Abstract
Systems biology approaches in virology aim to integrate viral and host biological networks, and thus model the infection process. The growing availability of high-throughput “-omics” techniques and datasets, as well as the ever-increasing sophistication of in silico modeling tools, has resulted in a corresponding rise in the complexity of the analyses that can be performed. The present study seeks to review and organize published evidence regarding virus-host interactions for the arenaviruses, from alterations in the host proteome during infection, to reported protein-protein interactions. In this way, we hope to provide an overview of the interplay between arenaviruses and the host cell, and lay the foundations for complementing current arenavirus research with a systems-level approach.
1. Introduction
1.1. Systems Biology as a Tool for Virus Research
Cells can be considered as complex circuits of interlinked molecular processes, whose activation or deactivation depends on stimulus-sensing, signaling, and pathway regulation through feedback loops. These processes and relationships can also be described mathematically in biological networks, using the concepts of graph theory, where cellular components such as proteins are represented as nodes or vertices, and biological relationships, for example protein-protein interactions, are indicated by connectors or edges. Recent technical advances that have allowed high-throughput and multivariate investigations of biological processes have generated a wealth of data about relationships in the cell that lends itself to such analyses. Systems biology refers to the study of how interactions between the components of a biological system give rise to the functions and behavior of that system, and can therefore be conceptually defined as a holistic approach to the investigation of biological processes.
Virus infection imposes new variables on the cell circuitry. On the one hand, viral components or ‘patterns’ are sensed by the infected cell, leading to the activation of distinct signaling cascades and alterations in cellular gene expression. In addition, the virus itself specifically targets cellular pathways as it attempts to deflect anti-viral attacks, and subvert the cellular machinery to complete its replication cycle. Such virus-induced modifications thus amount to a re-wiring of the biological circuitry of the host [1,2]. Of note, this re-wiring is often achieved by the virus targeting specific ‘hubs’ within the cell, defined as proteins with many interacting partners and/or that are central to several pathways, and which are identified in network analyses by parameters of connectivity and centrality [3,4]. In this way, even a small number of viral proteins can impact a large number of host processes and produce a complex phenotype that may be difficult to interpret without a systems-level approach.
As a practical consequence of these types of analyses, the modeling of virus-induced perturbations of the host network can be used to identify potential viral targets or hubs, which can then be experimentally tested. For example, network analysis predicted that enoyl-coA isomerase activity was important for HCV infection [5], and this was subsequently confirmed experimentally [6]. Similarly, networks can be used to predict additional consequences of any experimentally observed virus-host interaction, which may in turn provide an explanation for other observed pathogenic effects. In Coxsackie virus B3 infection, the contribution of an autocrine feedback circuit involving the proinflammatory cytokines TNF and IL-1 to virus-induced myocardial damage was initially predicted through network analysis [7]. Ultimately, identifying the most basic or direct levels of the host-virus interface in this way can suggest important targets for the development of antiviral therapies.
2. General Concepts in Systems Biology
2.1. Mathematical Concepts to Describe Biological Systems
A network is a collection of nodes or vertices connected by edges, which define pairwise relationships between the nodes. Networks are also referred to as graphs, with graph theory designating the mathematical field dedicated to the study of networks properties. Graph theory as a mathematical discipline is generally considered to date back to 1736, with the demonstration by Leonard Euler of the impossibility of finding a non-redundant path allowing the crossing of all seven bridges of Königsberg, which linked two islands to the rest of the city. By reformulating the problem in abstract terms, where land was represented as nodes and bridges as edges, Leonard Euler laid the foundations for graph theory [20]. However, despite attempts during the 20th century to introduce systems-level thinking into biology, notably with the work of Ludwig Von Bertalaffy [21,22], it is only in the early 2000’s with the development of high throughput molecular techniques, that systems biology has emerged as a significant field in biology. Attempts at modeling biological systems using existing mathematical tools revealed that, while some basic mathematical properties of networks could be ascribed biological relevance [23,24,25,26,27,28,29,30], novel analytic tools are needed in order to more aptly deal with the complexity of biological network architecture [31,32,33,34,35,36]. In this section, we will describe general network characteristics and their correlation to biological processes. For further information on systems-level thinking and mathematical concepts in systems biology, we refer the reader to the more comprehensive introductory books by Choi [37] and Alon [38].
2.2. Basic Principles in Biological Networks
In systems-level approaches to investigating cellular processes, the nodes within networks can represent any biological entity, including genes, proteins, complexes or small molecules. Edges represent the relationships between these entities, whether physical, for example receptor-ligand binding, or functional, such as the activation or inhibition of a given protein through phosphorylation (Figure 1A). A network can be composed of several connected components, i.e. groups of nodes can be connected to each other, but bear no common edges with the other subsets of nodes present within the network. A network is said to be fully connected if it is composed of a single connected component. Current knowledge of biological processes does not always allow the inclusion of specific nodes within the largest connected component of the network. These nodes may thus be found as single nodes, when no interaction has been characterized, or within a smaller connected component if information exists about their interaction with other biological elements, which also do not share an edge with any nodes of the largest connected component. It is important to note however, that this absence of connection of specific nodes to the largest connected component may not necessarily reflect biological properties, but rather may result from the lack of experimental data on the entity represented by that node [39].
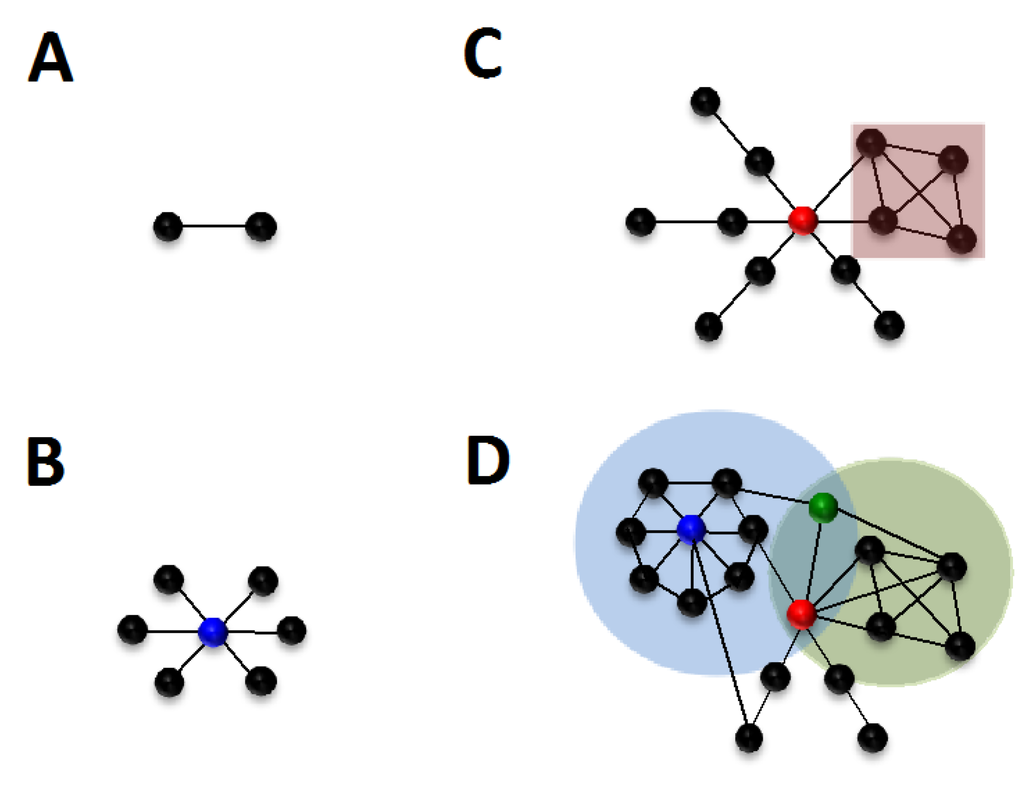
Figure 1.
Basic concepts in network analysis. (A) Two nodes are connected by an edge. (B) One node is connected to several nodes, the blue color denotes the highest connectivity (k) value within the network. (C) The red node displays high centrality (b). The red square designates a 4-node clique. (D) The network is more complex, the red node indicates the node with the highest centrality, but is not the node with the highest connectivity (blue node). The light green and blue circles show two communities in this network, and their shared nodes, the bridging node is shown as green.
