google Scholar] [CrossRef]
- Lin, M.H.; Hsu, H.J.; Bartenschlager, R.; Fischer, W.B. Membrane undulation induced by NS4A of Dengue virus: A molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2013. [Google Scholar] [CrossRef]
- Stern, O.; Hung, Y.F.; Valdau, O.; Yaffe, Y.; Harris, E.; Hoffmann, S.; Willbold, D.; Sklan, E.H. An N-terminal amphipathic helix in dengue virus nonstructural protein 4A mediates oligomerization and is essential for replication. J. Virol. 2013, 87, 4080–4085. [Google Scholar] [CrossRef]
- Westaway, E.G.; Khromykh, A.A.; Kenney, M.T.; Mackenzie, J.M.; Jones, M.K. Proteins C and NS4B of the flavivirus Kunjin translocate independently into the nucleus. Virology 1997, 234, 31–41. [Google Scholar] [CrossRef]
- Liu, W.J.; Wang, X.J.; Mokhonov, V.V.; Shi, P.Y.; Randall, R.; Khromykh, A.A. Inhibition of interferon signaling by the New York 99 strain and Kunjin subtype of West Nile virus involves blockage of STAT1 and STAT2 activation by nonstructural proteins. J. Virol. 2005, 79, 1934–1942. [Google Scholar] [CrossRef]
- Liu, W.J.; Chen, H.B.; Wang, X.J.; Huang, H.; Khromykh, A.A. Analysis of adaptive mutations in Kunjin virus replicon RNA reveals a novel role for the flavivirus nonstructural protein NS2A in inhibition of beta interferon promoter-driven transcription. J. Virol. 2004, 78, 12225–12235. [Google Scholar] [CrossRef]
- Liu, W.J.; Wang, X.J.; Clark, D.C.; Lobigs, M.; Hall, R.A.; Khromykh, A.A. A single amino acid substitution in the West Nile virus nonstructural protein NS2A disables its ability to inhibit alpha/beta interferon induction and attenuates virus virulence in mice. J. Virol. 2006, 80, 2396–2404. [Google Scholar] [CrossRef]
- Wicker, J.A.; Whiteman, M.C.; Beasley, D.W.; Davis, C.T.; Zhang, S.; Schneider, B.S.; Higgs, S.; Kinney, R.M.; Barrett, A.D. A single amino acid substitution in the central portion of the West Nile virus NS4B protein confers a highly attenuated phenotype in mice. Virology 2006, 349, 245–253. [Google Scholar] [CrossRef]
- Wicker, J.A.; Whiteman, M.C.; Beasley, D.W.C.; Davis, C.T.; McGee, C.E.; Lee, J.C.; Higgs, S.; Kinney, R.M.H.; Huang, C.Y.-H.; Barrett, A.D.T. Mutational analysis of the West Nile virus NS4B protein. Virology 2012, 426, 22–33. [Google Scholar] [CrossRef]
- Faggioni, G.; Ciammaruconi, A.; de Santis, R.; Pomponi, A.; Scicluna, M.T.; Barbaro, K.; Masuelli, L.; Autorino, G.; Bei, R.; Lista, F. Evidence of a humoral response to a novel protein WARF4 embedded in the West Nile virus NS4B gene encoded by an alternative open reading frame. Int. J. Mol. Med. 2009, 23, 509–512. [Google Scholar]
- Faggioni, G.; Pomponi, A.; de Santis, R.; Masuelli, L.; Ciammaruconi, A.; Monaco, F.; di Gennaro, A.; Marzocchella, L.; Sambri, V.; Lelli, R.; et al. West Nile alternative open reading frame (N-NS4B/WARF4) is produced in infected West Nile Virus (WNV) cells and induces humoral response in WNV infected individuals. Virol. J. 2012, 9. [Google Scholar] [CrossRef]
- Blitvich, B.J.; Scanlon, D.; Shiell, B.J.; Mackenzie, J.S.; Pham, K.; Hall, R.A. Determination of the intramolecular disulfide bond arrangement and biochemical identification of the glycosylation sites of the nonstructural protein NS1 of Murray Valley encephalitis virus. J. Gen. Virol. 2001, 82, 2251–2256. [Google Scholar]
- Wallis, T.P.; Huang, C.Y.; Nimkar, S.B.; Young, P.R.; Gorman, J.J. Determination of the disulfide bond arrangement of dengue virus NS1 protein. J. Biol. Chem. 2004, 279, 20729–20741. [Google Scholar] [CrossRef]
- Whiteman, M.C.; Wicker, J.A.; Kinney, R.M.; Huang, C.Y.; Solomon, T.; Barrett, A.D. Multiple amino acid changes at the first glycosylation motif in NS1 protein of West Nile virus are necessary for complete attenuation for mouse neuroinvasiveness. Vaccine 2011, 29, 9702–9710. [Google Scholar] [CrossRef]
- Falgout, B.; Chanock, R.; Lai, C.J. Proper processing of dengue virus nonstructural glycoprotein NS1 requires the N-terminal hydrophobic signal sequence and the downstream nonstructural protein NS2a. J. Virol. 1989, 63, 1852–1860. [Google Scholar]
- Winkler, G.; Maxwell, S.E.; Ruemmler, C.; Stollar, V. Newly synthesized dengue-2 virus nonstructural protein NS1 is a soluble protein but becomes partially hydrophobic and membrane-associated after dimerization. Virology 1989, 171, 302–305. [Google Scholar] [CrossRef]
- Winkler, G.; Randolph, V.B.; Cleaves, G.R.; Ryan, T.E.; Stollar, V. Evidence that the mature form of the flavivirus nonstructural protein NS1 is a dimer. Virology 1988, 162, 187–196. [Google Scholar] [CrossRef]
- Smith, G.W.; Wright, P.J. Synthesis of proteins and glycoproteins in dengue type 2 virus-infected vero and Aedes albopictus cells. J. Gen. Virol. 1985, 66, 559–571. [Google Scholar] [CrossRef]
- Flamand, M.; Megret, F.; Mathieu, M.; Lepault, J.; Rey, F.A.; Deubel, V. Dengue virus type 1 nonstructural glycoprotein NS1 is secreted from mammalian cells as a soluble hexamer in a glycosylation-dependent fashion. J. Virol. 1999, 73, 6104–6110. [Google Scholar]
