Regulation of Human Cytomegalovirus Transcription in Latency: Beyond the Major Immediate-Early Promoter


{kind=link}
Abstract
:1. Introduction
2. Histones, Chromatin and Gene Expression
3. Human Cytomegalovirus Latent Gene Products
4. The UL81-82 Antisense Transcript—LUNA
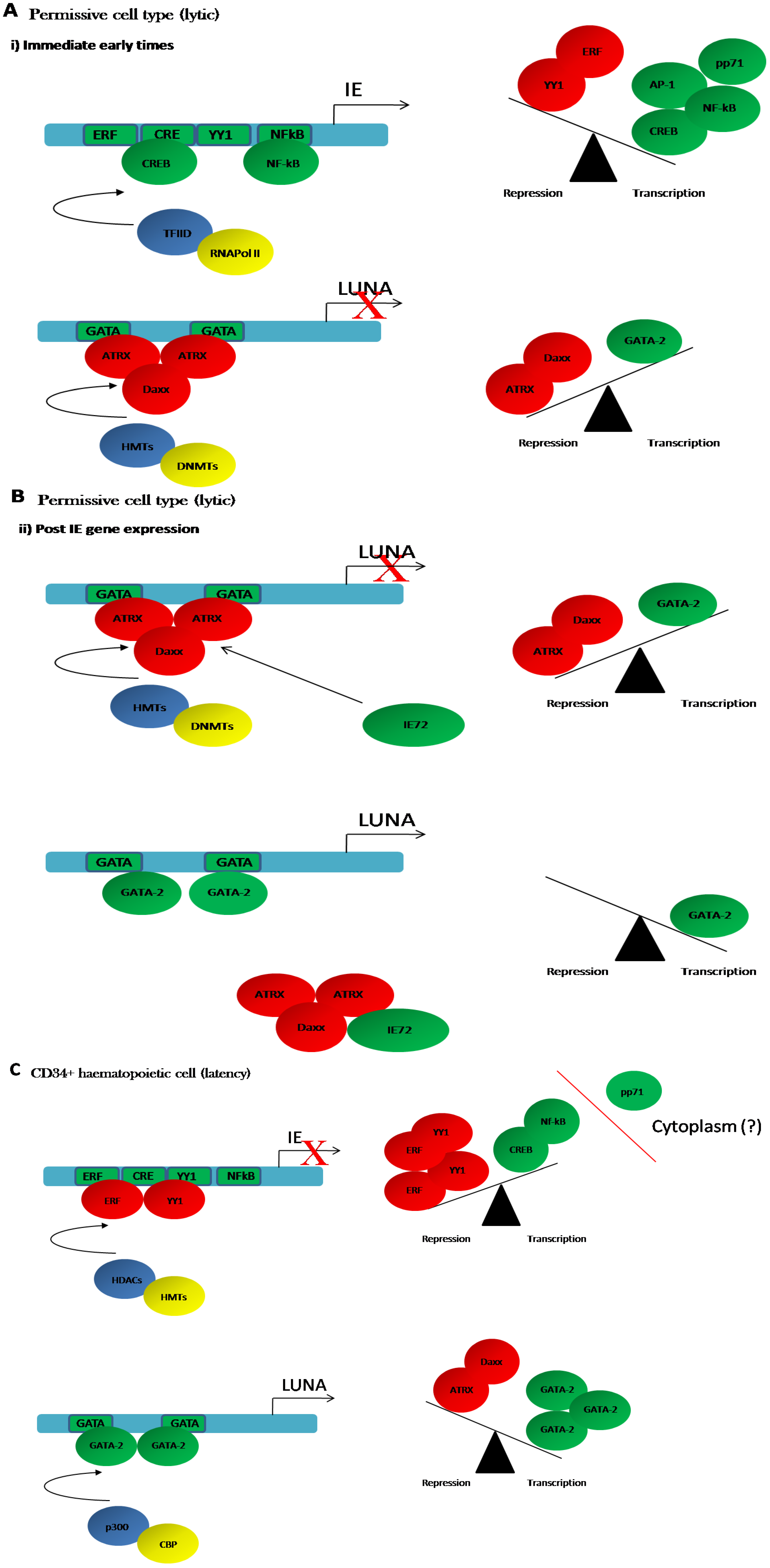
4.1. Regulation of LUNA during Latent Infection
4.2. Regulation of LUNA during Lytic Infection
4.3. LUNA Is Regulated by the GATA Transcription Factors during Latency
5. TNF Receptor Superfamily Member—UL144
5.1. UL144 Is Expressed during Latency in a Strain-Specific Manner
5.2. UL144 Expression during Latency Is Dependent on GATA-2 Binding Sites
6. Concluding Remarks
Acknowledgments
Conflict of Interest
References and Notes
- Pass, R.F.; Stagno, S.; Britt, W.J.; Alford, C.A. Specific cell-mediated immunity and the natural history of congenital infection with cytomegalovirus. J. Infect. Dis. 1983, 148, 953–961. [Google Scholar] [CrossRef]
- Zanghellini, F.; Boppana, S.B.; Emery, V.C.; Griffiths, P.D.; Pass, R.F. Asymptomatic primary cytomegalovirus infection: Virologic and immunologic features. J. Infect. Dis. 1999, 180, 702–707. [Google Scholar] [CrossRef]
- Griffiths, P.D. Cytomegalovirus in intensive care. Rev. Med. Virol. 2010, 20, 1–3. [Google Scholar] [CrossRef]
- Limaye, A.P.; Kirby, K.A.; Rubenfeld, G.D.; Leisenring, W.M.; Bulger, E.M.; Neff, M.J.; Gibran, N.S.; Huang, M.L.; Santo Hayes, T.K.; Corey, L.; et al. Cytomegalovirus reactivation in critically ill immunocompetent patients. JAMA 2008, 300, 413–422. [Google Scholar] [CrossRef]
- Crough, T.; Khanna, R. Immunobiology of human cytomegalovirus: From bench to bedside. Clin. Microbiol. Rev. 2009, 22, 76–98, Table of Contents. [Google Scholar] [CrossRef]
- Revello, M.G.; Gerna, G. Pathogenesis and prenatal diagnosis of human cytomegalovirus infection. J. Clin. Virol. 2004, 29, 71–83. [Google Scholar] [CrossRef]
- Kutza, A.S.; Muhl, E.; Hackstein, H.; Kirchner, H.; Bein, G. High incidence of active cytomegalovirus infection among septic patients. Clin. Infect. Dis. 1998, 26, 1076–1082. [Google Scholar]
- Heininger, A.; Jahn, G.; Engel, C.; Notheisen, T.; Unertl, K.; Hamprecht, K. Human cytomegalovirus infections in nonimmunosuppressed critically ill patients. Crit. Care Med. 2001, 29, 541–547. [Google Scholar] [CrossRef]
- Chilet, M.; Aguilar, G.; Benet, I.; Belda, J.; Tormo, N.; Carbonell, J.A.; Clari, M.A.; Costa, E.; Navarro, D. Virological and immunological features of active cytomegalovirus infection in nonimmunosuppressed patients in a surgical and trauma intensive care unit. J. Med. Virol. 2010, 82, 1384–1391. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Doenecke, D.; Gallwitz, D. Acetylation of histones in nucleosomes. Mol. Cell. Biochem. 1982, 44, 113–128. [Google Scholar]
- Lopez-Rodas, G.; Brosch, G.; Georgieva, E.I.; Sendra, R.; Franco, L.; Loidl, P. Histone deacetylase. A key enzyme for the binding of regulatory proteins to chromatin. FEBS Lett. 1993, 317, 175–180. [Google Scholar]
- Lusser, A. Acetylated, methylated, remodeled: Chromatin states for gene regulation. Curr. Opin. Plant Biol. 2002, 5, 437–443. [Google Scholar] [CrossRef]