2.3. Local Properties in Biological Networks
Local properties in biological networks refer to properties pertaining to a single node within the environment of its immediate neighbors. An elementary measure in networks is that of node degree or connectivity (k), which refers to the number of edges incident to that node (Figure 1B). Biological networks are usually inhomogeneous, and contain highly connected subnetworks [40], which can be defined at local or global levels. Locally, the clustering coefficient or transitivity defines the degree to which neighbors of a specific node are connected to each other [40,41]. A clique, or complete graph, designates a subset of nodes within the network which are fully connected, i.e. there is an edge connecting any two nodes within this subset [42] (Figure 1C). As such, the definition of cliques has been shown to strongly correlate with that of protein complexes or functional modules within networks [26].
2.4. Global Properties of Nodes and Edges
These properties describe information about the relative importance of network elements to the structure of the network. A common attribute within the network is the definition of the shortest path or geodesic, which is defined as the minimum distance between any two nodes in the network. From this the mean path length can be calculated by averaging all shortest paths for any two nodes in the network. In biological systems, path length can be used to understand the reactivity of pathways, for example a short path length within a signaling cascade ensures an efficient “information” flow, where only a small number of intermediate steps are required between the initial sensing of a given stimulus and the induction of a biological response, for example induction of gene expression [27].
Centrality or vertex betweenness (b) measures the number of shortest paths that pass through a given vertex (Figure 1C). This property theoretically describes the breadth of pathways a protein is involved with, and nodes with high scores are sometimes referred to as bottlenecks. High-betweenness or bottleneck-ness is a measure of the essentiality of a given protein to the biological system considered [28,29], and has been shown for protein-protein interaction networks to correlate with a regulatory function central to several pathways [30]. Centrality within the network can also be determined through edge betweenness, which calculates the number of shortest paths a specific edge participates in, and similarly to node betweenness, describes the essentiality of the biological relationship specified by the edge to the biological system considered.
2.5. Understanding Network Structure: Defining Communities
The goal of systems biology is to understand how the constitutive components of a network interact with each other. However, one of the main problem in systems biology is the mathematical characterization of such components in a way that is relevant to the elucidation of biological processes. In network theory, there are several ways to define subcomponents of a network, which we will refer to as communities, but which can also be designated as clusters, cohesive groups or modules. Most of the algorithms developed in graph theory rely on the definition of separated communities [43,44,45], which means that any node can only belong to one community. However, a biological element, for example a protein, can be part of several complexes or several pathways. In order to provide a better model for finding communities within biological networks, Palla and coworkers developed a novel algorithm, called the clique percolation method, which allows the identification of overlapping communities [31]. Analysis of a human infectome network using this algorithm revealed the unambiguous assignment of most communities to at least one cellular function [3], however reliable mathematical identification of complex biological associations, for example pathways, remains unresolved. Finally, the identification of shared nodes between communities also led to the characterization of bridging nodes, which, contrary to hubs which may be also shared by communities, display low local connectivity, but high centrality, and provide a link between one or more highly connected components [3] (Figure 1D).
3. Building and Analyzing an Arenavirus-Host Network
3.1. Datasets for the Construction of the Arenavirus-Host Network
Data pertaining to the arenavirus-host interplay was compiled from published studies on arenavirus-host protein-protein interactions [8,9,10,11,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62], viral requirements for specific host factors [63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83], viral inhibition of host proteins [84,85,86], kinomics and proteomics studies of cellular changes in response to infection [12,13,14,15,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107]. Data was also retrieved from studies describing co-localization of viral and host factors [108] or cellular factors that inhibit viral replication [109,110]. In total, 304 cellular proteins were identified as playing a role in arenavirus infection from this literature analysis (through September 2012). To simplify nomenclature, this primary set of 304 host proteins will be referred to as ‘host targets’ in this article. The proteins’ identities, as well as details of the nature of the virus-host interactions, and the specific viral systems in which the interaction was observed, are available in Table S1.
The usefulness of any network will of course only be as good as the primary data used to create it. Therefore, we have carefully curated the primary literature to establish that the experimental data reasonably supported any conclusions made in a publication about interactions, before including it in the dataset. However, we have not assigned any weight to a given interaction. Instead, details are provided that a user can access to determine for his or herself the strength of the data supporting a given interaction, and are available through the Description and References section within Table S1, or through the Description and Reference attribute within the network. In both cases, information is provided about whether the interaction was identified during virus infection, or through a sub-viral system such as the co-expression in cells of specific viral proteins.
A primary network was constructed, organized around the 4 arenaviral proteins and the 304 curated host targets. Protein-protein interactions and functional pathway information was then retrieved for each host target by searching the public databases available through Pathway Commons [111], and was merged with the primary network. Such functional enrichment of the primary network therefore allows a comprehensive overview of the host cell pathways affected by, or required during, arenavirus infection, based on current knowledge curated from the literature. A schematic of the network building process is shown in Figure 2, and the resulting arenavirus-host protein network is available as supplemental data (Supplemental Data—Arenavirus-Host Full Network.cys).
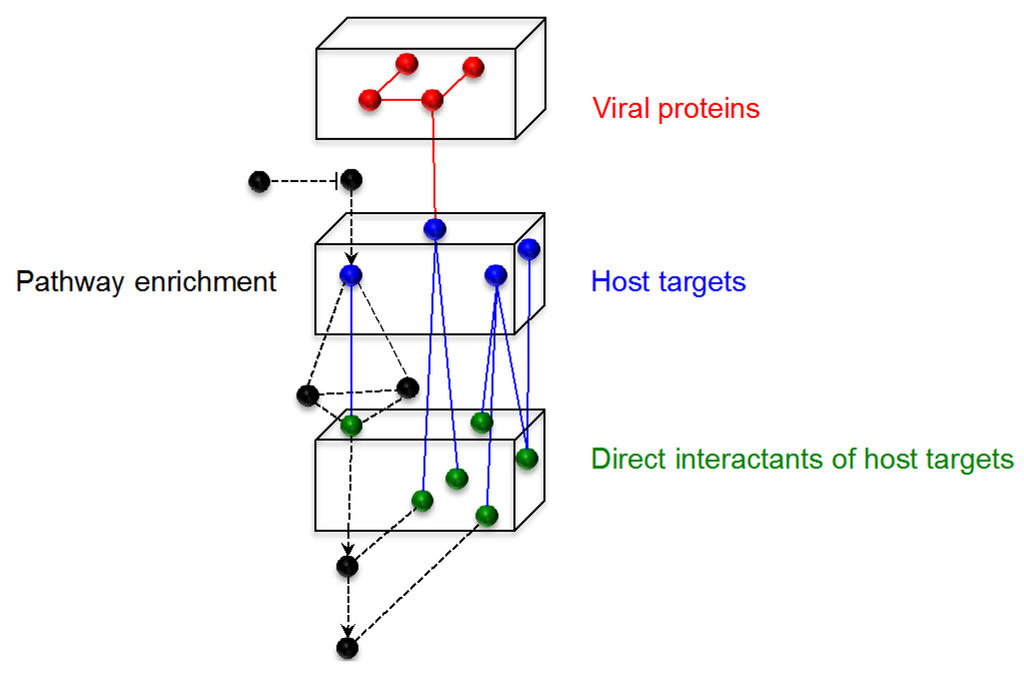
Figure 2.
Construction of the arenavirus-host network. Schematic representation of steps in the construction of the arenavirus-host network. The four arenavirus proteins are represented by red nodes, host targets identified in the literature are blue nodes, and the directly interacting partners of these host targets, obtained from databases, are represented by green nodes. Black nodes represent proteins uploaded into the network through pathway enrichment performed on the host targets, and may contain, in addition to protein-protein interaction data, additional information about functional interactions between proteins. Edges indicate a relationship between two proteins, such as a direct protein-protein interaction or a process of protein modification, such as phosphorylation or ubiquitination.
3.2. Programs for Building and Analyzing the Network
The arenavirus-host network was constructed in Cytoscape 2.8 [112,113], a freely available software for complex network visualization [114]. Retrieval of host protein-protein interactions, as well as pathway enrichment, was performed using the cPath software [115], which enabled access from the Cytoscape platform to the Pathway Commons Web Service [111], a web-based interface that allows the simultaneous mining of biological databases such as Reactome or BioGrid. Network visualization was also carried out in Cytoscape 2.8. Details on how to use the Cytoscape software, as well as links to advanced tutorials, are available in Supplemental File II.
General mathematical properties of the network, such as node connectivity and centrality, were determined using the R package igraph for network analysis [116]. A link to the igraph site, which includes information on how to download and use this software is provided in Supplemental File II.