- Lee, J.M.; Crooks, A.J.; Stephenson, J.R. The synthesis and maturation of a non-structural extracellular antigen from tick-borne encephalitis virus and its relationship to the intracellular NS1 protein. J. Gen. Virol. 1989, 70, 335–343. [Google Scholar] [CrossRef]
- Crooks, A.J.; Lee, J.M.; Easterbrook, L.M.; Timofeev, A.V.; Stephenson, J.R. The NS1 protein of tick-borne encephalitis virus forms multimeric species upon secretion from the host cell. J. Gen. Virol. 1994, 75, 3453–3460. [Google Scholar] [CrossRef]
- Mason, P.W. Maturation of Japanese encephalitis virus glycoproteins produced by infected mammalian and mosquito cells. Virology 1989, 169, 354–364. [Google Scholar] [CrossRef]
- Blitvich, B.J.; Scanlon, D.; Shiell, B.J.; Mackenzie, J.S.; Hall, R.A. Identification and analysis of truncated and elongated species of the flavivirus NS1 protein. Virus Res. 1999, 60, 67–79. [Google Scholar] [CrossRef]
- Mackenzie, J.M.; Jones, M.K.; Young, P.R. Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology 1996, 220, 232–240. [Google Scholar] [CrossRef]
- Westaway, E.G.; Mackenzie, J.M.; Kenney, M.T.; Jones, M.K.; Khromykh, A.A. Ultrastructure of Kunjin virus-infected cells: Colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J. Virol. 1997, 71, 6650–6661. [Google Scholar]
- Muylaert, I.R.; Chambers, T.J.; Galler, R.; Rice, C.M. Mutagenesis of the N-linked glycosylation sites of the yellow fever virus NS1 protein: Effects on virus replication and mouse neurovirulence. Virology 1996, 222, 159–168. [Google Scholar] [CrossRef]
- Muylaert, I.R.; Galler, R.; Rice, C.M. Genetic analysis of the yellow fever virus NS1 protein: Identification of a temperature-sensitive mutation which blocks RNA accumulation. J. Virol. 1997, 71, 291–298. [Google Scholar]
- Macdonald, J.; Tonry, J.; Hall, R.A.; Williams, B.; Palacios, G.; Ashok, M.S.; Jabado, O.; Clark, D.; Tesh, R.B.; Briese, T.; et al. NS1 protein secretion during the acute phase of West Nile virus infection. J. Virol. 2005, 79, 13924–13933. [Google Scholar] [CrossRef]
- Avirutnan, P.; Zhang, L.; Punyadee, N.; Manuyakorn, A.; Puttikhunt, C.; Kasinrerk, W.; Malasit, P.; Atkinson, J.P.; Diamond, M.S. Secreted NS1 of dengue virus attaches to the surface of cells via interactions with heparan sulfate and chondroitin sulfate E. PLoS Pathog. 2007, 3, e183. [Google Scholar] [CrossRef]
- Chung, K.M.; Nybakken, G.E.; Thompson, B.S.; Engle, M.J.; Marri, A.; Fremont, D.H.; Diamond, M.S. Antibodies against West Nile Virus nonstructural protein NS1 prevent lethal infection through Fc gamma receptor-dependent and -independent mechanisms. J. Virol. 2006, 80, 1340–1351. [Google Scholar] [CrossRef]
- Wilson, J.R.; de Sessions, P.F.; Leon, M.A.; Scholle, F. West Nile virus nonstructural protein 1 inhibits TLR3 signal transduction. J. Virol. 2008, 82, 8262–8271. [Google Scholar] [CrossRef]
- Baronti, C.; Sire, J.; de Lamballerie, X.; Querat, G. Nonstructural NS1 proteins of several mosquito-borne Flavivirus do not inhibit TLR3 signaling. Virology 2010, 404, 319–330. [Google Scholar] [CrossRef]
- Chung, K.M.; Liszewski, M.K.; Nybakken, G.; Davis, A.E.; Townsend, R.R.; Fremont, D.H.; Atkinson, J.P.; Diamond, M.S. West Nile virus nonstructural protein NS1 inhibits complement activation by binding the regulatory protein factor H. Proc. Natl. Acad. Sci. USA 2006, 103, 19111–19116. [Google Scholar] [CrossRef]
- Avirutnan, P.; Fuchs, A.; Hauhart, R.E.; Somnuke, P.; Youn, S.; Diamond, M.S.; Atkinson, J.P. Antagonism of the complement component C4 by flavivirus nonstructural protein NS1. J. Exp. Med. 2010, 207, 793–806. [Google Scholar] [CrossRef]
- Somnuke, P.; Hauhart, R.E.; Atkinson, J.P.; Diamond, M.S.; Avirutnan, P. N-linked glycosylation of dengue virus NS1 protein modulates secretion, cell-surface expression, hexamer stability, and interactions with human complement. Virology 2011, 413, 253–264. [Google Scholar] [CrossRef]
- Hall, R.A.; Khromykh, A.A.; Mackenzie, J.M.; Scherret, J.H.; Khromykh, T.I. Loss of dimerisation of the nonstructural protein NS1 of Kunjin virus delays viral replication and reduces virulence in mice, but still allows secretion of NS1. Virology 1999, 264, 66–75. [Google Scholar]
- Beasley, D.W.; Whiteman, M.C.; Zhang, S.; Huang, C.Y.; Schneider, B.S.; Smith, D.R.; Gromowski, G.D.; Higgs, S.; Kinney, R.M.; Barrett, A.D. Envelope protein glycosylation status influences mouse neuroinvasion phenotype of genetic lineage 1 West Nile virus strains. J. Virol. 2005, 79, 8339–8347. [Google Scholar] [CrossRef]
- Whiteman, M.C.; Li, L.; Wicker, J.A.; Kinney, R.M.; Huang, C.; Beasley, D.W.; Chung, K.M.; Diamond, M.S.; Solomon, T.; Barrett, A.D. Development and characterization of non-glycosylated E and NS1 mutant viruses as a potential candidate vaccine for West Nile virus. Vaccine 2010, 28, 1075–1083. [Google Scholar] [CrossRef]
- Firth, A.E.; Atkins, J.F. A conserved predicted pseudoknot in the NS2A-encoding sequence of West Nile and Japanese encephalitis flaviviruses suggests NS1' may derive from ribosomal frameshifting. Virol. J. 2009, 6. [Google Scholar] [CrossRef]
