Molecular Adjuvant Ag85A Enhances Protection against Influenza A Virus in Mice Following DNA Vaccination

Abstract
:1. Introduction
2. Results
2.1. Expression of Ag85A-sHA2 in Eukaryotic Cells Transfected with the Plasmid DNA Vectors
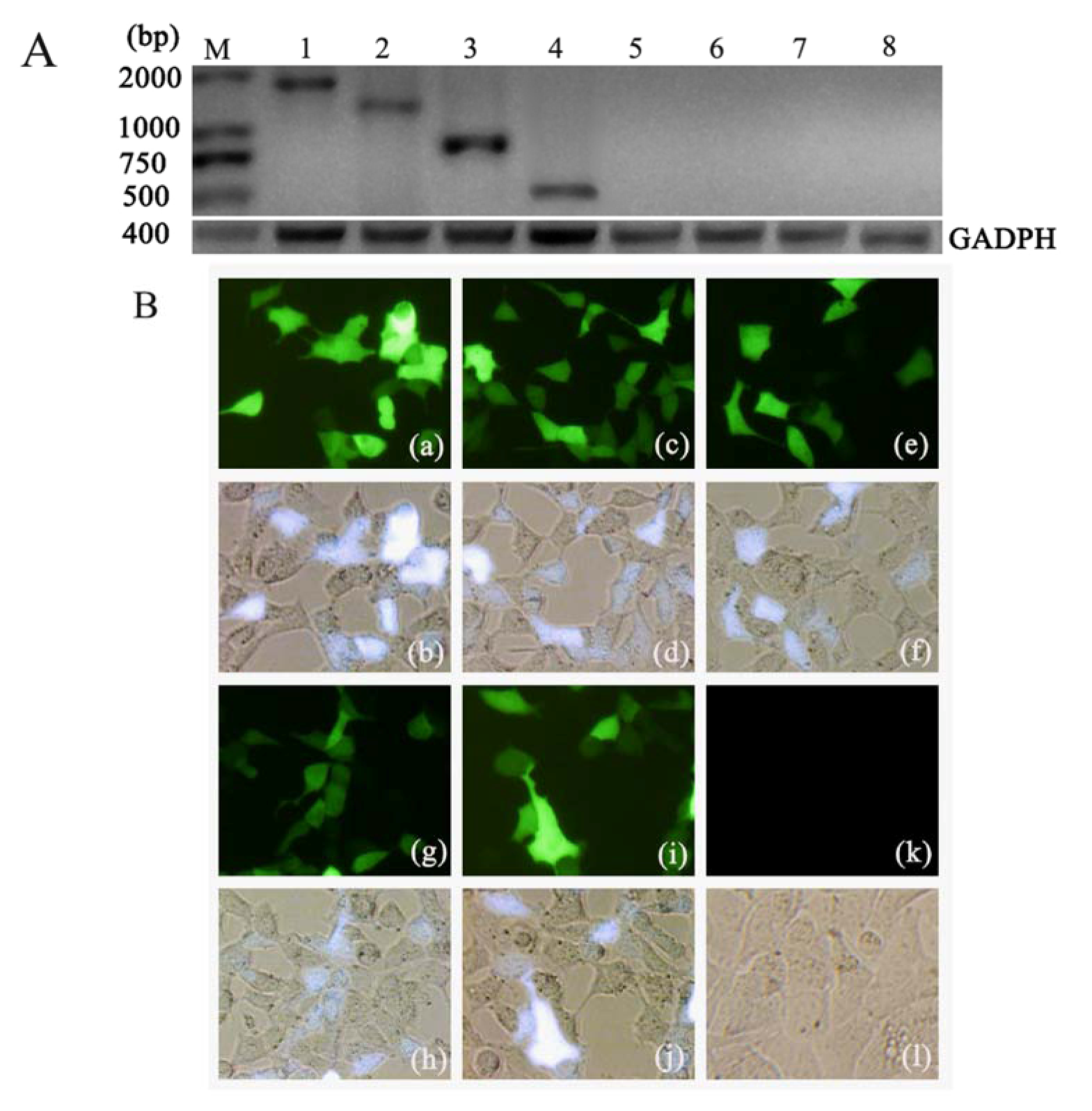
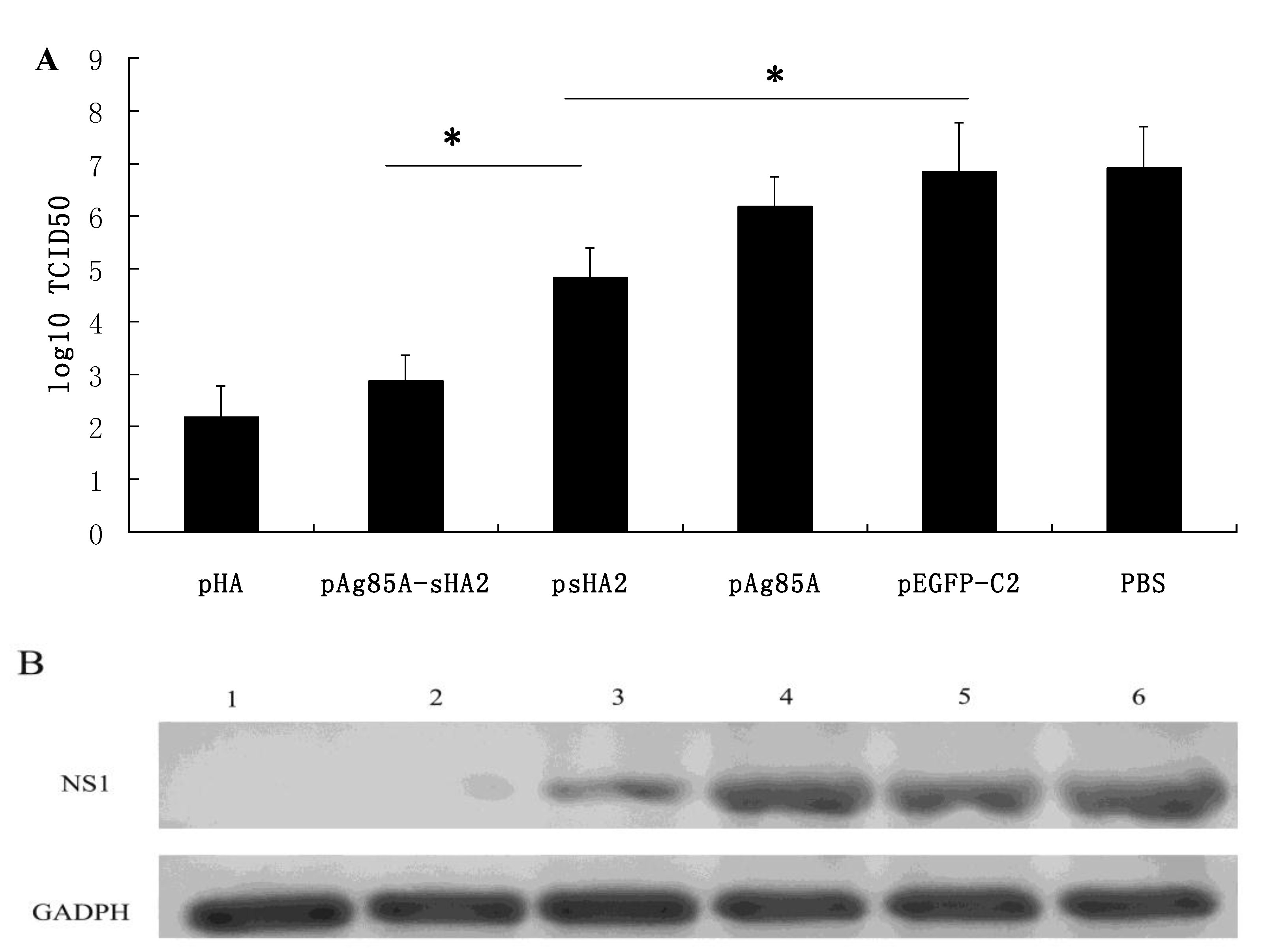
2.2. Vaccine-Mediated Induction of Functional Antibody Titers and Effect on IAV Challenge

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Primary response | Secondary response |
|---|---|---|
| pHA | 4.00 ± 0.82 | 5.75 ± 0.95 |
| pAg85A-sHA2 | 2.50 ± 0.58 | 2.75 ± 0.50 |
| pAg85A | 1.75 ± 0.96 | 1.75 ± 0.96 |
| psHA2 | 2.25 ± 0.50 | 2.50 ± 0.58 |
| pEGFP-C2 | 1.50 ± 0.58 | 1.50 ± 0.58 |
| PBS | 1.50 ± 0.58 | 1.50 ± 0.58 |
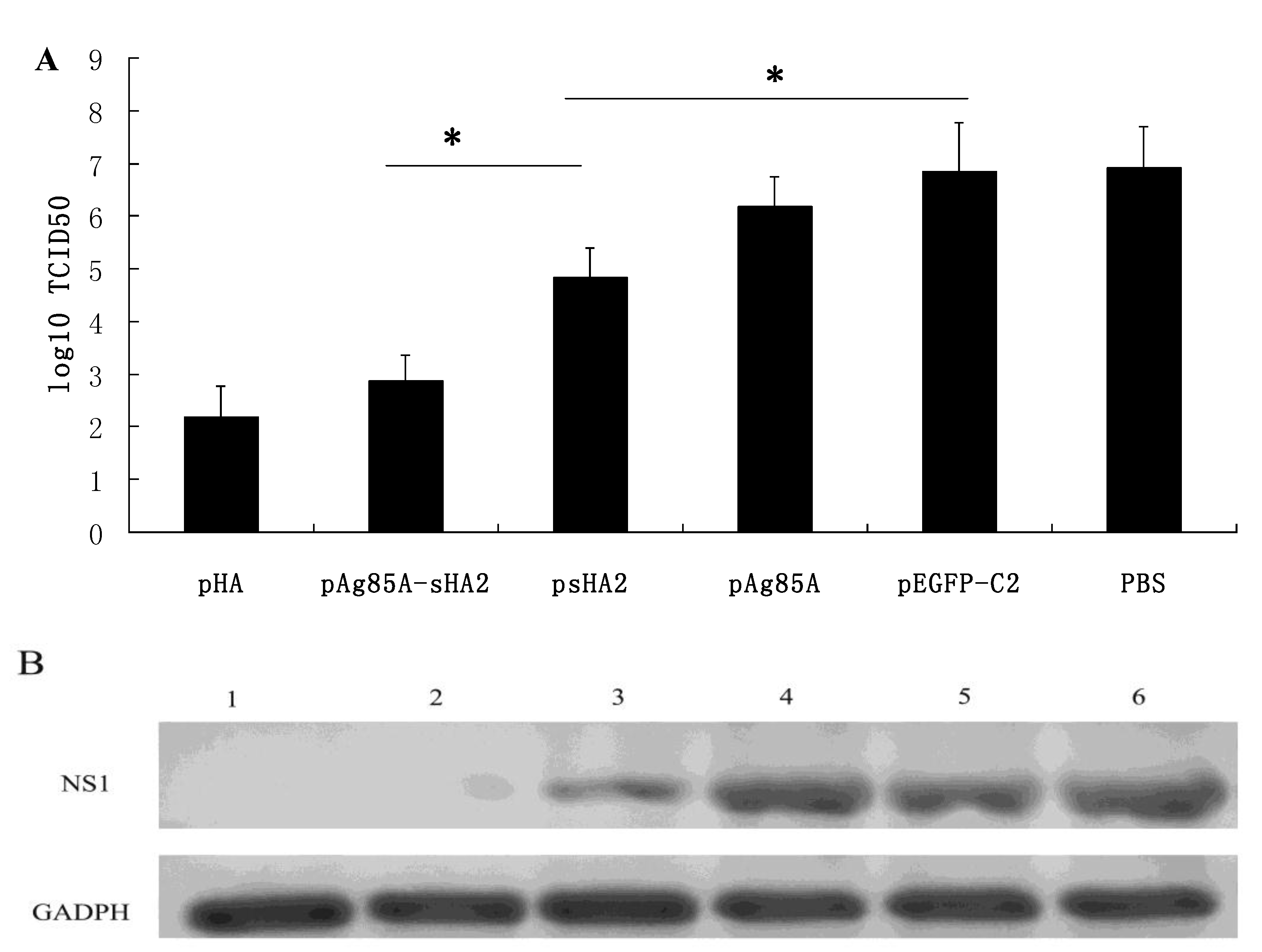
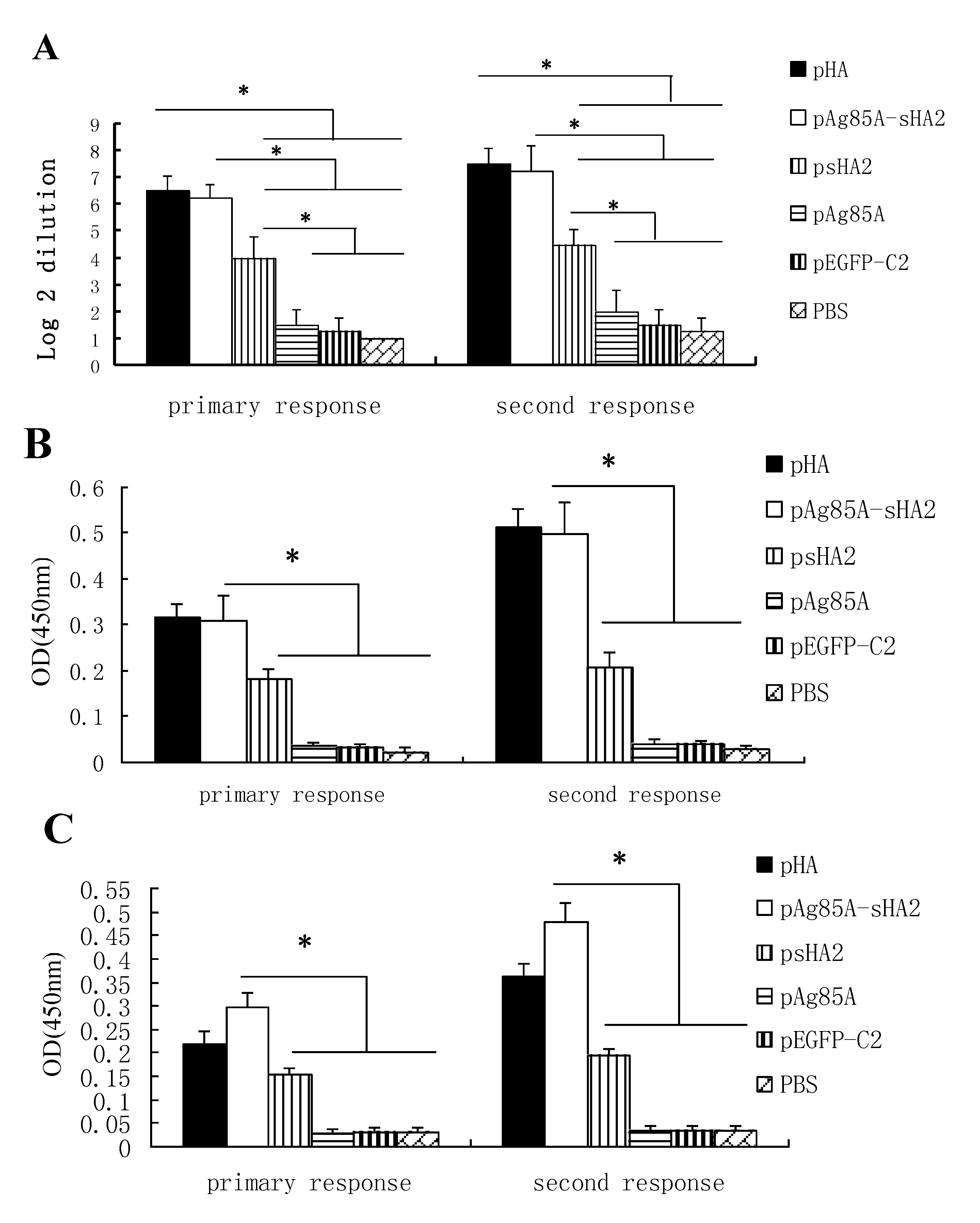
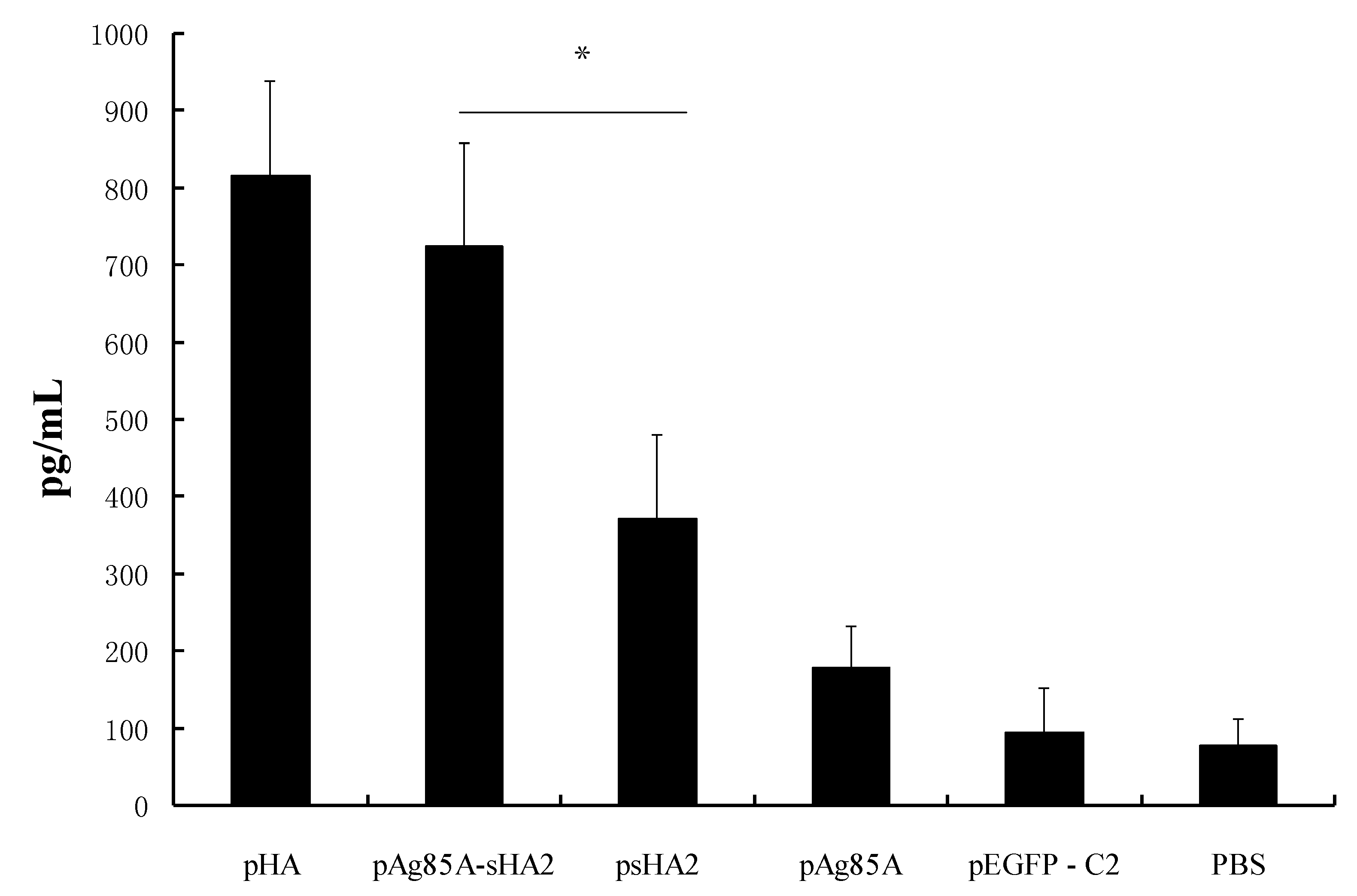
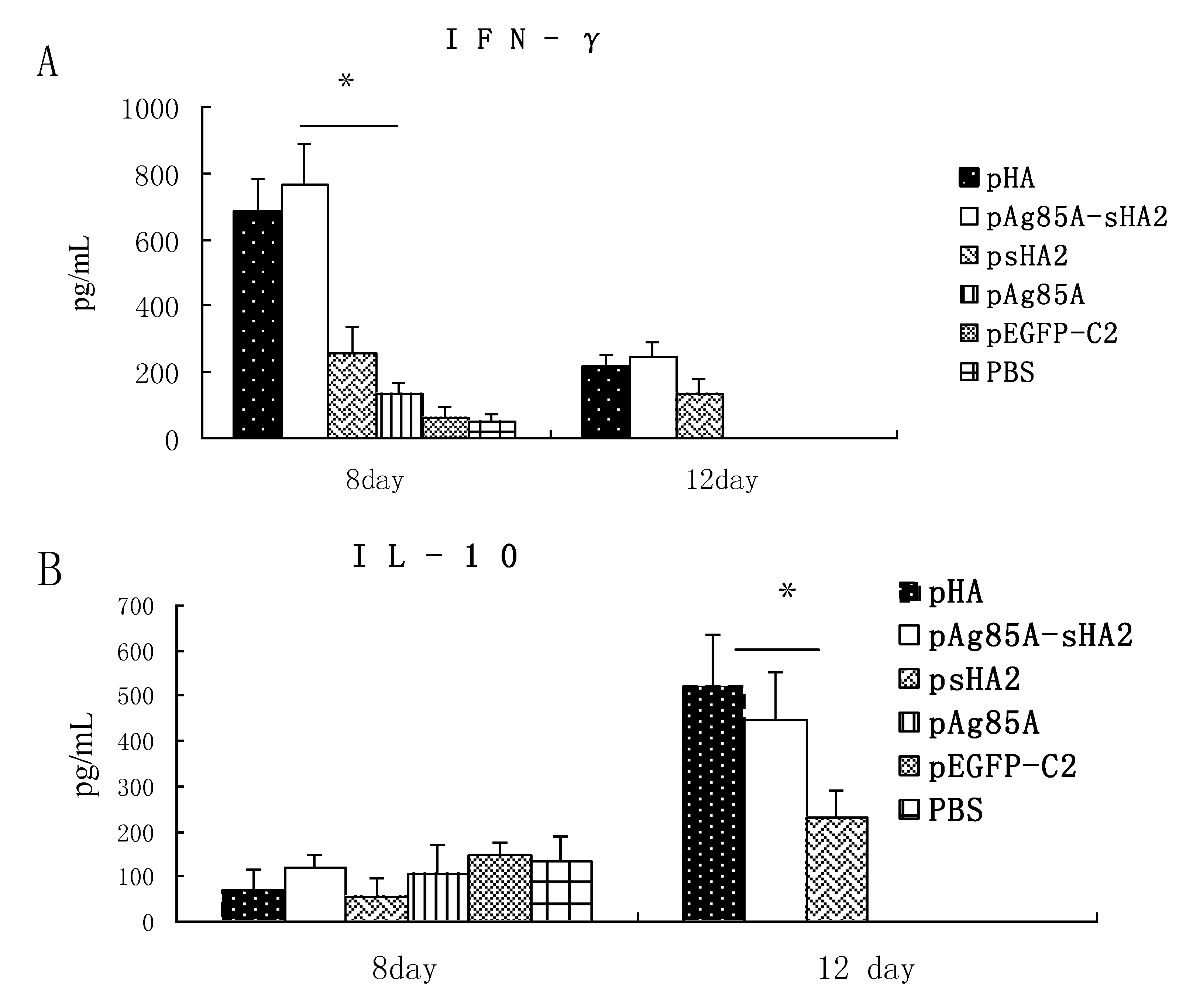
2.3. Cytokine Measurements

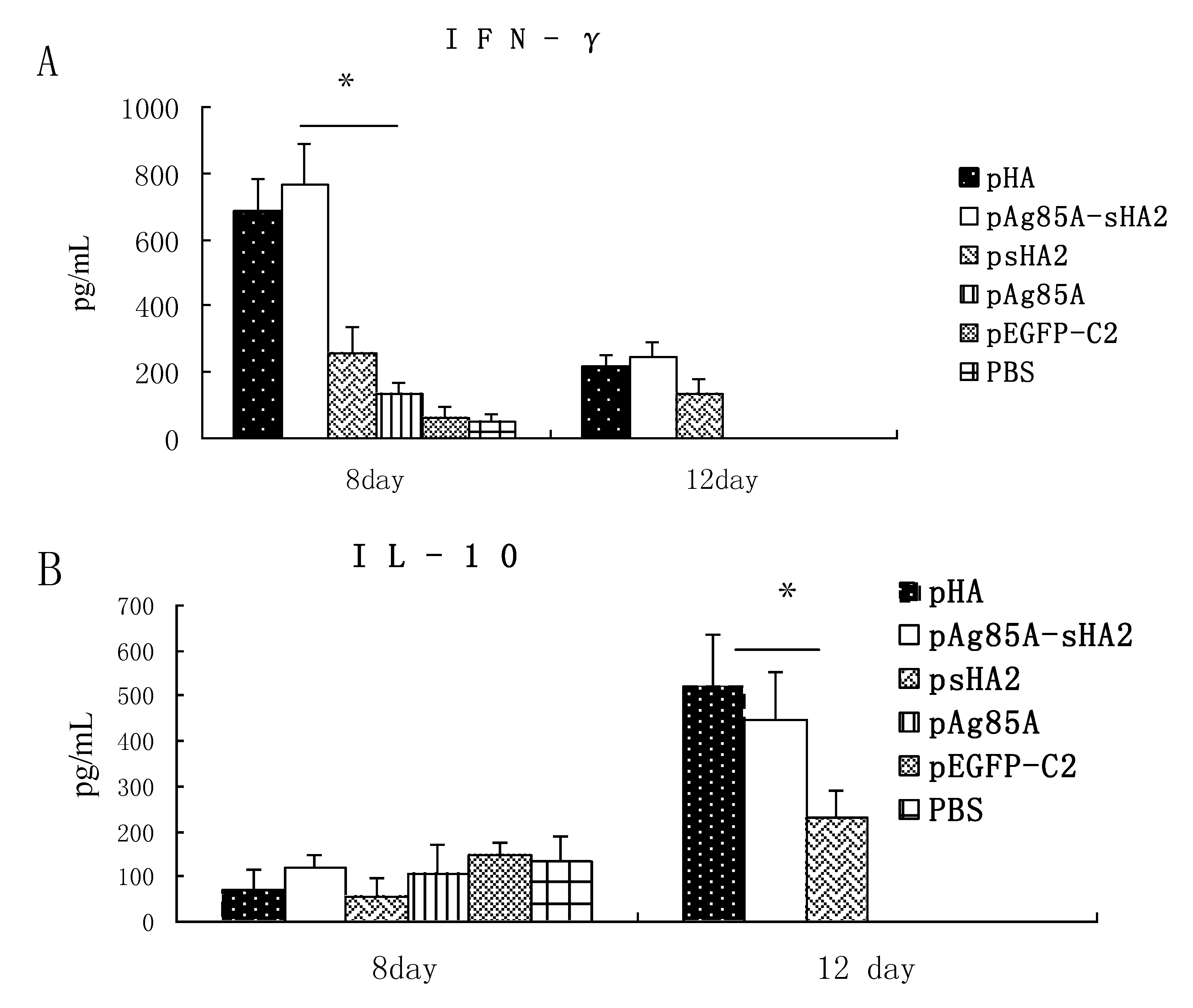
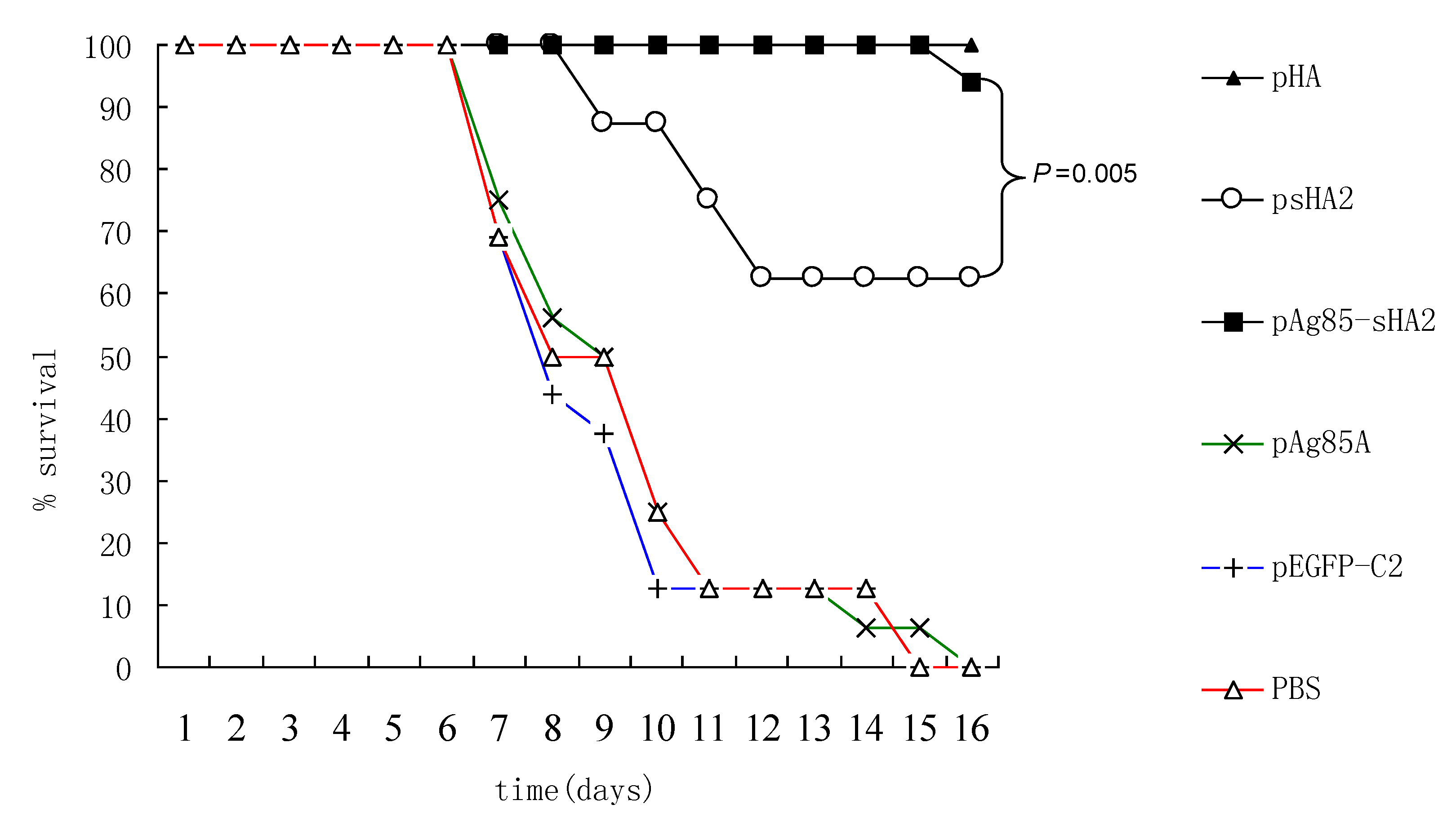
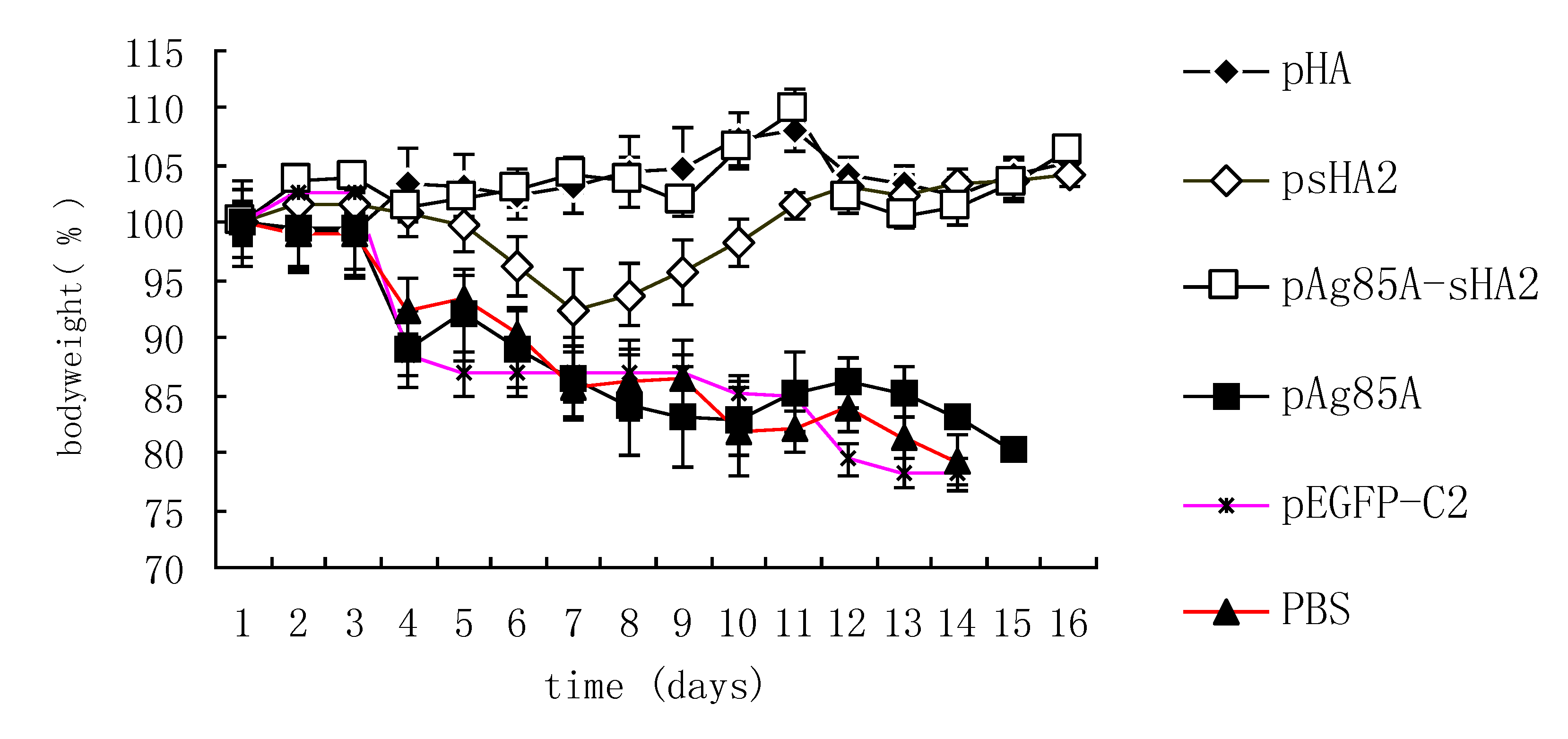
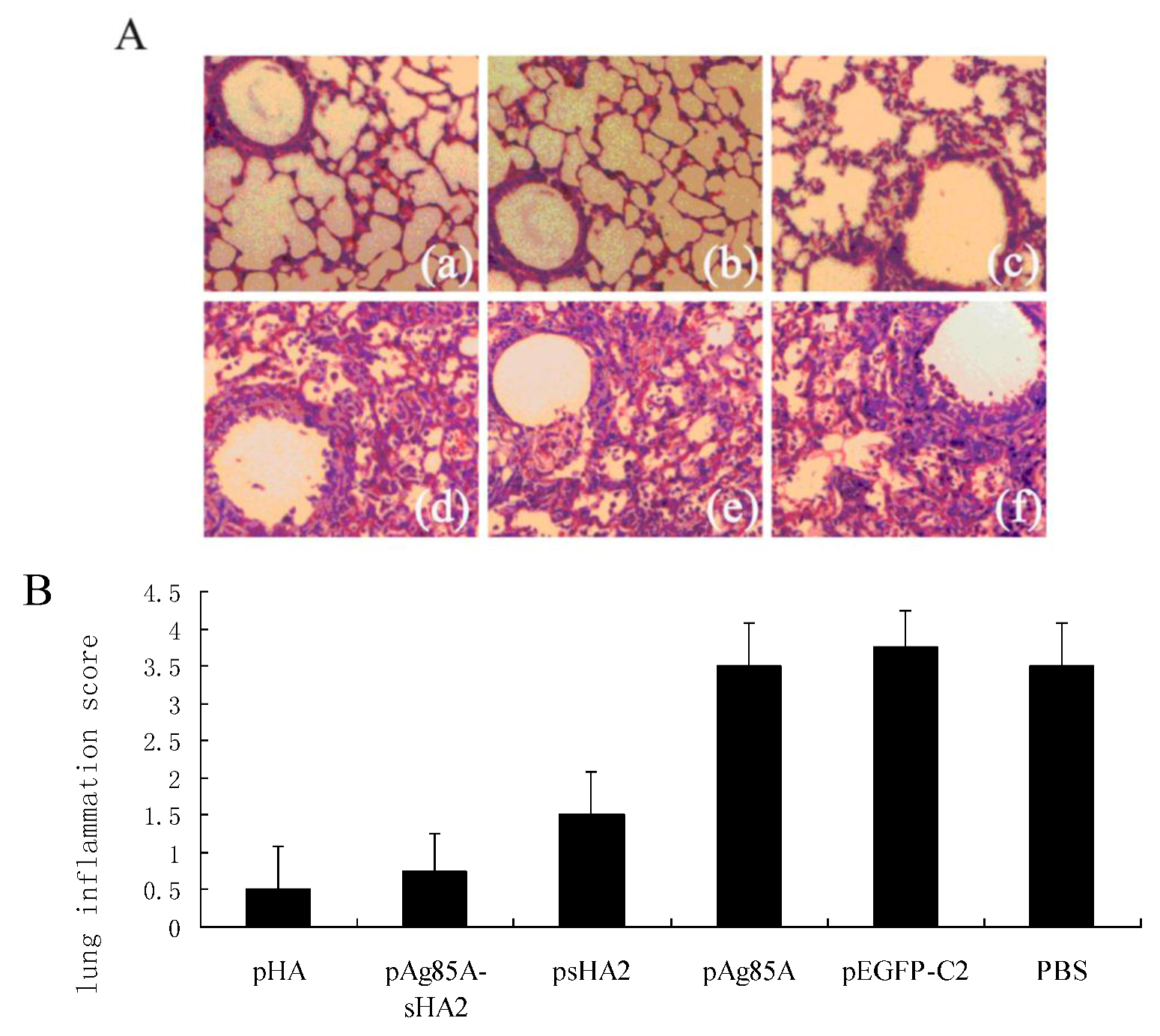
2.4. Comparison of Survival Rates and Histopathological Analyses