3.3. Embedding Information (Attributes) in the Network
Any information regarding nodes and edges is encoded within the network as attributes. Different categories of information, such as gene or protein name, role in arenavirus infection, or the pathways the protein is active in, are encoded as individual attributes. For example the gene name of any given node can be found under the attribute “biopax.xref.GENE_SYMBOL”. This information can be retrieved within Cytoscape by directly clicking on the node or edge of interest, which will result in the display of attributes values in the data panel. These attributes can also be used to search for specific elements within the network, such as nodes involved in a given biological process or pathway. For example, the user can configure Cytoscape search options, by clicking on the icon immediately to the right of the search box, to be based on the attribute category “nodes.ListPathway”. The user can then type in the desired request in the search box, such as the term “translation”, which will highlight all nodes involved in this pathway within the network, and provide a list of these nodes in the data panel. Attributes can also be used to specify differential displays within the network (such as the node color or size, but also the layout of the selected nodes), or can be used to create subnetworks through the use of filters (discussed in Section 3.5).
3.4. Virus-Specific Elements Encoded within the Network
The arenaviruses are distinguished serologically as belonging to either the Old World (LCMV complex) or the New World (Tacaribe complex) viruses, with the New World viruses being further divided into clades, A, B and C. The members of these groups have both common and distinct features, and pathogenic and non-pathogenic viruses are present in both of the two major serological complexes. The network we built includes all relevant information about the arenavirus-host relationships that we obtained from the literature, and includes observations about the following viruses: Old World (LASV, LCMV, MOPV, MOBV), New World clade A (PICV), clade B (JUNV, MACV, GTOV, AMAV, TCRV, SABV), clade C (OLVV, LATV), and clade A/B (WWAV). The network also contains information, where available, about different strains of viruses used, with human pathogenic strains denoted with a subscript ‘v’ and non-pathogenic strains with the subscript ‘a’, for example the attenuated PICV variant P2 is denoted PICVa, and the virulent PICV variant P18 is denoted as PICVv [117,118]. The information for each investigated virus is reported as string values (TRUE/ FALSE/ Not tested), indicating whether the interaction or the node has been confirmed for the specific virus indicated, or if no studies have yet evaluated the role of a host protein interaction for that arenavirus family member.
Additional information linking the interaction to a summary of findings, and a reference to the original published report for a specific virus-host relationship, is also accessible within the network through the attribute “nodes.Description and References”.
3.5. Using Filter-Set Subnetworks
The advantage of building a full network, that includes information from all published reports, is that the information pertaining to all arenaviruses is encoded in one single file. This full or master network should be considered a work in progress, and future experimental data will both confirm and add to the value of the relationships embedded in the present version (September 2012). The network is also paradoxical, since it includes data that is both true and false for different viral systems. For example, the binding of GP to two known receptors for the arenaviruses, human transferrin receptor 1 and alpha-dystroglycan [50,51,52,53,54,55,56,57], is both true and false, depending on the virus strain. Therefore, in order to perform relevant systems-level analyses for the arenaviruses, the network should be customized through the use of filters, from which a relevant subnetwork can be derived through user-driven curation, and depending on investigative needs.
Filters can be set within from within the Cytoscape software. The available filters can be applied to any attribute that is assigned to either nodes or edges (for example, to select only viral host targets), as well as topology (for example, to select neighbors at a given distance from nodes of interest). Multiple filters can be combined through the use of Boolean links: AND (–node or edge selected must pass both filters), OR (–node or edge selected must pass at least one filter) and NOT (–to exclude nodes or edges).
As a practical example, if a user wanted to generate a network displaying only the proteins that have been shown to play a role during infection by the pathogenic strains of JUNV, the following filter would be applied: nodes. JUNVv > TRUE. However, since this filter will only select a subset of the host target proteins, information and context could be lost. To put this data into a more relevant context, we have encoded a “Connect pathways” attribute. In this case, the user can select both the subset of viral host targets that are specific to pathogenic JUNV strains, as well as maintaining the integrity of the pathways that these proteins are involved in, by applying the following filter: nodes.Connect pathways >JUNVv. Finally, the specific subnetwork can be created and saved as a separate network through the path: File > New > Network > From selected nodes, All edges, and further analyzed using Cytoscape Network Analyzer, or other network analysis tools such as igraph.
3.6. Identifying Potential Viral Targets Through Centrality and Connectivity Values
Using the tools described above, we generated a subnetwork to investigate common viral host targets in arenavirus infection, and to analyze the role of these proteins in the context of the pathways they contribute to. As highlighted previously, viruses tend to target highly connected nodes within networks [3,4]. This can explain how viruses expressing only a small subset of proteins can subvert major cellular pathways, through the strategic targeting of these central elements. In order to identify potential hubs within the network, we plotted centrality values vs connectivity values for all nodes present in the common subnetwork (Figure 3B). The node with the highest connectivity value was found to be p53, a master regulator of cellular processes, whose steady-state levels are decreased during arenavirus infection [15], although no mechanism for this observation has yet been identified. Other hubs, which have already been identified as arenaviral host targets, include nucleophosmin [15], involved in ribosome biogenesis, and cytoskeletal elements such as vimentin [15], tubulin [70,71,72,77] and actin [13,70,71,72]. Amongst hub proteins that have not been previously identified as viral host targets, we found general adapters in signal transduction pathways such as YWHAZ or SHC1, as well as the master regulator AKT1 kinase, which is involved in many cellular processes such as metabolism, proliferation, cell survival and growth, and angiogenesis. These provide interesting leads for further investigation, since any direct effect on these proteins could potentially explain downstream phenotypes previously reported.
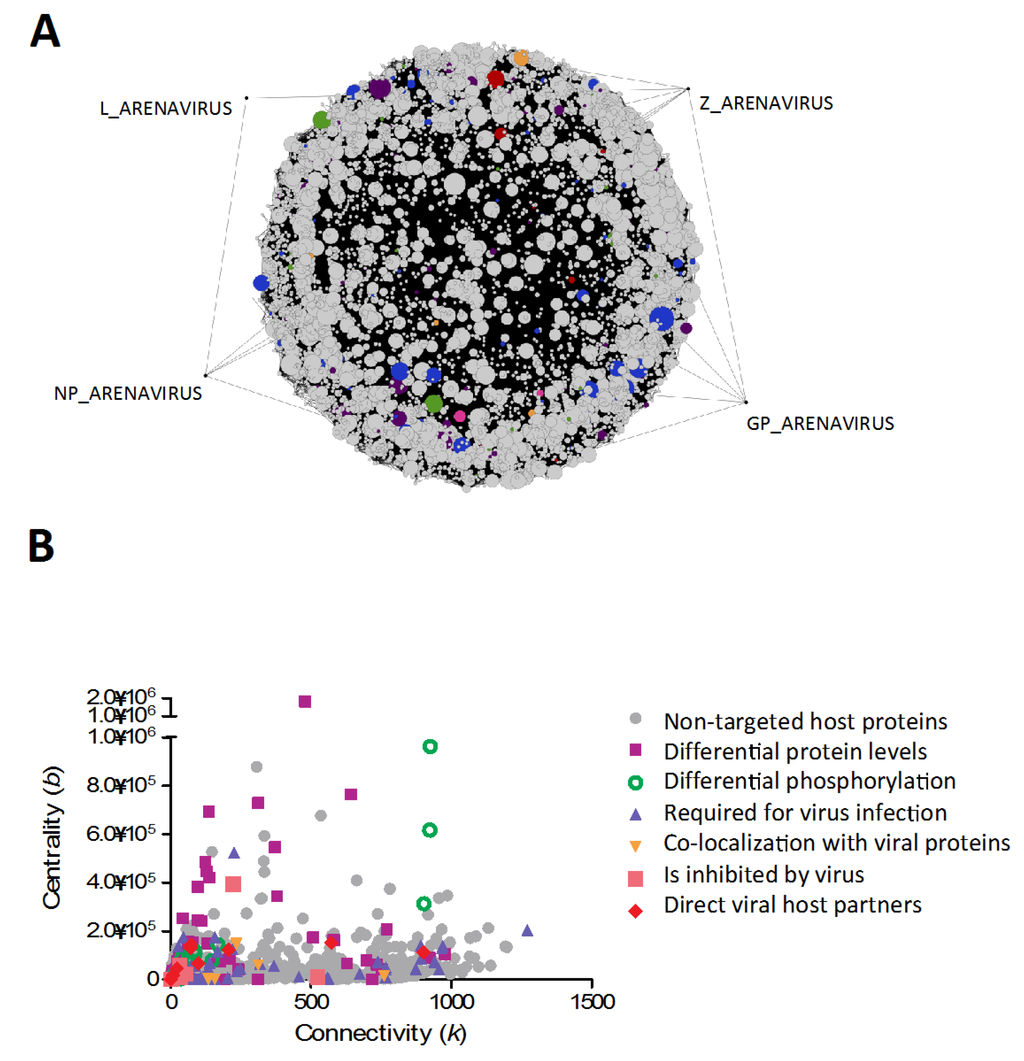
Figure 3.