- Melian, E.B.; Hinzman, E.; Nagasaki, T.; Firth, A.E.; Wills, N.M.; Nouwens, A.S.; Blitvich, B.J.; Leung, J.; Funk, A.; Atkins, J.F.; et al. NS1' of flaviviruses in the Japanese encephalitis virus serogroup is a product of ribosomal frameshifting and plays a role in viral neuroinvasiveness. J. Virol. 2010, 84, 1641–1647. [Google Scholar] [CrossRef]
- Satchidanandam, V.; Uchil, P.D.; Kumar, P. Organization of flaviviral replicase proteins in virus-induced membranes: A role for NS1' in Japanese encephalitis virus RNA synthesis. Novartis Found. Symp. 2006, 277, 136–145. [Google Scholar] [CrossRef]
- Young, L.B.; Melian, E.B.; Khromykh, A.A. NS1' colocalizes with NS1 and can substitute for NS1 in West Nile virus replication. J. Virol. 2013, 87, 9384–9390. [Google Scholar] [CrossRef]
- Ye, Q.; Li, X.F.; Zhao, H.; Li, S.H.; Deng, Y.Q.; Cao, R.Y.; Song, K.Y.; Wang, H.J.; Hua, R.H.; Yu, Y.X.; et al. A single nucleotide mutation in NS2A of Japanese encephalitis-live vaccine virus (SA14-14-2) ablates NS1' formation and contributes to attenuation. J. Gen. Virol. 2012, 93, 1959–1964. [Google Scholar] [CrossRef]
- Sun, J.; Yu, Y.; Deubel, V. Japanese encephalitis virus NS1' protein depends on pseudoknot secondary structure and is cleaved by caspase during virus infection and cell apoptosis. Microbes Infect. 2012, 14, 930–940. [Google Scholar] [CrossRef]
- Winkelmann, E.R.; Widman, D.G.; Suzuki, R.; Mason, P.W. Analyses of mutations selected by passaging a chimeric flavivirus identify mutations that alter infectivity and reveal an interaction between the structural proteins and the nonstructural glycoprotein NS1. Virology 2011, 421, 96–104. [Google Scholar] [CrossRef]
- Bazan, J.F.; Fletterick, R.J. Detection of a trypsin-like serine protease domain in flaviviruses and pestiviruses. Virology 1989, 171, 637–639. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Donchenko, A.P.; Koonin, E.V.; Blinov, V.M. N-terminal domains of putative helicases of flavi- and pestiviruses may be serine proteases. Nucleic Acids Res. 1989, 17, 3889–3897. [Google Scholar]
- Wengler, G.; Czaya, G.; Farber, P.M.; Hegemann, J.H. In vitro synthesis of West Nile virus proteins indicates that the amino-terminal segment of the NS3 protein contains the active centre of the protease which cleaves the viral polyprotein after multiple basic amino acids. J. Gen. Virol. 1991, 72, 851–858. [Google Scholar] [CrossRef]
- Yusof, R.; Clum, S.; Wetzel, M.; Murthy, H.M.; Padmanabhan, R. Purified NS2B/NS3 serine protease of dengue virus type 2 exhibits cofactor NS2B dependence for cleavage of substrates with dibasic amino acids in vitro. J. Biol. Chem. 2000, 275, 9963–9969. [Google Scholar] [CrossRef]
- Gayen, S.; Chen, A.S.; Huang, Q.; Kang, C. West Nile Virus (WNV) protease and membrane interactions revealed by NMR spectroscopy. Biochem. Biophys. Res. Commun. 2012, 423, 799–804. [Google Scholar] [CrossRef]
- Yu, L.; Takeda, K.; Markoff, L. Protein-protein interactions among West Nile non-structural proteins and transmembrane complex formation in mammalian cells. Virology 2013, 446, 365–377. [Google Scholar] [CrossRef]
- Robin, G.; Chappell, K.; Stoermer, M.J.; Hu, S.H.; Young, P.R.; Fairlie, D.P.; Martin, J.L. Structure of West Nile virus NS3 protease: Ligand stabilization of the catalytic conformation. J. Mol. Biol. 2009, 385, 1568–1577. [Google Scholar] [CrossRef]
- Aleshin, A.E.; Shiryaev, S.A.; Strongin, A.Y.; Liddington, R.C. Structural evidence for regulation and specificity of flaviviral proteases and evolution of the Flaviviridae fold. Protein Sci. 2007, 16, 795–806. [Google Scholar] [CrossRef]
- Kim, Y.M.; Gayen, S.; Kang, C.; Joy, J.; Huang, Q.; Chen, A.S.; Wee, J.L.; Ang, M.J.; Lim, H.A.; Hung, A.W.; et al. NMR analysis of a novel enzymatically active unlinked dengue NS2B-NS3 protease complex. J. Biol. Chem. 2013, 288, 12891–12900. [Google Scholar] [CrossRef]
- Assenberg, R.; Mastrangelo, E.; Walter, T.S.; Verma, A.; Milani, M.; Owens, R.J.; Stuart, D.I.; Grimes, J.M.; Mancini, E.J. Crystal structure of a novel conformational state of the flavivirus NS3 protein: Implications for polyprotein processing and viral replication. J. Virol. 2009, 83, 12895–12906. [Google Scholar]
- Luo, D.; Wei, N.; Doan, D.N.; Paradkar, P.N.; Chong, Y.; Davidson, A.D.; Kotaka, M.; Lescar, J.; Vasudevan, S.G. Flexibility between the protease and helicase domains of the dengue virus NS3 protein conferred by the linker region and its functional implications. J. Biol. Chem. 2010, 285, 18817–18827. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Koonin, E.V.; Donchenko, A.P.; Blinov, V.M. Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res. 1989, 17, 4713–4730. [Google Scholar] [CrossRef]
- Wengler, G.; Wengler, G. The NS 3 nonstructural protein of flaviviruses contains an RNA triphosphatase activity. Virology 1993, 197, 265–273. [Google Scholar] [CrossRef]