- Mizzen, C.A.; Allis, C.D. Linking histone acetylation to transcriptional regulation. Cell. Mol. Life Sci. 1998, 54, 6–20. [Google Scholar] [CrossRef]
- Zhang, Y.; Reinberg, D. Transcription regulation by histone methylation: Interplay between different covalent modifications of the core histone tails. Genes Dev. 2001, 15, 2343–2360. [Google Scholar] [CrossRef]
- Razin, A. CpG methylation, chromatin structure and gene silencing—A three-way connection. EMBO J. 1998, 17, 4905–4908. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef]
- Peters, A.H.; Mermoud, J.E.; O’Carroll, D.; Pagani, M.; Schweizer, D.; Brockdorff, N.; Jenuwein, T. Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat. Genet. 2002, 30, 77–80. [Google Scholar]
- Bannister, A.J.; Zegerman, P.; Partridge, J.F.; Miska, E.A.; Thomas, J.O.; Allshire, R.C.; Kouzarides, T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001, 410, 120–124. [Google Scholar] [CrossRef]
- Wang, H.; Huang, Z.Q.; Xia, L.; Feng, Q.; Erdjument-Bromage, H.; Strahl, B.D.; Briggs, S.D.; Allis, C.D.; Wong, J.; Tempst, P.; et al. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science 2001, 293, 853–857. [Google Scholar] [CrossRef]
- Sawicka, A.; Seiser, C. Histone H3 phosphorylation—A versatile chromatin modification for different occasions. Biochimie 2012, 94, 2193–2201. [Google Scholar]
- Reeves, M.B.; MacAry, P.A.; Lehner, P.J.; Sissons, J.G.; Sinclair, J.H. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carrier. Proc. Natl. Acad. Sci. USA 2005, 102, 4140–4145. [Google Scholar]
- Mendelson, M.; Monard, S.; Sissons, P.; Sinclair, J. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J. Gen. Virol. 1996, 77, 3099–3102. [Google Scholar]
- Hahn, G.; Jores, R.; Mocarski, E.S. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3937–3942. [Google Scholar]
- Taylor-Wiedeman, J.; Sissons, P.; Sinclair, J. Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J. Virol. 1994, 68, 1597–1604. [Google Scholar]
- Soderberg-Naucler, C.; Fish, K.N.; Nelson, J.A. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 1997, 91, 119–126. [Google Scholar]
- Stevens, J.G.; Wagner, E.K.; Devi-Rao, G.B.; Cook, M.L.; Feldman, L.T. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 1987, 235, 1056–1059. [Google Scholar]
- Perng, G.C.; Jones, C.; Ciacci-Zanella, J.; Stone, M.; Henderson, G.; Yukht, A.; Slanina, S.M.; Hofman, F.M.; Ghiasi, H.; Nesburn, A.B.; et al. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 2000, 287, 1500–1503. [Google Scholar]
- Mador, N.; Goldenberg, D.; Cohen, O.; Panet, A.; Steiner, I. Herpes simplex virus type 1 latency-associated transcripts suppress viral replication and reduce immediate-early gene mRNA levels in a neuronal cell line. J. Virol. 1998, 72, 5067–5075. [Google Scholar]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar]
- Thorley-Lawson, D.A. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar]
- Tierney, R.J.; Steven, N.; Young, L.S.; Rickinson, A.B. Epstein-Barr virus latency in blood mononuclear cells: Analysis of viral gene transcription during primary infection and in the carrier state. J. Virol. 1994, 68, 7374–7385. [Google Scholar]
