Biogenesis and Dynamics of the Coronavirus Replicative Structures

{kind=link}
{kind=link}
Abstract
:1. Introduction
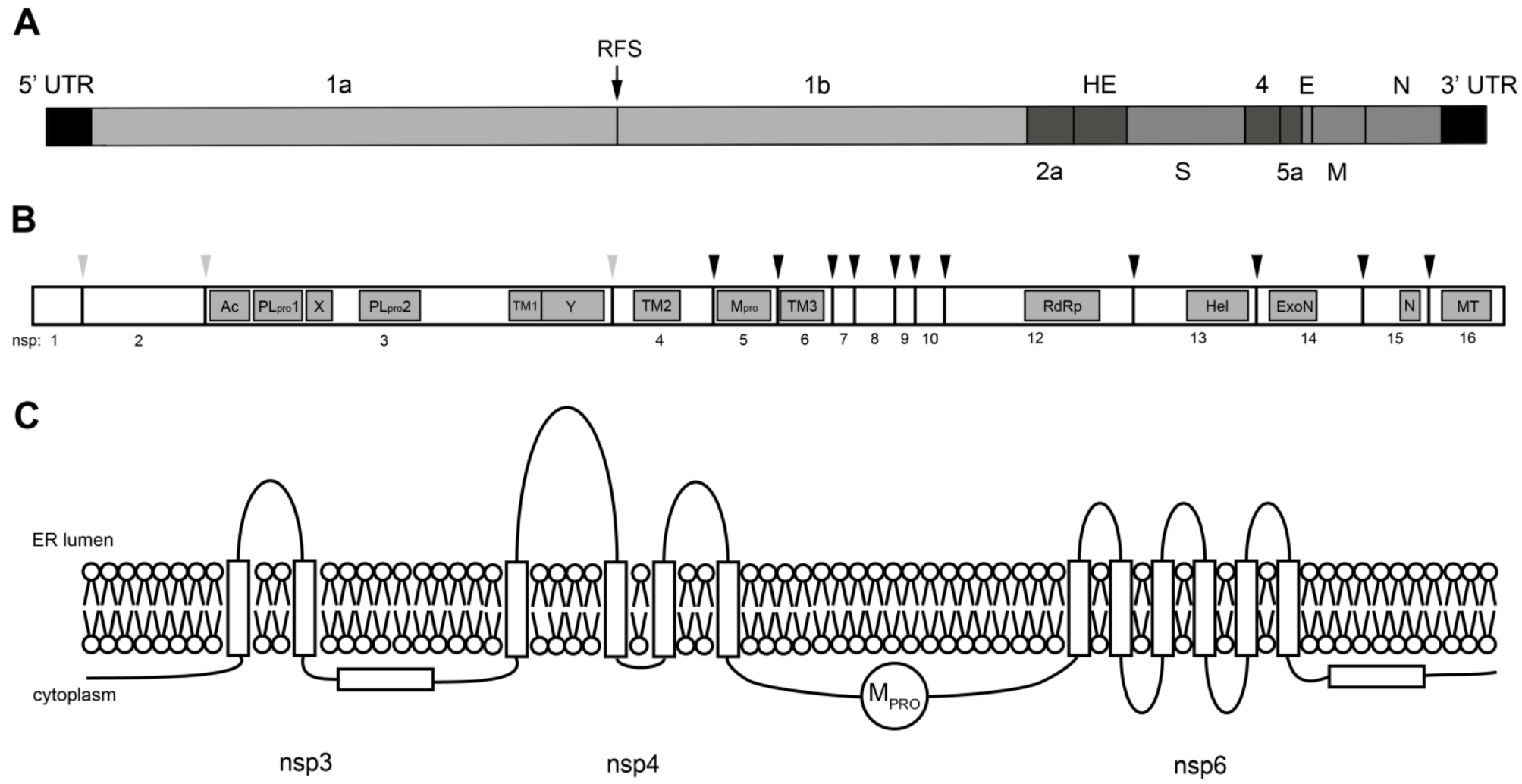
3. Coronavirus Nonstructural Proteins
3.1. Viral Proteinases