- Wu, J.; Bera, A.K.; Kuhn, R.J.; Smith, J.L. Structure of the Flavivirus helicase: Implications for catalytic activity, protein interactions, and proteolytic processing. J. Virol. 2005, 79, 10268–10277. [Google Scholar] [CrossRef]
- Xu, T.; Sampath, A.; Chao, A.; Wen, D.; Nanao, M.; Chene, P.; Vasudevan, S.G.; Lescar, J. Structure of the Dengue virus helicase/nucleoside triphosphatase catalytic domain at a resolution of 2.4 A. J. Virol. 2005, 79, 10278–10288. [Google Scholar] [CrossRef]
- Benarroch, D.; Selisko, B.; Locatelli, G.A.; Maga, G.; Romette, J.L.; Canard, B. The RNA helicase, nucleotide 5'-triphosphatase, and RNA 5'-triphosphatase activities of Dengue virus protein NS3 are Mg2+-dependent and require a functional Walker B motif in the helicase catalytic core. Virology 2004, 328, 208–218. [Google Scholar] [CrossRef]
- Wang, C.C.; Huang, Z.S.; Chiang, P.L.; Chen, C.T.; Wu, H.N. Analysis of the nucleoside triphosphatase, RNA triphosphatase, and unwinding activities of the helicase domain of dengue virus NS3 protein. FEBS Lett. 2009, 583, 691–696. [Google Scholar] [CrossRef]
- Luo, D.; Xu, T.; Watson, R.P.; Scherer-Becker, D.; Sampath, A.; Jahnke, W.; Yeong, S.S.; Wang, C.H.; Lim, S.P.; Strongin, A.; et al. Insights into RNA unwinding and ATP hydrolysis by the flavivirus NS3 protein. EMBO J. 2008, 27, 3209–3219. [Google Scholar] [CrossRef]
- Incicco, J.J.; Gebhard, L.G.; Gonzalez-Lebrero, R.M.; Gamarnik, A.V.; Kaufman, S.B. Steady-state NTPase activity of Dengue virus NS3: Number of catalytic sites, nucleotide specificity and activation by ssRNA. PLoS One 2013, 8, e58508. [Google Scholar]
- Shiryaev, S.A.; Chernov, A.V.; Aleshin, A.E.; Shiryaeva, T.N.; Strongin, A.Y. NS4A regulates the ATPase activity of the NS3 helicase: A novel cofactor role of the non-structural protein NS4A from West Nile virus. J. Gen. Virol. 2009, 90, 2081–2085. [Google Scholar] [CrossRef]
- Zhang, B.; Dong, H.; Zhou, Y.; Shi, P.Y. Genetic interactions among the West Nile virus methyltransferase, the RNA-dependent RNA polymerase, and the 5' stem-loop of genomic RNA. J. Virol. 2008, 82, 7047–7058. [Google Scholar] [CrossRef]
- Henderson, B.R.; Saeedi, B.J.; Campagnola, G.; Geiss, B.J. Analysis of RNA binding by the dengue virus NS5 RNA capping enzyme. PLoS One 2011, 6, e25795. [Google Scholar]
- Yap, L.J.; Luo, D.; Chung, K.Y.; Lim, S.P.; Bodenreider, C.; Noble, C.; Shi, P.Y.; Lescar, J. Crystal structure of the dengue virus methyltransferase bound to a 5'-capped octameric RNA. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Kamer, G.; Argos, P. Primary structural comparison of RNA-dependent polymerases from plant, animal and bacterial viruses. Nucleic Acids Res. 1984, 12, 7269–7282. [Google Scholar] [CrossRef]
- Koonin, E.V. The phylogeny of RNA-dependent RNA polymerases of positive-strand RNA viruses. J. Gen. Virol. 1991, 72, 2197–2206. [Google Scholar] [CrossRef]
- Brooks, A.J.; Johansson, M.; John, A.V.; Xu, Y.; Jans, D.A.; Vasudevan, S.G. The interdomain region of dengue NS5 protein that binds to the viral helicase NS3 contains independently functional importin beta 1 and importin alpha/beta-recognized nuclear localization signals. J. Biol. Chem. 2002, 277, 36399–36407. [Google Scholar]
- Johansson, M.; Brooks, A.J.; Jans, D.A.; Vasudevan, S.G. A small region of the dengue virus-encoded RNA-dependent RNA polymerase, NS5, confers interaction with both the nuclear transport receptor importin-beta and the viral helicase, NS3. J. Gen. Virol. 2001, 82, 735–745. [Google Scholar]
- Hannemann, H.; Sung, P.Y.; Chiu, H.C.; Yousuf, A.; Bird, J.; Lim, S.P.; Davidson, A.D. Serotype-specific differences in dengue virus non-structural protein 5 nuclear localization. J. Biol. Chem. 2013, 288, 22621–22635. [Google Scholar] [CrossRef]
- Kumar, A.; Buhler, S.; Selisko, B.; Davidson, A.; Mulder, K.; Canard, B.; Miller, S.; Bartenschlager, R. Nuclear localization of dengue virus nonstructural protein 5 does not strictly correlate with efficient viral RNA replication and inhibition of type I interferon signaling. J. Virol. 2013, 87, 4545–4557. [Google Scholar] [CrossRef]
- Malet, H.; Egloff, M.P.; Selisko, B.; Butcher, R.E.; Wright, P.J.; Roberts, M.; Gruez, A.; Sulzenbacher, G.; Vonrhein, C.; Bricogne, G.; Mackenzie, J.M.; Khromykh, A.A.; Davidson, A.D.; et al. Crystal structure of the RNA polymerase domain of the West Nile virus non-structural protein 5. J. Biol. Chem. 2007, 282, 10678–10689. [Google Scholar] [CrossRef]
- Yap, T.L.; Xu, T.; Chen, Y.L.; Malet, H.; Egloff, M.P.; Canard, B.; Vasudevan, S.G.; Lescar, J. Crystal structure of the dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J. Virol. 2007, 81, 4753–4765. [Google Scholar] [CrossRef]
- Zou, G.; Chen, Y.L.; Dong, H.; Lim, C.C.; Yap, L.J.; Yau, Y.H.; Shochat, S.G.; Lescar, J.; Shi, P.Y. Functional analysis of two cavities in flavivirus NS5 polymerase. J. Biol. Chem. 2011, 286, 14362–14372. [Google Scholar]