Common element network of arenavirus-host interactions, describing pathways targeted/perturbed during infection by all arenaviruses. (A) In this graphical representation, the arenaviral proteins (GP, NP, L and Z) are highlighted by dragging out of the main connected components, to facilitate visualization. Nodes are color-coded depending on role during arenavirus infection (see key in part b). Node size was correlated to node degree, to facilitate visualization of their relative importance within the network. Interested users can find the filter used to generate this network pre-loaded within the supplemental network file (Supplemental Data: Arenavirus-Host Full Network.cys; Filter name: Common elements of arenavirus replication).(B) Graphical representation of characteristics of nodes (connectivity and centrality values). Nodes of particular importance are revealed by high values in both parameters. The nodes are color-coded depending on their identified role during virus infection.
5. Convergence Between Different Virus-Host Networks
Systems biology can also be used to compare different viral-host systems, in an attempt to identify common targets of viral infection, and thus highlight convergent mechanisms of viral pathogenesis. For example, Bowick and McAuley reported on a systems-level meta-analysis of high-throughput datasets from hemorrhagic fever systems of various viral aetiologies [121]. The data analyzed included proteomics studies of responses to PICV infection [14] and transcriptomics analyses carried out during LCMV infection [16,17], both of which are used as models for Lassa fever, as well as microarray analyses from heterologous viral systems such as the filovirus Ebola virus (EBOV) [122], and bunyavirus Rift Valley Fever virus (RVFV) [123]. This analysis resulted in the identification of cyclooxygenase-2 (COX-2) as a common viral target, which was downregulated during LCMV and RVFV infections, but upregulated during infection by EBOV [121]. As noted by the authors, this result can further be extended to infection by the flavivirus Dengue virus, which was found to induce COX-2 expression [124]. COX-2 catalyzes the production of prostaglandin precursors, which are subsequently converted to active prostaglandin molecules such as prostacyclin PGI2, a vasodilator, by tissue-specific isomerases [125]. This finding of convergent targeting of prostaglandin pathways by hemorrhagic fever viruses from different families suggests an exciting new area of research towards unraveling the basis of hemorrhagic fever syndromes.
Systems-level combining of virus-host networks of different viral families has also been undertaken in recently published studies [3,126]. One study investigating common host targets of 70 viral proteins from 30 viruses uncovered the ubiquitous viral targeting of hnRNPU, phosphatidylinositol-3 OH kinase (PI3K), the WNK kinase family, and the ubiquitin-specific peptidase 19 [126]. Interestingly, PI3K has already been identified as essential for arenavirus infection [66,67,68], however, a direct interaction of PI3K with a viral partner has not yet been demonstrated. Moreover, hnRNPU, USP19 and WNK kinases 1 and 4 are also present in the arenavirus-host network, indicating their involvement in pathways identified for viral host targets.
In an another study, Navratil and coworkers developed a model containing virus-host interactions for 110 viruses from 8 different viral families, constituting the most comprehensive pan-viral “infectome” network available at present. Further annotation of host proteins regarding their known involvement in diseases revealed the significant association of 57 viruses with 34 diseases [3]. These studies are part of a current trend to investigate convergence in virus-host networks, and establish links through systems-level analysis between molecular characteristics and pathogenesis. One goal of these approaches is to generate a global viral infectome, with which to model general characteristics of the infected cell, and thereby identify suitable targets for the development of pan-viral therapeutic strategies.
6. Systems Biology to Suggest Therapeutic Targets
Therapeutic strategies that target cellular processes essential for virus infection are attractive since they are less likely to result in viral evolution towards drug-resistance [127]. In this way, biological networks can be used to identify candidate targets based on the mathematical properties of specific nodes. In network theory, situations where a small subset of proteins have a large number of interactions, while the majority of nodes have lower connectivity within the network, correlate with robustness against random attacks, which is characterized by the removal of any node or edge from the network. This robustness can be explained by the lower probability for a node with a higher number of interactions to be targeted in random attacks, meaning that the overall connectivity of the network is conserved. Analyses of these properties can be harnessed in biological networks towards the determination and testing of therapeutic targets in silico - effectively simulating an ‘attack’. In addition to hubs, bridging nodes (lower connectivity but connecting highly connected components), should be considered in attack analyses, since their removal could result in disconnection of the network.
Network analysis of large-scale RNAi screens has been used to identify a correlation between the preference for viruses to interact with highly connected host proteins and the functional essentiality of these proteins in virus infection [3]. Furthermore, amongst the lower-connected proteins represented in those screens, a predominance of bridging elements was observed. Interestingly, when targeting bridging nodes within the network, a lower impact on network topology was observed than when targeting central nodes [3]. It has been argued that targeting bridging proteins instead of highly connected nodes might result in lower toxicity within the host [3,128,129]. While it is true that extensive network disruption is likely to be detrimental to the host, only empirical testing will truly determine drug toxicity versus efficacy, and indeed whether a temporary toxicity can be tolerated in an attempt to thwart a severe viral disease such as the arenviral hemorrhagic fevers.
Systems biology is also used in pharmacogy for drug discovery or drug repositioning. By network analysis of phenotypic side-effect similarity of chemically dissimilar drugs, a recent demonstration was made of the power of systems biology to uncover shared cellular targets, and therefore potential alternative applications for existing drugs [130]. As networks grow in sophistication and power, reliable systems-level models of the infected cell or organism might not be so far ahead. And since the pathogenic arenaviruses require BSL4 containment, the pre-screening, in silico, of the antiviral potential of existing drugs appears all the more an attractive option.
7. Conclusions
Systems biology has been readily embraced by virologists, with the creation of databases entirely devoted to virus-host relationships such as VirhostNet [131] or VirusMint [132], as well as virus-specific ones such as HCVpro [133], and with the development of increasingly comprehensive networks being developed for several viruses [3,134,135]. In this study, we provide the first comprehensive synthesis of all published accounts of arenavirus-host protein interactions (September 2012) and have used this information to build a “starter kit” arenavirus-host network. It is expected that future unbiased studies of protein-protein interactions, including proteomics screens, will significantly improve its accuracy.
As -omics data become more abundant and refined, the next challenge for systems biology will be to integrate datasets pertaining to different biological entities (small molecules, proteins, genes) into a single network. At the same time, modeling of cellular processes, as well as viral infection, will need to take into account spatial (subcellular localization) and temporal (time post infection) parameters, in order to generate dynamic networks that more accurately reflect cellular processes. Also, in the (not-so-distant?) future, it might be possible to generate a reliable in silico model of arenavirus infection.