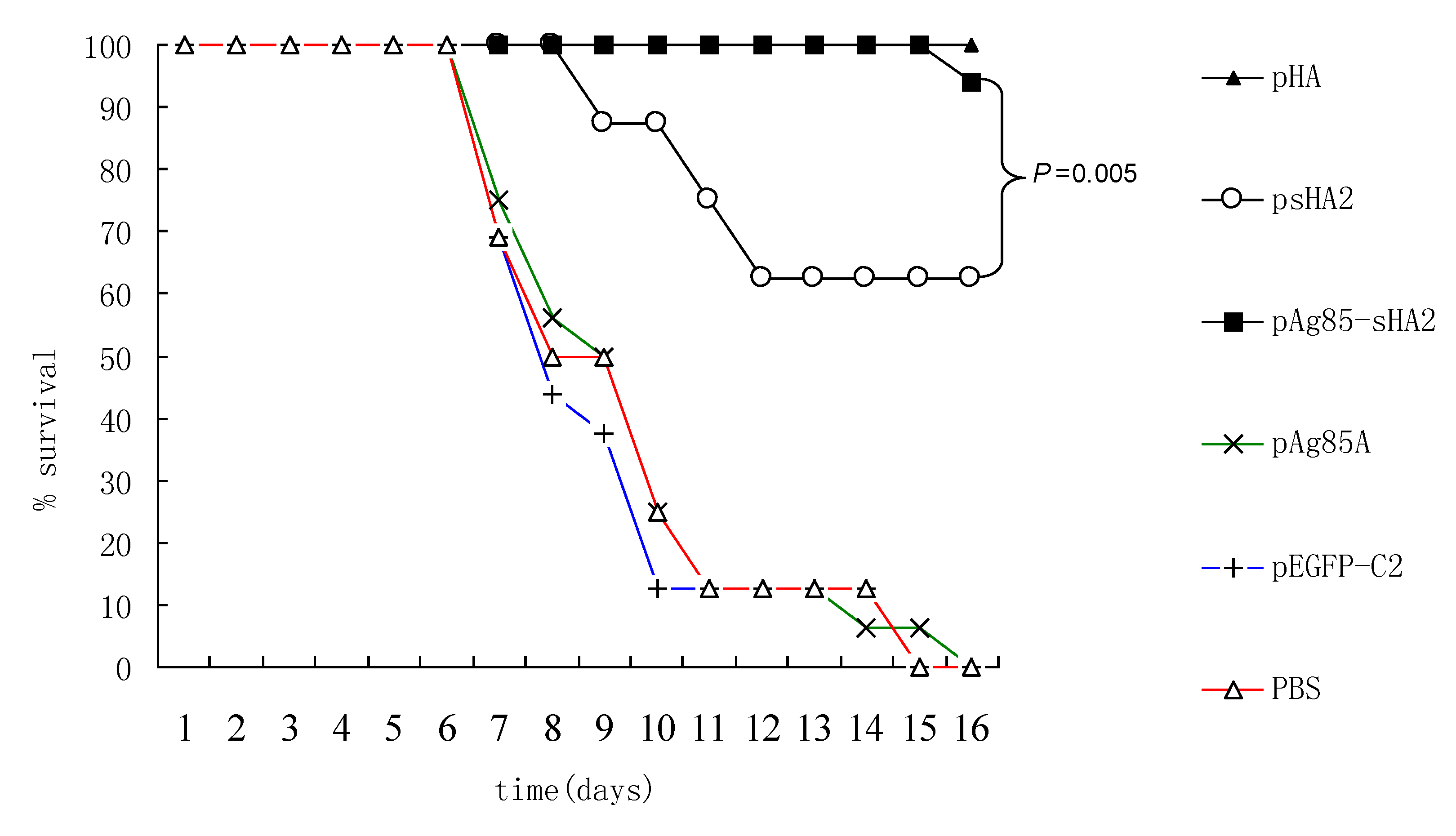
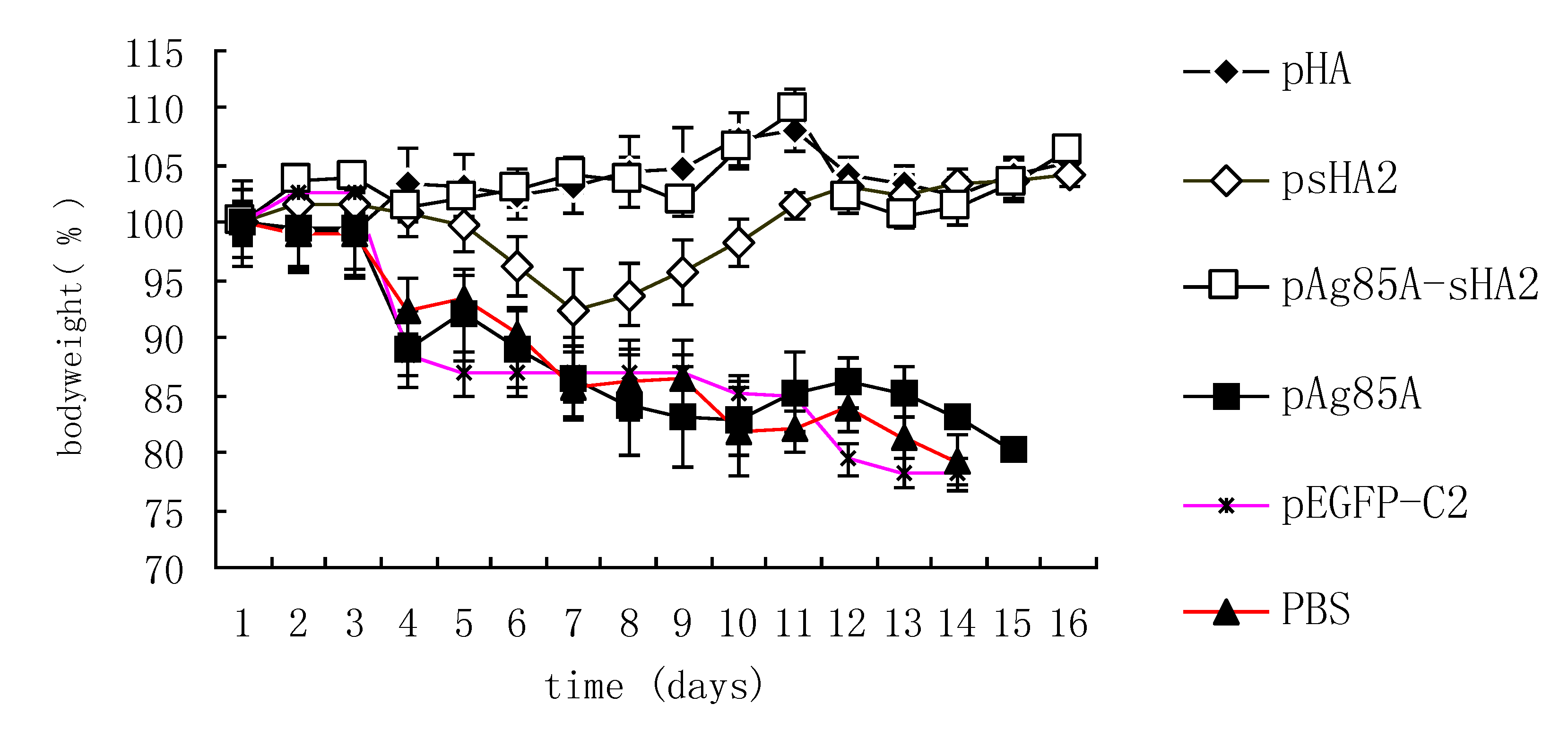
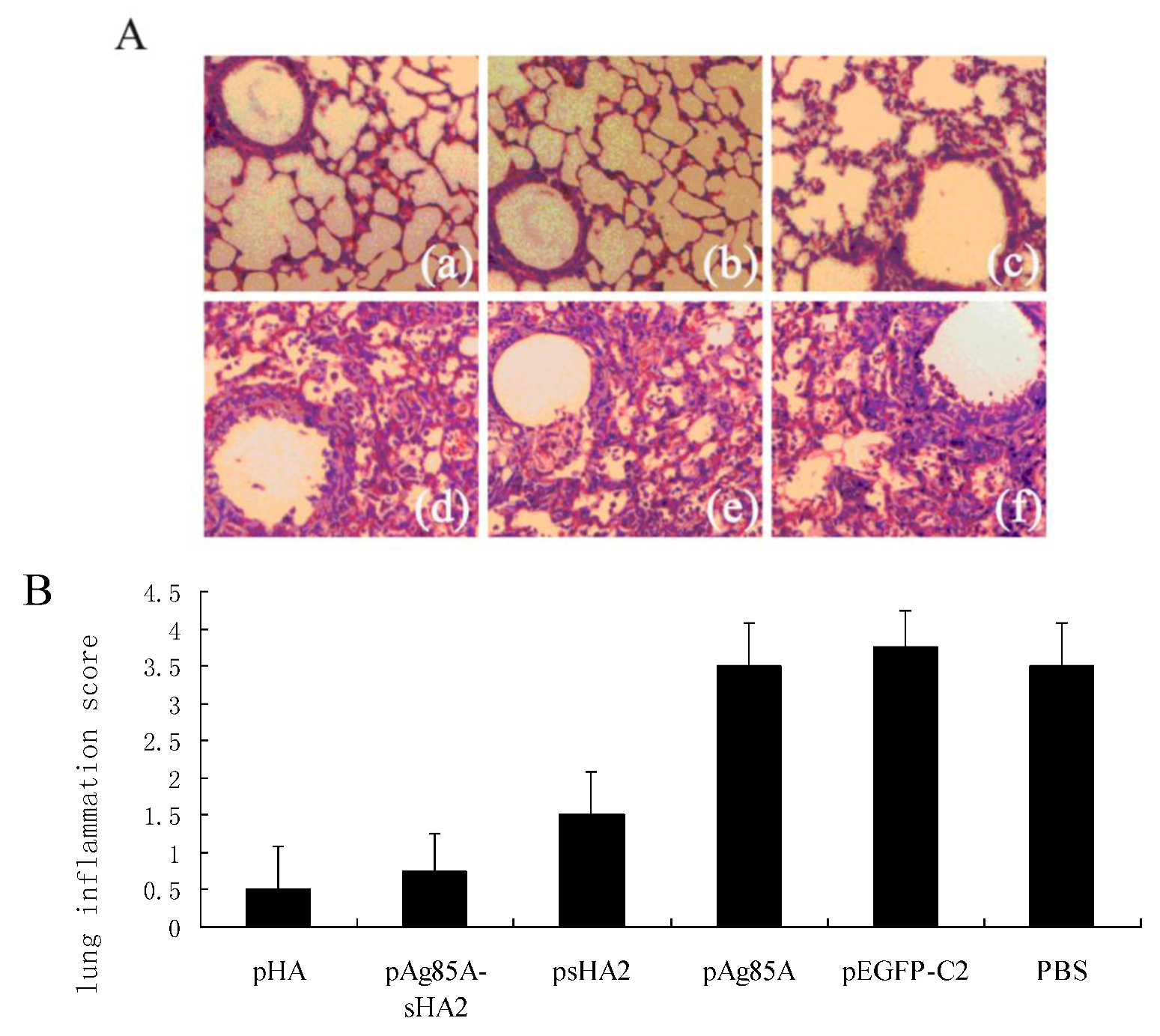
3. Discussion
4. Materials and Methods
4.1. Design of Epitopes
| Epitope | Region | Amino acid sequence |
|---|---|---|
| B epitope | H1HA368–375 | YHHQNEQG |
| B epitope | H1HA379–386 | YAADQKSTQ |
| B epitope | H1HA391–397, H5HA347–353 | GITNKVN |
| B epitope | H1HA453–460, H5HA455–462 | DFHDSNVK |
| B epitope | H1HA491–498, H5HA493–500 | ECMESVRN |
| T epitope | H1HA373–387 | EQGSGYAADQKSTQN |
| T epitope | H1HA439–453 | NAELLVLLENERTLD |
| T epitope | H1HA434–448 | DIWTYNAELLVLLEN |
4.2. Construction of Recombinant Eukaryotic Expression Plasmid Vaccine Vectors
4.3. Cell Transfection Assay
4.4. DNA Vaccination and Challenge Infection with the PR8 Virus
4.5. Specimen Collection
4.6. HA Inhibition Assay
4.7. Neutralization Assay
4.8. Serological Assays by ELISA
4.9. Cytokine Measurements
4.10. IAV Titrations
4.11. PCR-Amplification and Sequencing of PR8 Nucleic Acids Using RNA Extracted from Lungs of Infected Mice
4.12. Histopathological Analysis
4.13. Statistical Analysis
5. Conclusions
Acknowledgments
References and Notes