- Babcock, G.J.; Decker, L.L.; Freeman, R.B.; Thorley-Lawson, D.A. Epstein-barr virus-infected resting memory B cells, not proliferating lymphoblasts, accumulate in the peripheral blood of immunosuppressed patients. J. Exp. Med. 1999, 190, 567–576. [Google Scholar]
- Miyashita, E.M.; Yang, B.; Lam, K.M.; Crawford, D.H.; Thorley-Lawson, D.A. A novel form of Epstein-Barr virus latency in normal B cells in vivo. Cell 1995, 80, 593–601. [Google Scholar] [CrossRef]
- Hochberg, D.; Middeldorp, J.M.; Catalina, M.; Sullivan, J.L.; Luzuriaga, K.; Thorley-Lawson, D.A. Demonstration of the Burkitt’s lymphoma Epstein-Barr virus phenotype in dividing latently infected memory cells in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 239–244. [Google Scholar]
- Babcock, G.J.; Thorley-Lawson, D.A. Tonsillar memory B cells, latently infected with Epstein-Barr virus, express the restricted pattern of latent genes previously found only in Epstein-Barr virus-associated tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 12250–12255. [Google Scholar] [CrossRef]
- Babcock, G.J.; Hochberg, D.; Thorley-Lawson, A.D. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef]
- Kuppers, R. B cells under influence: Transformation of B cells by Epstein-Barr virus. Nat. Rev. Immunol. 2003, 3, 801–812. [Google Scholar] [CrossRef]
- Kondo, K.; Kaneshima, H.; Mocarski, E.S. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc. Natl. Acad. Sci. USA 1994, 91, 11879–11883. [Google Scholar]
- Kondo, K.; Xu, J.; Mocarski, E.S. Human cytomegalovirus latent gene expression in granulocyte-macrophage progenitors in culture and in seropositive individuals. Proc. Natl. Acad. Sci. USA 1996, 93, 11137–11142. [Google Scholar]
- Beisser, P.S.; Laurent, L.; Virelizier, J.L.; Michelson, S. Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected THP-1 monocytes. J. Virol. 2001, 75, 5949–5957. [Google Scholar]
- Goodrum, F.D.; Jordan, C.T.; High, K.; Shenk, T. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: A model for latency. Proc. Natl. Acad. Sci. USA 2002, 99, 16255–16260. [Google Scholar]
- Jenkins, C.; Abendroth, A.; Slobedman, B. A novel viral transcript with homology to human interleukin-10 is expressed during latent human cytomegalovirus infection. J. Virol. 2004, 78, 1440–1447. [Google Scholar] [CrossRef]
- Goodrum, F.; Reeves, M.; Sinclair, J.; High, K.; Shenk, T. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 2007, 110, 937–945. [Google Scholar] [CrossRef]
- Bego, M.; Maciejewski, J.; Khaiboullina, S.; Pari, G.; St Jeor, S. Characterization of an antisense transcript spanning the UL81-82 locus of human cytomegalovirus. J. Virol. 2005, 79, 11022–11034. [Google Scholar] [CrossRef]
- Reeves, M.B.; Sinclair, J.H. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J. Gen. Virol. 2010, 91, 599–604. [Google Scholar] [CrossRef]
- Poole, E.; Walther, A.; Raven, K.; Benedict, C.A.; Mason, G.M.; Sinclair, J. The myeloid transcription factor GATA-2 regulates the viral UL144 gene during human cytomegalovirus latency in an isolate-specific manner. J. Virol. 2013. [Google Scholar] [CrossRef]
- Hermiston, T.W.; Malone, C.L.; Witte, P.R.; Stinski, M.F. Identification and characterization of the human cytomegalovirus immediate-early region 2 gene that stimulates gene expression from an inducible promoter. J. Virol. 1987, 61, 3214–3221. [Google Scholar]