Acknowledgments
This work was supported by PHS grant U54 AI065359 (NIAD, NIH) to the Pacific Southwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Files
Supplementary File 1Supplementary File 2References and Notes
- Pawson, T.; Warner, N. Oncogenic re-wiring of cellular signaling pathways. Oncogene 2007, 26, 1268–1275. [Google Scholar] [CrossRef]
- Davey, N.E.; Trave, G.; Gibson, T.J. How viruses hijack cell regulation. Trends Biochem. Sci. 2011, 36, 159–169. [Google Scholar]
- Navratil, V.; de Chassey, B.; Rabourdin-Combe, C.; Lotteau, V. When the human viral infectome and diseasome networks collide: towards a systems biology platform for the aetiology of human diseases. BMC Syst. Biol. 2011, 5, 13. [Google Scholar] [CrossRef]
- Dyer, M.D.; Murali, T.M.; Sobral, B.W. The landscape of human proteins interacting with viruses and other pathogens. PLoSPathog. 2008, 4, e32. [Google Scholar]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010, 6, e1000719. [Google Scholar]
- Rasmussen, A.L.; Diamond, D.L.; McDermott, J.E.; Gao, X.; Metz, T.O.; Matzke, M.M.; Carter, V.S.; Belisle, S.E.; Korth, M.J.; Waters, K.M.; et al. Systems virology identifies a mitochondrial fatty acid oxidation enzyme, dodecenoyl coenzyme A delta isomerase, required for hepatitis C virus replication and likely pathogenesis. J. Virol. 2011, 85, 11646–11654. [Google Scholar]
- Garmaroudi, F.S.; Marchant, D.; Si, X.; Khalili, A.; Bashashati, A.; Wong, B.W.; Tabet, A.; Ng, R.T.; Murphy, K.; Luo, H.; et al. Pairwise network mechanisms in the host signaling response to coxsackievirus B3 infection. Proc. Natl. Acad. Sci. USA 2010, 107, 17053–17058. [Google Scholar]
- Borden, K.L.; Campbell Dwyer, E.J.; Salvato, M.S. An arenavirus RING (zinc-binding) protein binds the oncoproteinpromyelocyte leukemia protein (PML) and relocates PML nuclear bodies to the cytoplasm. J. Virol. 1998, 72, 758–766. [Google Scholar]
- Campbell Dwyer, E.J.; Lai, H.; MacDonald, R.C.; Salvato, M.S.; Borden, K.L. The lymphocytic choriomeningitis virus RING protein Z associates with eukaryotic initiation factor 4E and selectively represses translation in a RING-dependent manner. J. Virol. 2000, 74, 3293–3300. [Google Scholar] [CrossRef]
- Kentsis, A.; Dwyer, E.C.; Perez, J.M.; Sharma, M.; Chen, A.; Pan, Z.Q.; Borden, K.L. The RING domains of the promyelocytic leukemia protein PML and the arenaviral protein Z repress translation by directly inhibiting translation initiation factor eIF4E. J. Mol. Biol. 2001, 312, 609–623. [Google Scholar] [CrossRef]
- Borden, K.L.; Campbell-Dwyer, E.J.; Carlile, G.W.; Djavani, M.; Salvato, M.S. Two RING finger proteins, the oncoprotein PML and the arenavirus Z protein, colocalize with the nuclear fraction of the ribosomal P proteins. J. Virol. 1998, 72, 3819–3826. [Google Scholar]
- Bowick, G.C.; Soman, K.V.; Wang, H.; Aronson, J.F.; Luxon, B.A.; Lomas, L.O.; Gorenstein, D.G.; Herzog, N.K. Proteomic analysis of Pichinde virus infection identifies differential expression of prothymosin-alpha. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef]
- Bowick, G.C.; Spratt, H.M.; Hogg, A.E.; Endsley, J.J.; Wiktorowicz, J.E.; Kurosky, A.; Luxon, B.A.; Gorenstein, D.G.; Herzog, N.K. Analysis of the differential host cell nuclear proteome induced by attenuated and virulent hemorrhagic arenavirus infection. J. Virol. 2009, 83, 687–700. [Google Scholar]
- Bowick, G.C.; Fennewald, S.M.; Elsom, B.L.; Aronson, J.F.; Luxon, B.A.; Gorenstein, D.G.; Herzog, N.K. Differential signaling networks induced by mild and lethal hemorrhagic fever virus infections. J. Virol. 2006, 80, 10248–10252. [Google Scholar] [CrossRef]
- Bowick, G.C.; Fennewald, S.M.; Scott, E.P.; Zhang, L.H.; Elsom, B.L.; Aronson, J.F.; Spratt, H.M.; Luxon, B.A.; Gorenstein, D.G.; Herzog, N.K. Identification of differentially activated cell-signaling networks associated with Pichinde virus pathogenesis by using systems kinomics. J. Virol. 2007, 81, 1923–1933. [Google Scholar]
- Djavani, M.; Crasta, O.R.; Zhang, Y.; Zapata, J.C.; Sobral, B.; Lechner, M.G.; Bryant, J.; Davis, H.; Salvato, M.S. Gene expression in primate liver during viral hemorrhagic fever. Virol. J. 2009, 6, 20. [Google Scholar] [CrossRef]
- Djavani, M.M.; Crasta, O.R.; Zapata, J.C.; Fei, Z.; Folkerts, O.; Sobral, B.; Swindells, J.; Bryant, J.; Davis, H.; Pauza, C.D.; et al. Early blood profiles of virus infection in a monkey model for Lassa fever. J. Virol. 2007, 81, 7960–7973. [Google Scholar]
- Muller, S.; Geffers, R.; Gunther, S. Analysis of gene expression in Lassa virus-infected HuH-7 cells. J. Gen. Virol. 2007, 88, 1565–1575. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Kalisak, E.; Trauger, S.; Manchester, M.; Siuzdak, G. Response and recovery in the plasma metabolome tracks the acute LCMV-induced immune response. J. Proteome Res. 2009, 8, 3578–3587. [Google Scholar] [CrossRef]
- Biggs, N.; Lloyd, E.; Wilson, R. Graph Theory 1736–1936; Clarendon Press: Oxford, UK, 1976. [Google Scholar]
- von Bertalanffy, L. An outline of general system theory. Br. J. Philos. Sci. 1950, 1, 139–164. [Google Scholar]
- von Bertalanffy, L. General system theory—A new approach to unity of science. Hum. Biol. 1951, 23, 303–361. [Google Scholar]
- Wagner, A.; Fell, D.A. The small world inside large metabolic networks. Proc. Biol. Sci. 2001, 268, 1803–1810. [Google Scholar] [CrossRef]
- Jeong, H.; Tombor, B.; Albert, R.; Oltvai, Z.N.; Barabasi, A.L. The large-scale organization of metabolic networks. Nature 2000, 407, 651–654. [Google Scholar]
- Maslov, S.; Sneppen, K. Specificity and stability in topology of protein networks. Science 2002, 296, 910–913. [Google Scholar] [CrossRef]
- Spirin, V.; Mirny, L.A. Protein complexes and functional modules in molecular networks. Proc. Natl. Acad. Sci. USA 2003, 100, 12123–12128. [Google Scholar] [CrossRef]
- de la Fuente, A.; Fotia, G.; Maggio, F.; Mancosu, G.; Pieroni, E. Insights into biological information processing: Structural and dynamical analysis of a Human Protein Signaling network. J. Physics A 2008, 41, 224013. [Google Scholar] [CrossRef]
- Joy, M.P.; Brock, A.; Ingber, D.E.; Huang, S. High-betweenness proteins in the yeast protein interaction network. J. Biomed. Biotechnol. 2005, 96–103. [Google Scholar]
- Hahn, M.W.; Kern, A.D. Comparative genomics of centrality and essentiality in three eukaryotic protein-interaction networks. Mol. Biol. Evol. 2005, 22, 803–806. [Google Scholar] [CrossRef]
- Fox, A.D.; Hescott, B.J.; Blumer, A.C.; Slonin, D.K. Connectedness of PPI network neighborhood identifies regulatory hub proteins. Bioinformatics 2011, 27, 1135–1142. [Google Scholar] [CrossRef]
- Palla, G.; Dereny, I.; Farkas, I.; Vicsek, T. Uncovering the overlapping community structure of complex networks in nature and society. Nature 2005, 435, 814–818. [Google Scholar]
- Kim, W.; Li, M.; Wang, J.; Pan, Y. Biological network motif detection and evaluation. BMC Genom. 2010, 11, S10. [Google Scholar]
- Opgen-Rhein, R.; Strimmer, K. Learning causal networks from systems biology time course data: An effective model selection procedure for the vector autoregressive process. BMC Bioinformatics 2007, 8, S2–S3. [Google Scholar]