- Abdel-Motal, U.M.; Guay, H.M.; Wigglesworth, K.; Welsh, R.M.; Galili, U. Immunogenicity of influenza virus vaccine is increased by anti-gal-mediated targeting to antigen-presenting cells. J. Virol. 2007, 81, 9131–9141. [Google Scholar] [CrossRef]
- Gong, T.; Jiang, Y.; Wang, Y.; Yang, D.; Li, W.; Zhang, Q.; Feng, W.; Wang, B.; Jiang, Z.; Li, M. Recombinant mouse beta-defensin 2 inhibits infection by influenza A virus by blocking its entry. Arch. Virol. 2010, 155, 491–498. [Google Scholar] [CrossRef]
- Sahini, L.; Tempczyk-Russell, A.; Agarwa, R. Large-scale sequence analysis of hemagglutinin of influenza a virus identifies conserved regions suitable for targeting an anti-viral response. PLoS One 2010, 5, e9268. [Google Scholar]
- Tan, G.S.; Krammer, F.; Eggink, D.; Kongchanagul, A.; Moran, T.M.; Palese, P. A pan-H1 anti-hemagglutinin monoclonal antibody with potent broad-spectrum efficacy in vivo. J. Virol. 2012, 86, 6179–6188. [Google Scholar]
- Kim, J.H.; Skountzou, I.; Compans, R.; Jacob, J. Original antigenic sin responses to influenza viruses. J. Immunol. 2009, 183, 3294–3301. [Google Scholar] [CrossRef]