- Malone, C.L.; Vesole, D.H.; Stinski, M.F. Transactivation of a human cytomegalovirus early promoter by gene products from the immediate-early gene IE2 and augmentation by IE1: Mutational analysis of the viral proteins. J. Virol. 1990, 64, 1498–1506. [Google Scholar]
- Staprans, S.I.; Rabert, D.K.; Spector, D.H. Identification of sequence requirements and trans-acting functions necessary for regulated expression of a human cytomegalovirus early gene. J. Virol. 1988, 62, 3463–3473. [Google Scholar]
- Greaves, R.F.; Mocarski, E.S. Defective growth correlates with reduced accumulation of a viral DNA replication protein after low-multiplicity infection by a human cytomegalovirus ie1 mutant. J. Virol. 1998, 72, 366–379. [Google Scholar]
- Spector, D.H. Activation and regulation of human cytomegalovirus early genes. Intervirology 1996, 39, 361–377. [Google Scholar]
- Reeves, M.; Sinclair, J. Aspects of human cytomegalovirus latency and reactivation. Curr. Top. Microbiol. Immunol. 2008, 325, 297–313. [Google Scholar] [CrossRef]
- Reeves, M.; Woodhall, D.; Compton, T.; Sinclair, J. Human cytomegalovirus IE72 protein interacts with the transcriptional repressor hDaxx to regulate LUNA gene expression during lytic infection. J. Virol. 2010, 84, 7185–7194. [Google Scholar]
- Keyes, L.R.; Hargett, D.; Soland, M.; Bego, M.G.; Rossetto, C.C.; Almeida-Porada, G.; St Jeor, S. HCMV protein LUNA is required for viral reactivation from latently infected primary CD14(+) cells. PLoS One 2013, 7, e52827. [Google Scholar]
- Groves, I.J.; Reeves, M.B.; Sinclair, J.H. Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic ‘pre-immediate-early’ repression of viral gene expression mediated by histone post-translational modification. J. Gen. Virol. 2009, 90, 2364–2374. [Google Scholar] [CrossRef]
- Cuevas-Bennett, C.; Shenk, T. Dynamic histone H3 acetylation and methylation at human cytomegalovirus promoters during replication in fibroblasts. J. Virol. 2008, 82, 9525–9536. [Google Scholar] [CrossRef]
- Nitzsche, A.; Paulus, C.; Nevels, M. Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J. Virol. 2008, 82, 11167–11180. [Google Scholar] [CrossRef]
- Nevels, M.; Paulus, C.; Shenk, T. Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc. Natl. Acad. Sci. USA 2004, 101, 17234–17239. [Google Scholar] [CrossRef]
- Woodhall, D.L.; Groves, I.J.; Reeves, M.B.; Wilkinson, G.; Sinclair, J.H. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J. Biol. Chem. 2006, 281, 37652–37660. [Google Scholar]
- Ioudinkova, E.; Arcangeletti, M.C.; Rynditch, A.; de Conto, F.; Motta, F.; Covan, S.; Pinardi, F.; Razin, S.V.; Chezzi, C. Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 2006, 384, 120–128. [Google Scholar] [CrossRef]
- Saffert, R.T.; Kalejta, R.F. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 2006, 80, 3863–3871. [Google Scholar] [CrossRef]
- Lukashchuk, V.; McFarlane, S.; Everett, R.D.; Preston, C.M. Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J. Virol. 2008, 82, 12543–12554. [Google Scholar]