- Cui, J.; DeLuca, T.F.; Jung, J.Y.; Wall, D.P. Phylogenetically informed logic relationships improve detection of biological network organization. BMC Bioinformatics 2011, 12, 476. [Google Scholar] [CrossRef]
- Tang, B.; Chen, S.S.; Jin, V.X. Integrative identification of core genetic regulatory modules via a structural model-based clustering method. Int. J. Comput. Biol. Drug Des. 2011, 4, 127–146. [Google Scholar] [CrossRef]
- Marin-Sanguino, A.; Gupta, S.K.; Voit, E.O.; Vera, J. Biochemical pathway modeling tools for drug target detection in cancer and other complex diseases. Methods Enzymol. 2011, 487, 319–369. [Google Scholar]
- Choi, S. Introduction to Systems Biology; Humana press Inc.: New York, NY, USA, 2007. [Google Scholar]
- Alon, U. An Introduction to Systems Biology: Design Principles of Biological Circuits; Chapman and Hall/CRC: Oxford, UK, 2007. [Google Scholar]
- von Mering, D.J.; Krause, R.; Snel, B.; Cornell, M.; Oliver, S.G.; Fields, S.; Bork, P. Comparative assessment of large-scale data sets of protein-protein interactions. Nature 2002, 417, 399–403. [Google Scholar]
- Watts, D.J.; Strogatz, S.H. Collective dynamics of “small-world” networks. Nature 1998, 393, 440–442. [Google Scholar]
- Holland, P.W.; Leinhardt, S. Transitivity in structural models of small groups. Comparative Group Studies 1971, 2, 107–124. [Google Scholar]
- Luce, R.D.; Perry, A.D. A method of matrix analysis of group structure. Psychometrika 1949, 14, 95–116. [Google Scholar] [CrossRef]
- Newman, M.E.J. Detecting community structure in networks. Eur. Phys. J. B 2004, 38, 321–330. [Google Scholar] [CrossRef]
- Girvan, M.; Newman, M.E.J. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA. 2002, 99, 7821–7826. [Google Scholar] [CrossRef]
- Radicchi, F.; Castellano, C.; Cecconi, F.; Loreto, V.; Parisi, D. Defining and identifying communities in networks. Proc. Natl. Acad. Sci. USA. 2004, 101, 2658–2663. [Google Scholar] [CrossRef]
- Shtanko, O.; Watanabe, S.; Jasenosky, L.D.; Watanabe, T.; Kawaoka, Y. ALIX/AIP1 is required for NP incorporation into Mopeia virus Z-induced virus-like particles. J. Virol. 2011, 85, 3631–3641. [Google Scholar] [CrossRef]
- Volpon, L.; Osborne, M.J.; Capul, A.A.; de la Torre, J.C.; Borden, K.L. Structural characterization of the Z RING-eIF4E complex reveals a distinct mode of control for eIF4E. Proc. Natl. Acad. Sci. USA. 2010, 107, 5441–5446. [Google Scholar]
- Labudova, M.; Tomaskova, J.; Skultety, L.; Pastorek, J.; Pastorekova, S. The nucleoprotein of lymphocytic choriomeningitis virus facilitates spread of persistent infection through stabilization of the keratin network. J. Virol. 2009, 83, 7842–7849. [Google Scholar] [CrossRef]
- Pythoud, C.; Rodrigo, W.W.; Pasqual, G.; Rothenberger, S.; Martínez-Sobrido, L.; de la Torre, J.C.; Kunz, S. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase IKK(varepsilon). J. Virol. 2012, 86, 7728–7738. [Google Scholar]
- Radoshitzky, S.R.; Abraham, J.; Spiropoulou, J.F.; Kuhn, J.H.; Nguyen, D.; Li, W.; Nagel, J.; Schmidt, P.J.; Nunberg, J.H.; Andrews, N.C.; et al. Transferrin receptor 1 is a cellular receptor for New World hemorrhagic fever arenaviruses. Nature 2007, 446, 92–96. [Google Scholar]
- Flanagan, M.L; Oldenburg, J.; Reignier, T.; Holt, N.; Hamilton, G.A.; Martin, V.K.; Cannon, P.M. New world clade B arenaviruses can use transferrin receptor 1 (TfR1)-dependent and -independent entry pathways, and glycoproteins from human pathogenic strains are associated with the use of TfR1. J. Virol. 2008, 82, 938–948. [Google Scholar]
- Reignier, T.; Oldenburg, J.; Flanagan, M.L.; Hamilton, G.A.; Martin, V.K.; Cannon, P.M. Receptor use by the Whitewater Arroyo virus glycoprotein. Virology 2008, 371, 439–446. [Google Scholar] [CrossRef]
- Droniou-Bonzom, M.E.; Reignier, T.; Oldenburg, J.E.; Cox, A.U.; Exline, C.M.; Rathbun, J.Y.; Cannon, P.M. Substitutions in the glycoprotein (GP) of the Candid#1 vaccine strain of Junin virus increase dependence on human transferrin receptor 1 for entry and destabilize the metastable conformation of GP. J. Virol. 2011, 85, 13457–13462. [Google Scholar]
- Cao, W.; Henry, M.D.; Borrow, P.; Yamada, H.; Elder, J.H.; Ravkov, E.V; Nichol, S.T.; Compans, R.W.; Campbell, K.P.; Oldstone, M.B. Identification of alpha-dystroglycan as a receptor for lymphocytic chriomeningitis virus and Lassa fever virus. Science 1998, 282, 2079–2081. [Google Scholar]
- Spiropoulou, C.F.; Kunz, S.; Rollin, P.E.; Campbell, K.P.; Oldstone, M.B. New World arenavirusclade C, but not clade A and B viruses, utilizes α-dystroglycan as its major receptor. J. Virol. 2002, 76, 5140–5146. [Google Scholar] [CrossRef]
- Abraham, J.; Kwong, J.A.; Albariño, C.G.; Lu, J.G.; Radoshitzky, S.R.; Salazar-Bravo, J.; Farzan, M.; Spiropoulou, C.F.; Choe, H. Host-species transferrin receptor 1 orthologs are cellular receptors for non-pathogenic new World clade B arenaviruses. PLoS Pathog. 2009, 5, e1000358. [Google Scholar]
- Rojek, J.M.; Spiropoulou, C.F.; Kunz, S. Characterization of the cellular receptors for the South American hemorrhagic fever viruses Junin, Guanarito and Machupo. Virology 2006, 349, 476–491. [Google Scholar] [CrossRef]
- Shimojima, M.; Kawaoka, Y. Cell surface molecules involved in infection mediated by Lymphocytic choriomeningitis virus glycoprotein. J. Vet. Med. Sci. 2012, 10, 1363–1366. [Google Scholar] [CrossRef]
- Shimojima, M.; Ströher, U.; Ebihara, H.; Feldmann, H.; Kawaoka, Y. Identification of cell surface molecules involved in dystroglycan-independent Lassa virus cell entry. J. Virol. 2012, 86, 2067–2078. [Google Scholar]
- Martínez, M.G.; Prado Acosta, M.; Candurra, N.A.; Ruzal, S.M. S-layer proteins of Lactobacillus acidophilus inhibits JUNV infection. Biochem. Biophys. Res. Commun. 2012, 422, 590–595. [Google Scholar] [CrossRef]
- Fan, L.; Briese, T.; Lipkin, W.I. Z proteins of New World arenaviruses bind RIG-I and interfere with type I interferon induction. J. Virol. 2010, 84, 1785–1791. [Google Scholar] [CrossRef]
- Zhou, S.; Cerny, A.M.; Zacharia, A.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Finberg, R.W. Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomeningitis virus. J. Virol. 2010, 84, 9452–9462. [Google Scholar] [CrossRef]
- Perez, M.; Craven, R.C.; de la Torre, J.C. The small RING finger protein Z drives arenavirus budding: Implications for antiviral strategies. Proc. Natl. Acad. Sci. USA 2003, 100, 12978–12983. [Google Scholar]
- Urata, S.; Noda, T.; Kawaoka, Y.; Yokosawa, H.; Yasuda, J. Cellular factors required for Lassa virus budding. J. Virol. 2006, 80, 4191–4195. [Google Scholar] [CrossRef]
- Urata, S.; Yasuda, J.; de la Torre, J.C. The Z protein of the New World arenavirustacaribe virus has bonafide budding activity that does not depend on known late domain motifs. J. Virol. 2009, 83, 12651–12655. [Google Scholar] [CrossRef]
- Urata, S.; Ngo, N.; de la Torre, J.C. The PI3K/Akt pathway contributes to arenavirus budding. J. Virol. 2012, 86, 4578–4585. [Google Scholar] [CrossRef]
- Pasqual, G.; Rojek, J.M.; Masin, M.; Chatton, J.Y.; Kunz, S. Old World arenaviruses enter the host cell via the multivesicular body and depend on the endosomal sorting complex required for transport. PLoSPathog. 2011, 7, e1002232. [Google Scholar]
- Linero, F.N.; Scolaro, L.A. Participation of the phosphatidylinositol 3-kinase/Akt pathway in Junin virus replication in vitro. Virus Res. 2009, 145, 166–170. [Google Scholar] [CrossRef]