- Babon, J.A.; Cruz, J.; Ennis, F.A.; Yin, L.; Terajima, M. A human CD4+ T cell epitope in the influenza hemagglutinin is cross-reactive to influenza A subtypes and to influenza B virus. J. Virol. 2012. [Google Scholar] [CrossRef]
- Sui, J.; Hwang, W.C.; Perez, S.; Wei, G.; Aird, D.; Chen, L.M.; Santelli, E.; Stec, B.; Cadwell, G.; Ali, M.; et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza a viruses. Nat. Struct. Mol. Biol. 2009, 16, 265–273. [Google Scholar]
- Steel, J.; Lowen, A.C.; Wang, T.T.; Yondola, M.; Gao, Q.; Haye, K.; García-Sastre, A.; Palese, P. Influenza virus vaccine based on the conserved hemagglutinin stalk domain. Mbio 2010, 1, e00018-10. [Google Scholar]
- Ulmer, J.B.; Deck, R.R.; DeWitt, C.M.; Donnelly, J.J.; Friedman, A.; Montgomery, D.L.; Yawman, A.M.; Orme, I.M.; Denis, O.; Content, J.; et al. Induction of immunity by DNA vaccination: Application to influenza and tuberculosis. Behring Institute Mitteilungen 1997, 98, 79–86. [Google Scholar]
- Zykov, M.P.; Subbotina, T.I. Modulation of humoral immune response to influenza vaccines by BCG. Acta Virol. 1985, 29, 403–409. [Google Scholar]
- Masihi, K.N.; Brehmer, W.; Lange, W.; Ribi, E. Effects of mycobacterial fractions and muramyl dipeptide on the resistance of mice to aerogenic influenza virus infection. Int. J. Immunopharmacol. 1983, 5, 403–410. [Google Scholar]