- Iglesias, N.G.; Filomatori, C.V.; Gamarnik, A.V. The F1 motif of dengue virus polymerase NS5 is involved in promoter-dependent RNA synthesis. J. Virol. 2011, 85, 5745–5756. [Google Scholar] [CrossRef]
- Lu, G.; Gong, P. Crystal structure of the full-length Japanese encephalitis virus NS5 reveals a conserved methyltransferase-polymerase interface. PLoS Pathog. 2013, 9, e1003549. [Google Scholar] [CrossRef]
- Tan, C.S.; Hobson-Peters, J.M.; Stoermer, M.J.; Fairlie, D.P.; Khromykh, A.A.; Hall, R.A. An interaction between the methyltransferase and RNA dependent RNA polymerase domains of the West Nile virus NS5 protein. J. Gen. Virol. 2013, 94, 1961–1971. [Google Scholar] [CrossRef]
- Bussetta, C.; Choi, K.H. Dengue virus nonstructural protein 5 adopts multiple conformations in solution. Biochemistry 2012, 51, 5921–5931. [Google Scholar] [CrossRef]
- Ackermann, M.; Padmanabhan, R. De novo synthesis of RNA by the dengue virus RNA-dependent RNA polymerase exhibits temperature dependence at the initiation but not elongation phase. J. Biol. Chem. 2001, 276, 39926–39937. [Google Scholar] [CrossRef]
- Guyatt, K.J.; Westaway, E.G.; Khromykh, A.A. Expression and purification of enzymatically active recombinant RNA-dependent RNA polymerase (NS5) of the flavivirus Kunjin. J. Virol. Methods 2001, 92, 37–44. [Google Scholar] [CrossRef]
- Selisko, B.; Dutartre, H.; Guillemot, J.C.; Debarnot, C.; Benarroch, D.; Khromykh, A.; Despres, P.; Egloff, M.P.; Canard, B. Comparative mechanistic studies of de novo RNA synthesis by flavivirus RNA-dependent RNA polymerases. Virology 2006, 351, 145–158. [Google Scholar] [CrossRef]
- Grun, J.B.; Brinton, M.A. Characterization of West Nile virus RNA-dependent RNA polymerase and cellular terminal adenylyl and uridylyl transferases in cell-free extracts. J. Virol. 1986, 60, 1113–1124. [Google Scholar]
- Ranjith-Kumar, C.T.; Gajewski, J.; Gutshall, L.; Maley, D.; Sarisky, R.T.; Kao, C.C. Terminal nucleotidyl transferase activity of recombinant Flaviviridae RNA-dependent RNA polymerases: implication for viral RNA synthesis. J. Virol. 2001, 75, 8615–8623. [Google Scholar] [CrossRef]
- You, S.; Falgout, B.; Markoff, L.; Padmanabhan, R. In vitro RNA synthesis from exogenous dengue viral RNA templates requires long range interactions between 5'- and 3'-terminal regions that influence RNA structure. J. Biol. Chem. 2001, 276, 15581–15591. [Google Scholar] [CrossRef]
- Chu, P.W.; Westaway, E.G. Characterization of Kunjin virus RNA-dependent RNA polymerase: Reinitiation of synthesis in vitro. Virology 1987, 157, 330–337. [Google Scholar] [CrossRef]
- Lott, W.B.; Doran, M.R. Do RNA viruses require genome cyclisation for replication? Trends Biochem. Sci. 2013, 38, 350–355. [Google Scholar] [CrossRef]
- Keller, B.C.; Fredericksen, B.L.; Samuel, M.A.; Mock, R.E.; Mason, P.W.; Diamond, M.S.; Gale, M., Jr. Resistance to alpha/beta interferon is a determinant of West Nile virus replication fitness and virulence. J. Virol. 2006, 80, 9424–9434. [Google Scholar] [CrossRef]
- Laurent-Rolle, M.; Boer, E.F.; Lubick, K.J.; Wolfinbarger, J.B.; Carmody, A.B.; Rockx, B.; Liu, W.; Ashour, J.; Shupert, W.L.; Holbrook, M.R.; et al. The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J. Virol. 2010, 84, 3503–3515. [Google Scholar] [CrossRef]
- Pulit-Penaloza, J.A.; Scherbik, S.V.; Brinton, M.A. Type 1 IFN-independent activation of a subset of interferon stimulated genes in West Nile virus Eg101-infected mouse cells. Virology 2012, 425, 82–94. [Google Scholar] [CrossRef]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef]
- Mackenzie, J.M.; Khromykh, A.A.; Parton, R.G. Cholesterol manipulation by West Nile virus perturbs the cellular immune response. Cell Host Microbe 2007, 2, 229–239. [Google Scholar] [CrossRef]
- Beatman, E.; Oyer, R.; Shives, K.D.; Hedman, K.; Brault, A.C.; Tyler, K.L.; Beckham, J.D. West Nile virus growth is independent of autophagy activation. Virology 2012, 433, 262–272. [Google Scholar] [CrossRef]
- Vandergaast, R.; Fredericksen, B.L. West Nile virus (WNV) replication is independent of autophagy in mammalian cells. PLoS One 2012, 7, e45800. [Google Scholar] [CrossRef]
- McLean, J.E.; Wudzinska, A.; Datan, E.; Quaglino, D.; Zakeri, Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J. Biol. Chem. 2011, 286, 22147–22159. [Google Scholar] [CrossRef]
- Xu, Z.; Waeckerlin, R.; Urbanowski, M.D.; van Marle, G.; Hobman, T.C. West Nile virus infection causes endocytosis of a specific subset of tight junction membrane proteins. PLoS One 2012, 7, e37886. [Google Scholar]
- Medigeshi, G.R.; Hirsch, A.J.; Brien, J.D.; Uhrlaub, J.L.; Mason, P.W.; Wiley, C.; Nikolich-Zugich, J.; Nelson, J.A. West nile virus capsid degradation of claudin proteins disrupts epithelial barrier function. J. Virol. 2009, 83, 6125–6134. [Google Scholar] [CrossRef]
- Heaton, N.S.; Perera, R.; Berger, K.L.; Khadka, S.; Lacount, D.J.; Kuhn, R.J.; Randall, G. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 17345–17350. [Google Scholar]
- Courtney, S.C.; Brinton, M.A. Georgia State University: Atlanta, GA, USA, Unpublished work. 2012.