- Panda, D.; Das, A.; Dinh, P.X.; Subramaniam, S.; Nayak, D.; Barrows, N.J.; Pearson, J.L.; Thompson, J.; Kelly, D.L.; Ladunga, I.; et al. RNAi screening reveals requirement for host cell secretory pathway in infection by diverse families of negative-strand RNA viruses. Proc. Natl. Acad. of Sci. USA 2011, 47, 19036–19041. [Google Scholar]
- Candurra, N.A.; Lago, M.J.; Maskin, L.; Damonte, E.B. Involvement of the cytoskeleton in Junin virus multiplication. J. Gen. Virol. 1999, 80, 147–156. [Google Scholar]
- Cordo, S.M.; Candurra, N.A. Intermediate filament integrity is required for Junin virus replication. Virus Res. 2003, 97, 47–55. [Google Scholar] [CrossRef]
- Martinez, M.G.; Cordo, S.M.; Candurra, N.A. Involvement of cytoskeleton in Junin virus entry. Virus Res. 2008, 138, 17–25. [Google Scholar] [CrossRef]
- Martinez, M.G.; Cordo, S.M.; Candurra, N.A. Characterization of Juninarenavirus cell entry. J. Gen. Virol. 2007, 88, 1776–1784. [Google Scholar] [CrossRef]
- Martinez, M.G.; Forlenza, M.B.; Candurra, N.A. Involvement of cellular proteins in Junin arenavirus entry. Biotechnology J. 2009, 4, 866–870. [Google Scholar]
- Borrow, P.; Oldstone, M.B. Mechanism of lymphocytic choriomeningitis virus entry into cells. Virology 1994, 198, 1–9. [Google Scholar] [CrossRef]
- Rojek, J.M.; Perez, M.; Kunz, S. Cellular entry of lymphocytic choriomeningitis virus. J. Virol. 2008, 82, 1505–1517. [Google Scholar] [CrossRef]
- Rojek, J.M.; Sanchez, A.B.; Nguyen, N.T.; de la Torre, J.C.; Kunz, S. Different mechanisms of cell entry by human pathogenic Old World and New World arenaviruses. J. Virol. 2008, 82, 7677–7687. [Google Scholar] [CrossRef]
- Castilla, V.; Palermo, L.M.; Coto, C.E. Involvement of vacuolar proton ATPase in Junin virus multiplication. Arch. Virol. 2001, 146, 251–263. [Google Scholar] [CrossRef]
- Lenz, O.; ter Meulen, J.; Klenk, H.D.; Seidah, N.G.; Garten, W. The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc. Natl. Acad. Sci. USA 2001, 98, 12701–12705. [Google Scholar] [CrossRef]
- Beyer, W.R.; Pöpplau, D.; Garten, W.; von Laer, D.; Lenz, O. Endoproteolytic processing of the lymphocytic choriomeningitis virus glycoprotein by the subtilase SKI-1/S1P. J. Virol. 2003, 77, 2866–2872. [Google Scholar] [CrossRef]
- Kunz, S.; Edelmann, K.H.; de la Torre, J.C.; Gorney, R.; Oldstone, M.B. Mechanisms for lymphocytic choriomeningitis virus glycoprotein cleavage, transport, and incorporation into virions. Virology 2003, 314, 168–178. [Google Scholar] [CrossRef]
- Rojek, J.M.; Lee, A.M.; Nguyen, N.; Spiropoulou, C.F.; Kunz, S. Site 1 protease is required for proteolytic processing of the glycoproteins of the South American hemorrhagic fever viruses Junin, Machupo and Guanarito. J. Virol. 2008, 82, 6045–6051. [Google Scholar]
- Pasquato, A.; Burri, D.J.; Traba, E.G.; Hanna-El-Daher, L.; Seidah, N.G.; Kunz, S. Arenavirus envelope glycoproteins mimic autoprocessing sites of the cellular proproteinconvertasesubtilisinkexin isozyme-1/ site-1 protease. Virology 2011, 417, 18–26. [Google Scholar]
- Martinez-Sobrido, L.; Zúñiga, E.I.; Rosario, D.; Garcia-Sastre, A.; de la Torre, J.C. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2006, 80, 9192–9199. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Giannakas, P.; Cubitt, B; Garcia-Sastre, A.; de la Torre, J.C. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J. Virol. 2007, 81, 12696–12703. [Google Scholar] [CrossRef]
- Rodrigo, W.W.; Ortiz-Riaño, E.; Pythoud, C.; Kunz, S.; de la Torre, J.C.; Martínez-Sobrido, L. Arenavirus nucleoproteins prevent activation of nuclear factor kappa B. J. Virol. 2012, 86, 8185–8197. [Google Scholar]
- Gomez, R.M.; Pozner, R.G.; Lazzari, M.A.; D’Atri, L.P; Negrotto, S.; Chudzinski-Tavassi, A.M.; Berria, M.I.; Schattner, M. Endothelial cell function alteration after Junin virus infection. Thromb. Haemost. 2003, 90, 326–333. [Google Scholar]
- Molinas, F.C.; de Bracco, M.M.; Maitzegui, J.I. Hemostasis and the complement system in Argentine hemorrhagic fever. Rev. Infect. Dis. 1989, 11, S762–S770. [Google Scholar]
- Bowick, G.C.; Fennewald, S.M.; Zhang, L.; Yang, X.; Aronson, J.F.; Shope, R.E.; Luxon, B.A.; Gorenstein, D.G.; Herzog, N.K. Attenuated and lethal variants of Pichinde virus induce differential patterns of NF-kappaB activation suggesting a potential target for novel therapeutics. Viral Immunol. 2009, 22, 457–462. [Google Scholar] [CrossRef]
- Fennewald, S.M.; Aronson, J.F.; Zhang, L.; Herzog, N.K. Alterations in NK-kappaB and RBP-Jkappa by arenavirus infection of macrophages in vitro and in vivo. J. Virol. 2002, 76, 1154–1162. [Google Scholar] [CrossRef]
- Hayes, M.W.; Carrion, R., Jr.; Nunneley, J.; Medvedev, A.E.; Salvato, M.S.; Lukashevich, I.S. Pathogenic Old World arenaviruses inhibit TLR2/Mal-dependent proinflammatory cytokines in vitro. J. Virol. 2012, 86, 7216–7226. [Google Scholar] [CrossRef]
- Rojek, J.M.; Campbell, K.P.; Oldstone, M.B.; Kunz, S. Old World arenavirus infection interferes with the expression of functional alpha-dystroglycan in the host cell. Mol. Biol. Cell 2007, 18, 4493–4507. [Google Scholar] [CrossRef]
- Aronson, J.F.; Herzog, N.K.; Jerrells, T.R. Tumor-necrosis-factor and the pathogenesis of Pichinde virus infection in guinea pigs. Am. J. Trop. Med. Hyg. 1995, 52, 262–269. [Google Scholar]
- Marta, R.F.; Montero, V.S.; Hack, C.E.; Sturk, A.; Maiztegui, J.I.; Molinas, F.C. Proinflammatory cytokines and elastase-α-1-antitrypsin in Argentine hemorrhagic fever. Am. J. Trop. Med. Hyg. 1999, 60, 85–89. [Google Scholar]
- Djavani, M.; Topisirovic, I.; Zapata, J.C.; Sadowska, M.; Yang, Y.; Rodas, J.; Lukashevich, I.S.; Bogue, C.W.; Pauza, C.D.; Borden, K.L.; et al. The proline-rich homeodomain (PRH/HEX) protein is down-regulated in liver during infection with lymphocytic choriomeningitis virus. J. Virol. 2005, 79, 2461–2473. [Google Scholar]
- Topcu, Z.; Mack, D.L; Hromas, R.A.; Borden, K.L.B. The promyelocytic leukemia protein PML interacts with the proline-rich homeodomain protein PRH: a RING may link hematopoiesis and growth control. Oncogene 1999, 18, 7091–7100. [Google Scholar] [CrossRef]
- Jaquenod De Giusti, C.; Alberdi, L.; Frik, J.; Ferrer, M.F.; Scharrig, E.; Schattner, M.; Gomwez, R.M. Galectin-3 is upregulated in activated glia during Junin virus-induced murine encephalitis. Neurosci. Lett. 2011, 501, 163–166. [Google Scholar] [CrossRef]
- Huang, C.; Kolokoltsova, O.A.; Yun, N.E.; Seregin, A.V.; Poussard, A.L.; Walker, A.G.; Brasier, A.R.; Zhao, Y.; Tian, B.; de la Torre, J.C.; et al. Junin virus infection activates the type I interferon pathway in a RIG-I-dependent manner. PLoS Negl. Trop. Dis. 2012, 6, e1659. [Google Scholar] [CrossRef]
- Cuevas, C.D.; Lavanaya, M.; Wnag, E.; Ross, S.R. Junin virus infects mouse cells and induces innate immune responses. J. Virol. 2011, 85, 11058–11068. [Google Scholar] [CrossRef]
- Dejean, C.B.; Oubina, J.R.; Carballal, G.; Teyssie, A.R. Circulating interferon in the guinea pig infected with the XJ prototype Junin virus strain. J. Med. Virol. 1988, 24, 97–99. [Google Scholar]
- Dejean, C.B.; Ayerra, B.L.; Teyssie, A.R. Interferon response in the guinea pig infected with Junin virus. J. Med. Virol. 1987, 23, 83–91. [Google Scholar] [CrossRef]