- Romano, M.; Roupie, V.; Hamard, M.; Huygen, K. Evaluation of the immunogenicity of pBudCE4.1 plasmids encoding mycolyl-transferase Ag85A and phosphate transport receptor PstS-3 from Mycobacterium tuberculosis. Vaccine 2006, 24, 4640–4643. [Google Scholar] [CrossRef]
- Harris, K.; Ream, R.; Gao, J.; Eichelberger, M.C. Intramuscular immunization of mice with live influenza virus is more immunogenic and offers greater protection than immunization with inactivated virus. Virol. J. 2011, 8, 251–261. [Google Scholar] [CrossRef]
- Denis, O.; Tanghe, A.; Palfliet, K.; Jurion, F.; van den Berg, T.-P.; Vanonckelen, A.; Ooms, J.; Saman, E.; Ulmer, J.B.; Content, J.; et al. Vaccination with plasmid DNA encoding mycobacterial antigen 85A stimulates a CD41 and CD81 T-cell epitopic repertoire broader than that stimulated by Mycobacterium tuberculosis H37Rv Infection. Infect. Immun. 1998, 66, 1527–1533. [Google Scholar]
- Borremans, M.; Wit, L.D.; Volckaert, G.; Ooms, J.; Bruyn, J.D.; Huygen, K.; van Vooren, J.; Stelander, M.; Verhofstadt, R.; Content, J. Cloning, sequence determination, and expression of a 32-kilodalton-protein gene of Mycobacterium tuberculosis. Infect. Immun. 1989, 57, 3123–3130. [Google Scholar]