- Hofmann, H.; Sindre, H.; Stamminger, T. Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx. J. Virol. 2002, 76, 5769–5783. [Google Scholar] [CrossRef]
- Cantrell, S.R.; Bresnahan, W.A. Interaction between the human cytomegalovirus UL82 gene product (pp71) and hDaxx regulates immediate-early gene expression and viral replication. J. Virol. 2005, 79, 7792–7802. [Google Scholar] [CrossRef]
- Tsai, K.; Thikmyanova, N.; Wojcechowskyj, J.A.; Delecluse, H.J.; Lieberman, P.M. EBV tegument protein BNRF1 disrupts DAXX-ATRX to activate viral early gene transcription. PLoS Pathog. 2011, 7, e1002376. [Google Scholar] [CrossRef]
- Bresnick, E.H.; Katsumura, K.R.; Lee, H.Y.; Johnson, K.D.; Perkins, A.S. Master regulatory GATA transcription factors: Mechanistic principles and emerging links to hematologic malignancies. Nucleic Acids Res. 2012, 40, 5819–5831. [Google Scholar] [CrossRef]
- Tsai, F.Y.; Keller, G.; Kuo, F.C.; Weiss, M.; Chen, J.; Rosenblatt, M.; Alt, F.W.; Orkin, S.H. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature 1994, 371, 221–226. [Google Scholar] [CrossRef]
- Ling, K.W.; Ottersbach, K.; van Hamburg, J.P.; Oziemlak, A.; Tsai, F.Y.; Orkin, S.H.; Ploemacher, R.; Hendriks, R.W.; Dzierzak, E. GATA-2 plays two functionally distinct roles during the ontogeny of hematopoietic stem cells. J. Exp. Med. 2004, 200, 871–882. [Google Scholar]
- Slobedman, B.; Mocarski, E.S.; Arvin, A.M.; Mellins, E.D.; Abendroth, A. Latent cytomegalovirus down-regulates major histocompatibility complex class II expression on myeloid progenitors. Blood 2002, 100, 2867–2873. [Google Scholar] [CrossRef]
- Boyes, J.; Byfield, P.; Nakatani, Y.; Ogryzko, V. Regulation of activity of the transcription factor GATA-1 by acetylation. Nature 1998, 396, 594–598. [Google Scholar] [CrossRef]
- Hayakawa, F.; Towatari, M.; Ozawa, Y.; Tomita, A.; Privalsky, M.L.; Saito, H. Functional regulation of GATA-2 by acetylation. J. Leukoc. Biol. 2004, 75, 529–540. [Google Scholar]
- Hung, H.L.; Lau, J.; Kim, A.Y.; Weiss, M.J.; Blobel, G.A. CREB-Binding protein acetylates hematopoietic transcription factor GATA-1 at functionally important sites. Mol. Cell. Biol. 1999, 19, 3496–3505. [Google Scholar]
- Ozawa, Y.; Towatari, M.; Tsuzuki, S.; Hayakawa, F.; Maeda, T.; Miyata, Y.; Tanimoto, M.; Saito, H. Histone deacetylase 3 associates with and represses the transcription factor GATA-2. Blood 2001, 98, 2116–2123. [Google Scholar] [CrossRef]
- Poole, E.; McGregor Dallas, S.R.; Colston, J.; Joseph, R.S.; Sinclair, J. Virally induced changes in cellular microRNAs maintain latency of human cytomegalovirus in CD34(+) progenitors. J. Gen. Virol. 2011, 92, 1539–1549. [Google Scholar] [CrossRef]
- Wright, E.; Bain, M.; Teague, L.; Murphy, J.; Sinclair, J. Ets-2 repressor factor recruits histone deacetylase to silence human cytomegalovirus immediate-early gene expression in non-permissive cells. J. Gen. Virol. 2005, 86, 535–544. [Google Scholar] [CrossRef]
- Sinclair, J.H.; Baillie, J.; Bryant, L.A.; Taylor-Wiedeman, J.A.; Sissons, J.G. Repression of human cytomegalovirus major immediate early gene expression in a monocytic cell line. J. Gen. Virol. 1992, 73, 433–435. [Google Scholar] [CrossRef]