- Hsu, N.Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef]
- Martin-Acebes, M.A.; Blazquez, A.B.; Jimenez de Oya, N.; Escribano-Romero, E.; Saiz, J.C. West Nile virus replication requires fatty acid synthesis but is independent on phosphatidylinositol-4-phosphate lipids. PLoS One 2011, 6, e24970. [Google Scholar]
- Courtney, S.C.; Scherbik, S.V.; Stockman, B.M.; Brinton, M.A. West nile virus infections suppress early viral RNA synthesis and avoid inducing the cell stress granule response. J. Virol. 2012, 86, 3647–3657. [Google Scholar]
- Tu, Y.C.; Yu, C.Y.; Liang, J.J.; Lin, E.; Liao, C.L.; Lin, Y.L. Blocking double-stranded RNA-activated protein kinase PKR by Japanese encephalitis virus nonstructural protein 2A. J. Virol. 2012, 86, 10347–10358. [Google Scholar] [CrossRef]
- Elbahesh, H.; Scherbik, S.V.; Brinton, M.A. West Nile virus infection does not induce PKR activation in rodent cells. Virology 2011, 421, 51–60. [Google Scholar] [CrossRef]
- Katoh, H.; Okamoto, T.; Fukuhara, T.; Kambara, H.; Morita, E.; Mori, Y.; Kamitani, W.; Matsuura, Y. Japanese encephalitis virus core protein inhibits stress granule formation through an interaction with Caprin-1 and facilitates viral propagation. J. Virol. 2013, 87, 489–502. [Google Scholar]
- Villas-Boas, C.S.; Conceicao, T.M.; Ramirez, J.; Santoro, A.B.; Da Poian, A.T.; Montero-Lomeli, M. Dengue virus-induced regulation of the host cell translational machinery. Braz. J. Med. Biol. Res. 2009, 42, 1020–1026. [Google Scholar]
- Edgil, D.; Polacek, C.; Harris, E. Dengue virus utilizes a novel strategy for translation initiation when cap-dependent translation is inhibited. J. Virol. 2006, 80, 2976–2986. [Google Scholar] [CrossRef]
- Hoenninger, V.M.; Rouha, H.; Orlinger, K.K.; Miorin, L.; Marcello, A.; Kofler, R.M.; Mandl, C.W. Analysis of the effects of alterations in the tick-borne encephalitis virus 3'-noncoding region on translation and RNA replication using reporter replicons. Virology 2008, 377, 419–430. [Google Scholar] [CrossRef]
- Polacek, C.; Friebe, P.; Harris, E. Poly(A)-binding protein binds to the non-polyadenylated 3' untranslated region of dengue virus and modulates translation efficiency. J. Gen. Virol. 2009, 90, 687–692. [Google Scholar] [CrossRef]
- Wei, Y.; Qin, C.; Jiang, T.; Li, X.; Zhao, H.; Liu, Z.; Deng, Y.; Liu, R.; Chen, S.; Yu, M.; et al. Translational regulation by the 3' untranslated region of the dengue type 2 virus genome. Am. J. Trop. Med. Hyg. 2009, 81, 817–824. [Google Scholar] [CrossRef]
- Chien, H.L.; Liao, C.L.; Lin, Y.L. FUSE binding protein 1 interacts with untranslated regions of Japanese encephalitis virus RNA and negatively regulates viral replication. J. Virol. 2011, 85, 4698–4706. [Google Scholar] [CrossRef]
- Fan, Y.H.; Nadar, M.; Chen, C.C.; Weng, C.C.; Lin, Y.T.; Chang, R.Y. Small noncoding RNA modulates Japanese encephalitis virus replication and translation in trans. Virol. J. 2011, 8. [Google Scholar] [CrossRef]
- Ambrose, R.L.; Mackenzie, J.M. ATF6 signaling is required for efficient West Nile virus replication by promoting cell survival and inhibition of innate immune responses. J. Virol. 2013, 87, 2206–2214. [Google Scholar] [CrossRef]
- Ambrose, R.L.; Mackenzie, J.M. West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J. Virol. 2011, 85, 2723–2732. [Google Scholar] [CrossRef]
- Medigeshi, G.R.; Lancaster, A.M.; Hirsch, A.J.; Briese, T.; Lipkin, W.I.; Defilippis, V.; Fruh, K.; Mason, P.W.; Nikolich-Zugich, J.; Nelson, J.A. West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J. Virol. 2007, 81, 10849–10860. [Google Scholar] [CrossRef]
- Blackwell, J.L.; Brinton, M.A. Translation elongation factor-1 alpha interacts with the 3' stem-loop region of West Nile virus genomic RNA. J. Virol. 1997, 71, 6433–6444. [Google Scholar]
- De Nova-Ocampo, M.; Villegas-Sepulveda, N.; del Angel, R.M. Translation Elongation Factor-1α, La, and PTB Interact with the 3' Untranslated Region of Dengue 4 Virus RNA. Virology 2002, 295, 337–347. [Google Scholar] [CrossRef]
- Kim, S.M.; Jeong, Y.S. Polypyrimidine tract-binding protein interacts with the 3' stem-loop region of Japanese encephalitis virus negative-strand RNA. Virus Res. 2006, 115, 131–140. [Google Scholar] [CrossRef]
- Paranjape, S.M.; Harris, E. Y box-binding protein-1 binds to the dengue virus 3'-untranslated region and mediates antiviral effects. J. Biol. Chem. 2007, 282, 30497–30508. [Google Scholar] [CrossRef]
- Ta, M.; Vrati, S. Mov34 protein from mouse brain interacts with the 3' noncoding region of Japanese encephalitis virus. J. Virol. 2000, 74, 5108–5115. [Google Scholar] [CrossRef]
- Yocupicio-Monroy, M.; Padmanabhan, R.; Medina, F.; del Angel, R.M. Mosquito La protein binds to the 3' untranslated region of the positive and negative polarity dengue virus RNAs and relocates to the cytoplasm of infected cells. Virology 2007, 357, 29–40. [Google Scholar] [CrossRef]
- Li, C.; Ge, L.L.; Li, P.P.; Wang, Y.; Sun, M.X.; Huang, L.; Ishag, H.; Di, D.D.; Shen, Z.Q.; Fan, W.X.; et al. The DEAD-box RNA helicase DDX5 acts as a positive regulator of Japanese encephalitis virus replication by binding to viral 3' UTR. Antivir. Res. 2013, 100, 487–499. [Google Scholar] [CrossRef]
- Gomila, R.C.; Martin, G.W.; Gehrke, L. NF90 binds the dengue virus RNA 3' terminus and is a positive regulator of dengue virus replication. PLoS One 2011, 6, e16687. [Google Scholar] [CrossRef]
- Yang, S.H.; Liu, M.L.; Tien, C.F.; Chou, S.J.; Chang, R.Y. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) interaction with 3' ends of Japanese encephalitis virus RNA and colocalization with the viral NS5 protein. J. Biomed. Sci. 2009, 16. [Google Scholar] [CrossRef]
- Pastorino, B.; Nougairede, A.; Wurtz, N.; Gould, E.; de Lamballerie, X. Role of host cell factors in flavivirus infection: Implications for pathogenesis and development of antiviral drugs. Antivir. Res. 2010, 87, 281–294. [Google Scholar] [CrossRef]
- Blackwell, J.L.; Brinton, M.A. BHK cell proteins that bind to the 3' stem-loop structure of the West Nile virus genome RNA. J. Virol. 1995, 69, 5650–5658. [Google Scholar]
- Riis, B.; Rattan, S.I.; Clark, B.F.; Merrick, W.C. Eukaryotic protein elongation factors. Trends Biochem. Sci. 1990, 15, 420–424. [Google Scholar] [CrossRef]
- Davis, W.G.; Blackwell, J.L.; Shi, P.Y.; Brinton, M.A. Interaction between the cellular protein eEF1A and the 3'-terminal stem-loop of West Nile virus genomic RNA facilitates viral minus-strand RNA synthesis. J. Virol. 2007, 81, 10172–10187. [Google Scholar] [CrossRef]
- Li, W.; Li, Y.; Kedersha, N.; Anderson, P.; Emara, M.; Swiderek, K.M.; Moreno, G.T.; Brinton, M.A. Cell proteins TIA-1 and TIAR interact with the 3' stem-loop of the West Nile virus complementary minus-strand RNA and facilitate virus replication. J. Virol. 2002, 76, 11989–12000. [Google Scholar]
- Dember, L.M.; Kim, N.D.; Liu, K.Q.; Anderson, P. Individual RNA recognition motifs of TIA-1 and TIAR have different RNA binding specificities. J. Biol. Chem. 1996, 271, 2783–2788. [Google Scholar] [CrossRef]
- Taupin, J.L.; Tian, Q.; Kedersha, N.; Robertson, M.; Anderson, P. The RNA-binding protein TIAR is translocated from the nucleus to the cytoplasm during Fas-mediated apoptotic cell death. Proc. Natl. Acad. Sci. USA 1995, 92, 1629–1633. [Google Scholar] [CrossRef]
- Dixon, D.A.; Balch, G.C.; Kedersha, N.; Anderson, P.; Zimmerman, G.A.; Beauchamp, R.D.; Prescott, S.M. Regulation of cyclooxygenase-2 expression by the translational silencer TIA-1. J. Exp. Med. 2003, 198, 475–481. [Google Scholar] [CrossRef]
- Cok, S.J.; Acton, S.J.; Sexton, A.E.; Morrison, A.R. Identification of RNA-binding proteins in RAW 264.7 cells that recognize a lipopolysaccharide-responsive element in the 3-untranslated region of the murine cyclooxygenase-2 mRNA. J. Biol. Chem. 2004, 279, 8196–8205. [Google Scholar]
- Liu, H.; Brinton, M.A. Georgia State University: Atlanta, GA, USA, Unpublished work. 2013.