- Smith, S.M.; Brookes, R.; Klein, M.R.; Malin, A.S.; Lukey, P.T.; King, A.S.; Ogg, G.S.; Hill, A.V.S.; Dockrell, H.M. Human CD81 CTL specific for the mycobacterial major secreted Antigen 85A. J. Immunol. 2000, 165, 7088–7095. [Google Scholar]
- Zhang, W.; Li, W.; Li, Y.; Li, H.; Wang, B.; Wang, F.; Zhu, Y.; Jiang, Z.; Li, L.Z.M. Immune effects against influenza a virus and a novel DNA vaccine with co-expression of haemagglutinin- and neuraminidase-encoding genes. J. Med. Microbiol. 2009, 58, 845–854. [Google Scholar] [CrossRef]
- Tamura, S.I.; Asanuma, H.; Ito, Y.; Hirabayashi, Y.; Suzuki, Y.; Nagamine, T.; Aizawa, C.; Kurata, T.; Oya, A. Superior cross-protective effect of nasal vaccination to subcutaneous inoculation with influenza hemagglutinin vaccine. Eur. J. Immunol. 1992, 22, 477–481. [Google Scholar] [CrossRef]
- Dessing, M.C.; van der Sluijs, K.F.; Florquin, S.; Akira, S.; van der Poll, T. Toll-like receptor 2 does not contribute to host response during postinfluenza pneumococcal pneumonia. Am. J. Respir. Cell Mol. Biol. 2007, 36, 609–614. [Google Scholar]