- Shelbourn, S.L.; Kothari, S.K.; Sissons, J.G.; Sinclair, J.H. Repression of human cytomegalovirus gene expression associated with a novel immediate early regulatory region binding factor. Nucleic Acids Res. 1989, 17, 9165–9171. [Google Scholar] [CrossRef]
- Liu, R.; Baillie, J.; Sissons, J.G.; Sinclair, J.H. The transcription factor YY1 binds to negative regulatory elements in the human cytomegalovirus major immediate early enhancer/promoter and mediates repression in non-permissive cells. Nucleic Acids Res. 1994, 22, 2453–2459. [Google Scholar] [CrossRef]
- Zweidler-Mckay, P.A.; Grimes, H.L.; Flubacher, M.M.; Tsichlis, P.N. Gfi-1 encodes a nuclear zinc finger protein that binds DNA and functions as a transcriptional repressor. Mol. Cell. Biol. 1996, 16, 4024–4034. [Google Scholar]
- Slobedman, B.; Stern, J.L.; Cunningham, A.L.; Abendroth, A.; Abate, D.A.; Mocarski, E.S. Impact of human cytomegalovirus latent infection on myeloid progenitor cell gene expression. J. Virol. 2004, 78, 4054–4062. [Google Scholar]
- Browne, E.P.; Wing, B.; Coleman, D.; Shenk, T. Altered cellular mRNA levels in human cytomegalovirus-infected fibroblasts: Viral block to the accumulation of antiviral mRNAs. J. Virol. 2001, 75, 12319–12330. [Google Scholar] [CrossRef]
- Keller, M.J.; Wu, A.W.; Andrews, J.I.; McGonagill, P.W.; Tibesar, E.E.; Meier, J.L. Reversal of human cytomegalovirus major immediate-early enhancer/promoter silencing in quiescently infected cells via the cyclic AMP signaling pathway. J. Virol. 2007, 81, 6669–6681. [Google Scholar]
- Yuan, J.; Liu, X.; Wu, A.W.; McGonagill, P.W.; Keller, M.J.; Galle, C.S.; Meier, J.L. Breaking human cytomegalovirus major immediate-early gene silence by vasoactive intestinal peptide stimulation of the protein kinase A-CREB-TORC2 signaling cascade in human pluripotent embryonal NTera2 cells. J. Virol. 2009, 83, 6391–6403. [Google Scholar] [CrossRef]
- Prosch, S.; Wuttke, R.; Kruger, D.H.; Volk, H.D. NF-kappaB—A potential therapeutic target for inhibition of human cytomegalovirus (re)activation? Biol. Chem. 2002, 383, 1601–1609. [Google Scholar]
- Dhayalan, A.; Tamas, R.; Bock, I.; Tattermusch, A.; Dimitrova, E.; Kudithipudi, S.; Ragozin, S.; Jeltsch, A. The ATRX-ADD domain binds to H3 tail peptides and reads the combined methylation state of K4 and K9. Hum. Mol. Genet. 2011, 20, 2195–2203. [Google Scholar] [CrossRef]
- Tang, J.; Wu, S.; Liu, H.; Stratt, R.; Barak, O.G.; Shiekhattar, R.; Picketts, D.J.; Yang, X. A novel transcription regulatory complex containing death domain-associated protein and the ATR-X syndrome protein. J. Biol. Chem. 2004, 279, 20369–20377. [Google Scholar]
- De La Fuente, R.; Baumann, C.; Viveiros, M.M. Role of ATRX in chromatin structure and function: Implications for chromosome instability and human disease. Reproduction 2011, 142, 221–234. [Google Scholar] [CrossRef]
- Argentaro, A.; Yang, J.C.; Chapman, L.; Kowalczyk, M.S.; Gibbons, R.J.; Higgs, D.R.; Neuhaus, D.; Rhodes, D. Structural consequences of disease-causing mutations in the ATRX-DNMT3-DNMT3L (ADD) domain of the chromatin-associated protein ATRX. Proc. Natl. Acad. Sci. USA 2007, 104, 11939–11944. [Google Scholar]