- Levis, S.C.; Saavedra, M.C.; Ceccoli, C.; Falcoff, E.; Feuillade, M.R.; Enria, D.A.; Maitzegui, J.I.; Falcoff, R. Endogenous interferon in Argentine hemorrhagic fever. J. Infect. Dis. 1984, 149, 428–433. [Google Scholar] [CrossRef]
- Groseth, A.; Hoenen, T.; Weber, M.; Wolff, S.; Herwig, A.; Kaufmann, A.; Becker, S. Tacaribe virus but not junin virus infection induces cytokine release from primary human monocytes and macrophages. PLoSNegl. Trop. Dis. 2011, 5, e1137. [Google Scholar] [CrossRef]
- Marta, R.F.; Enria, D.; Molinas, F.C. Relationship between hematopoietic growth factors levels and hematological parameters in Argentine hemorrhagic fever. Am. J. Hematol. 2000, 64, 1–6. [Google Scholar] [CrossRef]
- Mahanty, S.; Bausch, D.G.; Thomas, R.L.; Goba, A.; Bah, A.; Peters, C.J.; Rollin, P.E. Low levels of interleukin-8 and interferon-inducible protein-10 in serum are associated with fatal infections in acute Lassa fever. J. Infect. Dis. 2001, 183, 1713–1721. [Google Scholar]
- Baize, S.; Marianneau, P.; Loth, P.; Reynard, S.; Journeaux, A.; Chevallier, M.; Tordo, N.; Deubel, V.; Contamin, H. Early and strong immune responses are associated with control of viral replication and recovery in lassa-virus-infected cynomolgus monkeys. J. Virol. 2009, 83, 5890–5903. [Google Scholar]
- Zhou, S.; Halle, A.; Kurt-Jones, E.A.; Cerny, A.M.; Porpiglia, E.; Rogers, M.; Golenbock, D.T.; Finberg, R.W. Lymphocytic choriomeningitis virus (LCMV) infection of CNS glial cells results in TLR2-MyD88/Mal-dependent inflammatory responses. J. Neuroimmunol. 2008, 194, 70–82. [Google Scholar] [CrossRef]
- Baird, N.L.; York, J.; Nunberg, J.H. Arenavirus infection induces discrete cytosolic structures for RNA replication. J. Virol. 2012, 86, 11301–11310. [Google Scholar] [CrossRef]
- Radoshitzky, S.R.; Dong, L.; Chi, X.; Clester, J.C.; Retterer, C.; Spurgers, K.; Kuhn, J.H.; Sandwick, S.; Ruthel, G.; Kota, K.; et al. Infectious Lassa virus, but not filoviruses, is restricted by BST-2/ tetherin. J. Virol. 2010, 84, 10569–10580. [Google Scholar]
- Sakuma, T.; Noda, T.; Urata, S.; Kawaoka, Y.; Yasuda, J. Inhibition of Lassa and Marburg virus production by tetherin. J. Virol. 2009, 83, 2382–2385. [Google Scholar] [CrossRef]
- Cerami, E.G.; Gross, B.E.; Demir, E.; Rodchenkov, I.; Babur, O.; Anwar, N.; Schultz, N.; Bader, G.D.; Sander, C. Pathway Commons, a web ressource for biological pathway data. Nucleic. Acids Res. 2011, 39, D685–D690. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef]
- Cytoscape. Available online: www.cytoscape.org (accessed on 25 July 2012).
- Cerami, E.G.; Bader, G.D.; Gross, B.E.; Sander, C. cPath: Open source software for collecting, storing, and querying biological pathways. BMC Bioinformatics 2006, 7, 497. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal 2006, 1695. [Google Scholar]
- Jahrling, P.B.; Hesse, R.A.; Rhoderick, J.B.; Elwell, M.A.; Moe, J.B. Pathogenesis of a Pichinde virus strain adapted to produce lethal infections in guinea pigs. Infect. Immunol. 1981, 32, 872–880. [Google Scholar]
- Aronson, J.F.; Herzog, N.K.; Jerrels, T.R. Pathological and virological features of arenavirus disease in guinea pigs. Comparison of two Pichinde virus strains. Am. J. Pathol. 1994, 145, 228–235. [Google Scholar]
- Walker, D.H.; McCormick, J.B.; Johnson, K.M.; Webb, P.A.; Komba-Kono, G.; Elliott, L.H.; Gardner, J.J. Pathologic and virologic study of fatal Lassa fever in man. Am. J. Pathol. 1982, 107, 349–356. [Google Scholar]
- Moraz, M.L.; Kunz, S. Pathogenesis of arenavirus hemorrhagic fevers. Expert Rev. Anti. Infect. Ther. 2011, 9, 49–59. [Google Scholar]
- Bowick, G.C.; McAuley, A.J. Meta-analysis of high-throughput datasets reveals cellular responses following hemorrhagic fever virus infection. Viruses 2011, 3, 613–619. [Google Scholar] [CrossRef]
- Hartman, A.L.; Ling, L.; Nichol, S.T.; Hibberd, M.L. Whole-genome expression profiling reveals that inhibition of host innate immune response pathways by Ebola virus can be reversed by a single amino acid change in the VP35 protein. J. Virol. 2008, 82, 5348–5358. [Google Scholar] [CrossRef]
- do Valle, T.Z.; Billecocq, A.; Guillemot, L.; Alberts, R.; Gommet, C.; Geffers, R.; Calabrese, K.; Schughart, K.; Bouloy, M.; Montagutelli, X.; et al. A new mouse model reveals a critical role for host innate immunity in resistance to Rift Valley fever. J. Immunol. 2010, 185, 6146–6156. [Google Scholar] [CrossRef]
- Wu, W.L.; Ho, L.J.; Chang, D.M.; Chen, C.H.; Lai, J.H. Triggering of DC migration by dengue virus stimulation of COX-2-dependent signaling cascades in vitro highlights the significance of these cascades beyond inflammation. Europ. J. Immunol. 2009, 39, 3413–3422. [Google Scholar] [CrossRef]
- O’Banion, M.K. Cyclooxygenase-2: Molecular biology, pharmacology, and neurobiology. Crit. Rev. Neurobiol. 1999, 13, 45–82. [Google Scholar]
- Pichlmair, A.; Kandasamy, K.; Alvisi, G.; Mulhern, O.; Sacco, R.; Habjan, M.; Binder, M.; Stefanovic, A.; Eberle, C.A.; Goncalves, A.; et al. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature 2012, 26, 486–490. [Google Scholar]
- Holt, N.; Wang, J.; Kim, K.; Friedman, G.; Wang, X.; Taupin, V.; Crooks, G.M.; Kohn, D.B.; Gregory, P.D.; Holmes, M.C.; et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 2010, 28, 839–847. [Google Scholar] [CrossRef]
- Spiro, Z.; Kovacs, I.A.; Csermely, P. Drug-therapy networks and the prediction of novel drug targets. J. Biol. 2008, 7, 20. [Google Scholar] [CrossRef]
- Faustino, R.S.; Terzic, A. Bioinformatic networks: molecular reticles for pinpointing pharmalogical target selection. Clin. Pharmacol. Ther. 2008, 5, 543–545. [Google Scholar] [CrossRef]
- Campillos, M.; Kuhn, M.; Gavin, A.C.; Jensen, L.J.; Bork, P. Drug target identification using side-effect similarity. Science 2008, 321, 263–266. [Google Scholar] [CrossRef]
- Navratil, V.; de Chassey, B.; Meyniel, L.; Delmotte, S.; Gautier, C.; Andre, P.; Lotteau, V.; Rabourdin-Combe, C. VirHostNet: A knowledge base for the management and the analysis of proteome-wide virus-host interaction networks. Nucleic Acids Res. 2009, 37, D661–D668. [Google Scholar]
- Chatr-aryamontri, A.; Ceol, A.; Peluso, D.; Nardozza, A.; Panni, S.; Sacco, F.; Tinti, M.; Smolyar, A.; Castagnoli, L.; Vidal, M.; et al. VirusMINT: a viral protein interaction database. Nucleic Acids Res. 2009, 37, D669–D673. [Google Scholar] [CrossRef]
- Kwofie, S.K.; Schaefer, U.; Sundararajan, V.S.; Bajic, V.B.; Christoffels, A. HCVpro: Hepatitis C virus protein interaction database. Infect. Genet. Evol. 2011, 11, 1971–1977. [Google Scholar] [CrossRef]
- Shoemaker, J.E.; Fukuyama, S.; Eisfeld, A.J.; Muramoto, Y.; Watanabe, S.; Watanabe, T.; Matsuoka, Y.; Kitano, H.; Kawaoka, Y. Integrated network analysis reveals a novel role for the cell cycle in 2009 pandemic influenza virus-induced inflammation in macaque lungs. BMC Syst. Biol. 2012, 6, 117. [Google Scholar] [CrossRef]
- McDermott, J.E.; Diamond, D.L.; Corley, C.; Rasmussen, A.L.; Katze, M.G.; Waters, K.M. Topological analysis of protein co-abundance networks identified novel host targets important for HCV infection and pathogenesis. BMC Syst. Biol. 2012, 6, 28. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).