- Tisoncik, J.R.; Billharz, R.; Burmakina, S.; Belisle, S.E.; Proll, S.C.; Korth, M.J.; Garcíia-Sastre, A.; Katze, M.G. The NS1 protein of influenza a virus suppresses interferon-regulated activation of antigen-presentation and immune-proteasome pathways. J. Gen. Virol. 2011, 92, 2093–2104. [Google Scholar] [CrossRef]
- Antunes, I.; Kassiotis, G. Suppression of innate immune pathology by regulatory T cells during Influenza a virus infection of immunodeficient mice. J. Virol. 2010, 84, 12564–12575. [Google Scholar]
- Viallard, J.F.; Pellegrin, J.L.; Ranchin, V.; Schaeverbeke, T.; Dehais, J.; Longy-Boursier, M.; Ragnaud, J.M.; Leng, B.; Moreau, J.F. Th1 (IL-2, interferon-gamma (IFN-γ)) and Th2 (IL-10, IL-4) cytokine production by peripheral blood mononuclear cells (PBMC) from patients with systemic lupus erythematosus (SLE). Clin. Exp. Immunol. 1999, 115, 189–195. [Google Scholar]
- Bcepred. Available online: http://www.imtech.res.in/raghava/bcepred/ (accessed on 10 August 2010).
- SYFPEITHI. Available online: http://www.syfpeithi.de/ (accessed on 12 August 2010).
- Standard Protein BLAST. Available online: http://blast.ncbi.nlm.nih.gov/Blast.cgi/ (accessed on 20 August 2010).