- Benedict, C.A.; Butrovich, K.D.; Lurain, N.S.; Corbeil, J.; Rooney, I.; Schneider, P.; Tschopp, J.; Ware, C.F. Cutting edge: A novel viral TNF receptor superfamily member in virulent strains of human cytomegalovirus. J. Immunol. 1999, 162, 6967–6970. [Google Scholar]
- Montgomery, R.I.; Warner, M.S.; Lum, B.J.; Spear, P.G. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 1996, 87, 427–436. [Google Scholar] [CrossRef]
- Cheung, T.C.; Humphreys, I.R.; Potter, K.G.; Norris, P.S.; Shumway, H.M.; Tran, B.R.; Patterson, G.; Jean-Jacques, R.; Yoon, M.; Spear, P.G.; et al. Evolutionarily divergent herpesviruses modulate T cell activation by targeting the herpesvirus entry mediator cosignaling pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 13218–13223. [Google Scholar] [CrossRef]
- Poole, E.; Groves, I.; MacDonald, A.; Pang, Y.; Alcami, A.; Sinclair, J. Identification of TRIM23 as a cofactor involved in the regulation of NF-kappaB by human cytomegalovirus. J. Virol. 2009, 83, 3581–3590. [Google Scholar] [CrossRef]
- Poole, E.; King, C.A.; Sinclair, J.H.; Alcami, A. The UL144 gene product of human cytomegalovirus activates NFkappaB via a TRAF6-dependent mechanism. EMBO J. 2006, 25, 4390–4399. [Google Scholar] [CrossRef]
- Charlton, B.; Lafferty, K.J. The Th1/Th2 balance in autoimmunity. Curr. Opin. Immunol. 1995, 7, 793–798. [Google Scholar]
- Nakayama, T.; Hieshima, K.; Nagakubo, D.; Sato, E.; Nakayama, M.; Kawa, K.; Yoshie, O. Selective induction of Th2-attracting chemokines CCL17 and CCL22 in human B cells by latent membrane protein 1 of Epstein-Barr virus. J. Virol. 2004, 78, 1665–1674. [Google Scholar] [CrossRef]
- Stanton, R.J.; Baluchova, K.; Dargan, D.J.; Cunningham, C.; Sheehy, O.; Seirafian, S.; McSharry, B.P.; Neale, M.L.; Davies, J.A.; Tomasec, P.; et al. Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J. Clin. Invest. 2010, 120, 3191–3208. [Google Scholar] [CrossRef]
- Reeves, M.B. Chromatin-mediated regulation of cytomegalovirus gene expression. Virus Res. 2011, 157, 134–143. [Google Scholar] [CrossRef]
- Murphy, J.C.; Fischle, W.; Verdin, E.; Sinclair, J.H. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J. 2002, 21, 1112–1120. [Google Scholar] [CrossRef]
- Sinclair, J. Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim. Biophys. Acta 2009, 1799, 286–295. [Google Scholar]
- Reeves, M.; Murphy, J.; Greaves, R.; Fairley, J.; Brehm, A.; Sinclair, J. Autorepression of the human cytomegalovirus major immediate-early promoter/enhancer at late times of infection is mediated by the recruitment of chromatin remodeling enzymes by IE86. J. Virol. 2006, 80, 9998–10009. [Google Scholar] [CrossRef]
- Reeves, M.B.; Lehner, P.J.; Sissons, J.G.; Sinclair, J.H. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J. Gen. Virol. 2005, 86, 2949–2954. [Google Scholar] [CrossRef]
- Varnum, S.M.; Streblow, D.N.; Monroe, M.E.; Smith, P.; Auberry, K.J.; Pasa-Tolic, L.; Wang, D.; Camp, D.G., 2nd; Rodland, K.; Wiley, S.; et al. Identification of proteins in human cytomegalovirus (HCMV) particles: The HCMV proteome. J. Virol. 2004, 78, 10960–10966. [Google Scholar] [CrossRef]
- Reeves, R.; Gorman, C.M.; Howard, B. Minichromosome assembly of non-integrated plasmid DNA transfected into mammalian cells. Nucleic Acids Res. 1985, 13, 3599–3615. [Google Scholar] [CrossRef]