- Emara, M.M.; Brinton, M.A. Mutation of mapped TIA-1/TIAR binding sites within the West Nile virus 3' terminal minus-strand RNA sequence in an infectious clone negatively affects viral plus-strand synthesis. J. Virol. 2008, 82, 10657–10670. [Google Scholar] [CrossRef]
- Kapoor, M.; Zhang, L.; Ramachandra, M.; Kusukawa, J.; Ebner, K.E.; Padmanabhan, R. Association between NS3 and NS5 proteins of dengue virus type 2 in the putative RNA replicase is linked to differential phosphorylation of NS5. J. Biol. Chem. 1995, 270, 19100–19106. [Google Scholar] [CrossRef]
- Cui, T.; Sugrue, R.J.; Xu, Q.; Lee, A.K.; Chan, Y.C.; Fu, J. Recombinant dengue virus type 1 NS3 protein exhibits specific viral RNA binding and NTPase activity regulated by the NS5 protein. Virology 1998, 246, 409–417. [Google Scholar] [CrossRef]
- Yon, C.; Teramoto, T.; Mueller, N.; Phelan, J.; Ganesh, V.K.; Murthy, K.H.; Padmanabhan, R. Modulation of the nucleoside triphosphatase/RNA helicase and 5'-RNA triphosphatase activities of Dengue virus type 2 nonstructural protein 3 (NS3) by interaction with NS5, the RNA-dependent RNA polymerase. J. Biol. Chem. 2005, 280, 27412–27419. [Google Scholar]
- Lindenbach, B.D.; Rice, C.M. Genetic interaction of flavivirus nonstructural proteins NS1 and NS4A as a determinant of replicase function. J. Virol. 1999, 73, 4611–4621. [Google Scholar]
- Youn, S.; Li, T.; McCune, B.T.; Edeling, M.A.; Fremont, D.H.; Cristea, I.M.; Diamond, M.S. Evidence for a genetic and physical interaction between nonstructural proteins NS1 and NS4B that modulates replication of West Nile virus. J. Virol. 2012, 86, 7360–7371. [Google Scholar] [CrossRef]
- Scherbik, S.V.; Pulit-Penaloza, J.A.; Basu, M.; Courtney, S.C.; Brinton, M.A. Increased early RNA replication by chimeric West Nile virus W956IC leads to IPS-1 mediated activation of NF-kappaB and insufficient virus-mediated counteraction of the resulting canonical type I interferon signaling. J. Virol. 2013, 87, 7952–7965. [Google Scholar] [CrossRef]
- Yamshchikov, V.F.; Wengler, G.; Perelygin, A.A.; Brinton, M.A.; Compans, R.W. An infectious clone of the West Nile flavivirus. Virology 2001, 281, 294–304. [Google Scholar] [CrossRef]
- Van Slyke, G.A.; Ciota, A.T.; Willsey, G.G.; Jaeger, J.; Shi, P.Y.; Kramer, L.D. Point mutations in the West Nile virus (Flaviviridae; Flavivirus) RNA-dependent RNA polymerase alter viral fitness in a host-dependent manner in vitro and in vivo. Virology 2012, 427, 18–24. [Google Scholar] [CrossRef]
- Mackenzie, J.M.; Kenney, M.T.; Westaway, E.G. West Nile virus strain Kunjin NS5 polymerase is a phosphoprotein localized at the cytoplasmic site of viral RNA synthesis. J. Gen. Virol. 2007, 88, 1163–1168. [Google Scholar] [CrossRef]
- Reed, K.E.; Gorbalenya, A.E.; Rice, C.M. The NS5A/NS5 proteins of viruses from three genera of the family flaviviridae are phosphorylated by associated serine/threonine kinases. J. Virol. 1998, 72, 6199–6206. [Google Scholar]
- Laurent-Rolle, M.; Boer, E.F.; Lubick, K.J.; Wolfinbarger, J.B.; Carmody, A.B.; Rockx, B.; Liu, W.; Ashour, J.; Shupert, W.L.; Holbrook, M.R.; et al. The NS5 protein of thevirulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAKSTAT signaling. J. Virol. 2010, 84, 3503–3515. [Google Scholar] [CrossRef]
- Morais, A.; Terzian, A.C.; Duarte, D.V.; Bronzoni, R.V.; Madrid, M.C.; Gavioli, A.F.; Gil, L.H.; Oliveira, A.G.; Zanelli, C.F.; Valentini, S.R.; et al. The eukaryotic translation initiation factor 3 subunit L protein interacts with Flavivirus NS5 and may modulate yellow fever virus replication. Virol. J. 2013, 10. [Google Scholar] [CrossRef]
- Wang, R.Y.; Huang, Y.R.; Chong, K.M.; Hung, C.Y.; Ke, Z.L.; Chang, R.Y. DNAJ homolog Hdj2 facilitates Japanese encephalitis virus replication. Virol. J. 2011, 8. [Google Scholar] [CrossRef]
- Melik, W.; Ellencrona, K.; Wigerius, M.; Hedstrom, C.; Elvang, A.; Johansson, M. Two PDZ binding motifs within NS5 have roles in Tick-borne encephalitis virus replication. Virus Res. 2012, 169, 54–62. [Google Scholar] [CrossRef]
- Le Breton, M.; Meyniel-Schicklin, L.; Deloire, A.; Coutard, B.; Canard, B.; de Lamballerie, X.; Andre, P.; Rabourdin-Combe, C.; Lotteau, V.; Davoust, N. Flavivirus NS3 and NS5 proteins interaction network: A high-throughput yeast two-hybrid screen. BMC Microbiol. 2011, 11. [Google Scholar] [CrossRef]