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dai, J.; Pei, D.; Wang, B.; Kuang, Y.; Ren, L.; Cao, K.; Wang, H.; Zuo, B.; Shao, J.; Li, S.; et al. Molecular Adjuvant Ag85A Enhances Protection against Influenza A Virus in Mice Following DNA Vaccination. Viruses 2012, 4, 3606-3624. https://doi.org/10.3390/v4123606
Dai J, Pei D, Wang B, Kuang Y, Ren L, Cao K, Wang H, Zuo B, Shao J, Li S, et al. Molecular Adjuvant Ag85A Enhances Protection against Influenza A Virus in Mice Following DNA Vaccination. Viruses. 2012; 4(12):3606-3624. https://doi.org/10.3390/v4123606
Chicago/Turabian StyleDai, Jun, Decui Pei, Baoning Wang, Yu Kuang, Laifeng Ren, Kang Cao, Huan Wang, Bin Zuo, Jingjing Shao, Sha Li, and et al. 2012. "Molecular Adjuvant Ag85A Enhances Protection against Influenza A Virus in Mice Following DNA Vaccination" Viruses 4, no. 12: 3606-3624. https://doi.org/10.3390/v4123606
APA StyleDai, J., Pei, D., Wang, B., Kuang, Y., Ren, L., Cao, K., Wang, H., Zuo, B., Shao, J., Li, S., Li, H., & Li, M. (2012). Molecular Adjuvant Ag85A Enhances Protection against Influenza A Virus in Mice Following DNA Vaccination. Viruses, 4(12), 3606-3624. https://doi.org/10.3390/v4123606