- Riu, E.; Chen, Z.Y.; Xu, H.; He, C.Y.; Kay, M.A. Histone modifications are associated with the persistence or silencing of vector-mediated transgene expression in vivo. Mol. Ther. 2007, 15, 1348–1355. [Google Scholar] [CrossRef]
- Maul, G.G. Initiation of cytomegalovirus infection at ND10. Curr. Top. Microbiol. Immunol. 2008, 325, 117–132. [Google Scholar] [CrossRef]
- Tavalai, N.; Stamminger, T. Intrinsic cellular defense mechanisms targeting human cytomegalovirus. Virus Res. 2011, 157, 128–133. [Google Scholar] [CrossRef]
- Ishov, A.M.; Vladimirova, O.V.; Maul, G.G. Daxx-mediated accumulation of human cytomegalovirus tegument protein pp71 at ND10 facilitates initiation of viral infection at these nuclear domains. J. Virol. 2002, 76, 7705–7712. [Google Scholar] [CrossRef]
- Wilkinson, G.W.; Kelly, C.; Sinclair, J.H.; Rickards, C. Disruption of PML-associated nuclear bodies mediated by the human cytomegalovirus major immediate early gene product. J. Gen. Virol. 1998, 79, 1233–1245. [Google Scholar]
- Tavalai, N.; Papior, P.; Rechter, S.; Leis, M.; Stamminger, T. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J. Virol. 2006, 80, 8006–8018. [Google Scholar] [CrossRef]
- Ahn, J.H.; Brignole, E.J., 3rd; Hayward, G.S. Disruption of PML subnuclear domains by the acidic IE1 protein of human cytomegalovirus is mediated through interaction with PML and may modulate a RING finger-dependent cryptic transactivator function of PML. Mol. Cell. Biol. 1998, 18, 4899–4913. [Google Scholar]
- Ahn, J.H.; Hayward, G.S. The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J. Virol. 1997, 71, 4599–4613. [Google Scholar]
- Saffert, R.T.; Kalejta, R.F. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J. Virol. 2007, 81, 9109–9120. [Google Scholar] [CrossRef]
- Groves, I.J.; Sinclair, J.H. Knockdown of hDaxx in normally non-permissive undifferentiated cells does not permit human cytomegalovirus immediate-early gene expression. J. Gen. Virol. 2007, 88, 2935–2940. [Google Scholar] [CrossRef]
- Saffert, R.T.; Penkert, R.R.; Kalejta, R.F. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J. Virol. 2010, 84, 5594–5604. [Google Scholar] [CrossRef]
- Hargett, D.; Shenk, T.E. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 20039–20044. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Reeves, M.; Sinclair, J. Regulation of Human Cytomegalovirus Transcription in Latency: Beyond the Major Immediate-Early Promoter. Viruses 2013, 5, 1395-1413. https://doi.org/10.3390/v5061395
Reeves M, Sinclair J. Regulation of Human Cytomegalovirus Transcription in Latency: Beyond the Major Immediate-Early Promoter. Viruses. 2013; 5(6):1395-1413. https://doi.org/10.3390/v5061395
Chicago/Turabian StyleReeves, Matthew, and John Sinclair. 2013. "Regulation of Human Cytomegalovirus Transcription in Latency: Beyond the Major Immediate-Early Promoter" Viruses 5, no. 6: 1395-1413. https://doi.org/10.3390/v5061395
APA StyleReeves, M., & Sinclair, J. (2013). Regulation of Human Cytomegalovirus Transcription in Latency: Beyond the Major Immediate-Early Promoter. Viruses, 5(6), 1395-1413. https://doi.org/10.3390/v5061395