- Miorin, L.; Romero-Brey, I.; Maiuri, P.; Hoppe, S.; Krijnse-Locker, J.; Bartenschlager, R.; Marcello, A. Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. J. Virol. 2013, 87, 6469–6481. [Google Scholar] [CrossRef]
- Dokland, T.; Walsh, M.; Mackenzie, J.M.; Khromykh, A.A.; Ee, K.H.; Wang, S. West Nile virus core protein; tetramer structure and ribbon formation. Structure 2004, 12, 1157–1163. [Google Scholar] [CrossRef]
- Kofler, R.M.; Heinz, F.X.; Mandl, C.W. Capsid protein C of tick-borne encephalitis virus tolerates large internal deletions and is a favorable target for attenuation of virulence. J. Virol. 2002, 76, 3534–3543. [Google Scholar] [CrossRef]
- Khromykh, A.A.; Westaway, E.G. RNA binding properties of core protein of the flavivirus Kunjin. Arch. Virol. 1996, 141, 685–699. [Google Scholar] [CrossRef]
- Heinz, F.X.; Auer, G.; Stiasny, K.; Holzmann, H.; Mandl, C.; Guirakhoo, F.; Kunz, C. The interactions of the flavivirus envelope proteins: implications for virus entry and release. Arch. Virol. 1994, 9, 339–348. [Google Scholar]
- Zhang, Y.; Corver, J.; Chipman, P.R.; Zhang, W.; Pletnev, S.V.; Sedlak, D.; Baker, T.S.; Strauss, J.H.; Kuhn, R.J.; Rossmann, M.G. Structures of immature flavivirus particles. EMBO J. 2003, 22, 2604–2613. [Google Scholar] [CrossRef]
- Stadler, K.; Allison, S.L.; Schalich, J.; Heinz, F.X. Proteolytic activation of tick-borne encephalitis virus by furin. J. Virol. 1997, 71, 8475–8481. [Google Scholar]
- Kuhn, R.J.; Zhang, W.; Rossmann, M.G.; Pletnev, S.V.; Corver, J.; Lenches, E.; Jones, C.T.; Mukhopadhyay, S.; Chipman, P.R.; Strauss, E.G.; et al. Structure of dengue virus: Implications for flavivirus organization, maturation, and fusion. Cell 2002, 108, 717–725. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Kim, B.S.; Chipman, P.R.; Rossmann, M.G.; Kuhn, R.J. Structure of West Nile virus. Science 2003, 302. [Google Scholar] [CrossRef]
- Zhang, Y.; Kaufmann, B.; Chipman, P.R.; Kuhn, R.J.; Rossmann, M.G. Structure of immature West Nile virus. J. Virol. 2007, 81, 6141–6144. [Google Scholar] [CrossRef]
- Russell, P.K.; Brandt, W.E.; Dalrymple, J.M. Chemical and antigenic structure of flaviviruses. In The Togaviruses; Schlesinger, R.W., Ed.; Academic Press: New York, NY, USA, 1980; pp. 503–529. [Google Scholar]
- Allison, S.L.; Tao, Y.J.; O'Riordain, G.; Mandl, C.W.; Harrison, S.C.; Heinz, F.X. Two distinct size classes of immature and mature subviral particles from tick-borne encephalitis virus. J. Virol. 2003, 77, 11357–11366. [Google Scholar] [CrossRef]
- Xu, Z.; Anderson, R.; Hobman, T.C. The capsid-binding nucleolar helicase DDX56 is important for infectivity of West Nile virus. J. Virol. 2011, 85, 5571–5580. [Google Scholar] [CrossRef]
- Xu, Z.; Hobman, T.C. The helicase activity of DDX56 is required for its role in assembly of infectious West Nile virus particles. Virology 2012, 433, 226–235. [Google Scholar] [CrossRef]
- Hirsch, A.J.; Medigeshi, G.R.; Meyers, H.L.; DeFilippis, V.; Fruh, K.; Briese, T.; Lipkin, W.I.; Nelson, J.A. The Src family kinase c-Yes is required for maturation of West Nile virus particles. J. Virol. 2005, 79, 11943–11951. [Google Scholar] [CrossRef]
- Carpp, L.N.; Galler, R.; Bonaldo, M.C. Interaction between the yellow fever virus nonstructural protein NS3 and the host protein Alix contributes to the release of infectious particles. Microbes Infect. 2011, 13, 85–95. [Google Scholar] [CrossRef]
- Agrawal, T.; Schu, P.; Medigeshi, G.R. Adaptor protein complexes-1 and 3 are involved at distinct stages of flavivirus life-cycle. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef]
- Groat-Carmona, A.M.; Orozco, S.; Friebe, P.; Payne, A.; Kramer, L.; Harris, E. A novel coding-region RNA element modulates infectious dengue virus particle production in both mammalian and mosquito cells and regulates viral replication in Aedes aegypti mosquitoes. Virology 2012, 432, 511–526. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Brinton, M.A. Replication Cycle and Molecular Biology of the West Nile Virus. Viruses 2014, 6, 13-53. https://doi.org/10.3390/v6010013
Brinton MA. Replication Cycle and Molecular Biology of the West Nile Virus. Viruses. 2014; 6(1):13-53. https://doi.org/10.3390/v6010013
Chicago/Turabian StyleBrinton, Margo A. 2014. "Replication Cycle and Molecular Biology of the West Nile Virus" Viruses 6, no. 1: 13-53. https://doi.org/10.3390/v6010013
APA StyleBrinton, M. A. (2014). Replication Cycle and Molecular Biology of the West Nile Virus. Viruses, 6(1), 13-53. https://doi.org/10.3390/v6010013