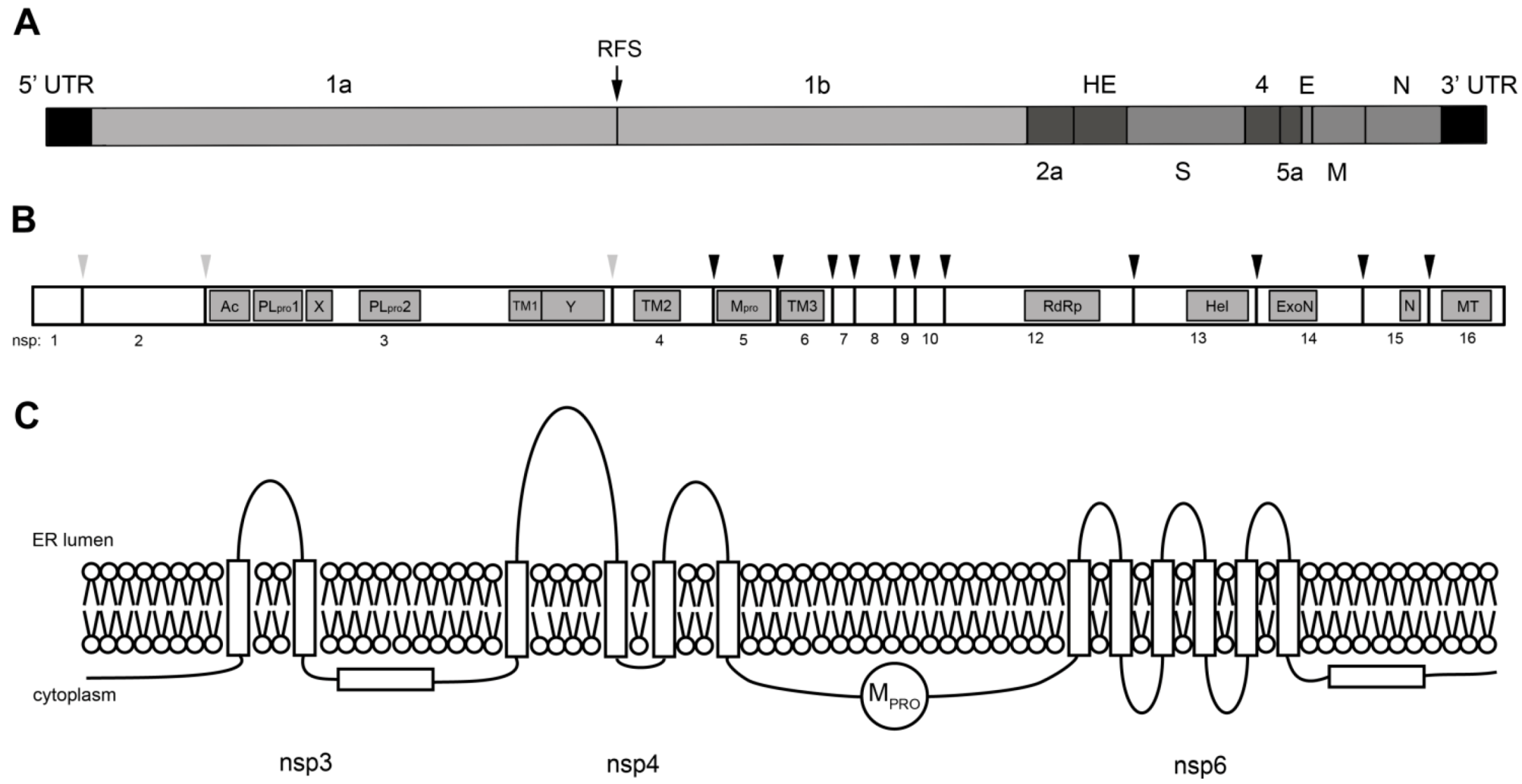
3.2. RNA-modifying Enzymes
3.3. Nonstructural Transmembrane Proteins
3.4. Nonstructural Proteins with Other Functions
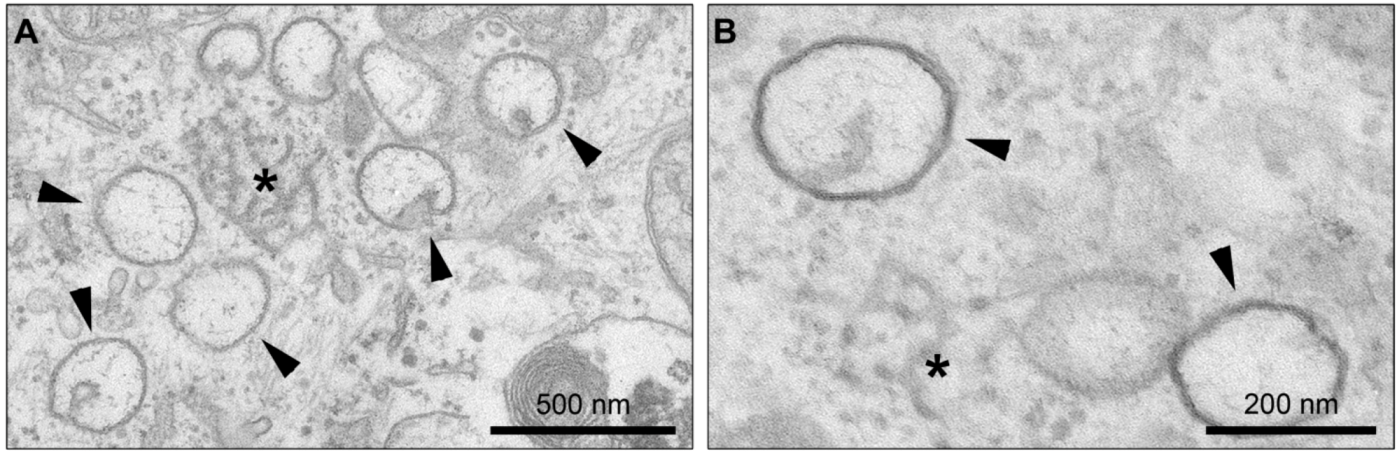
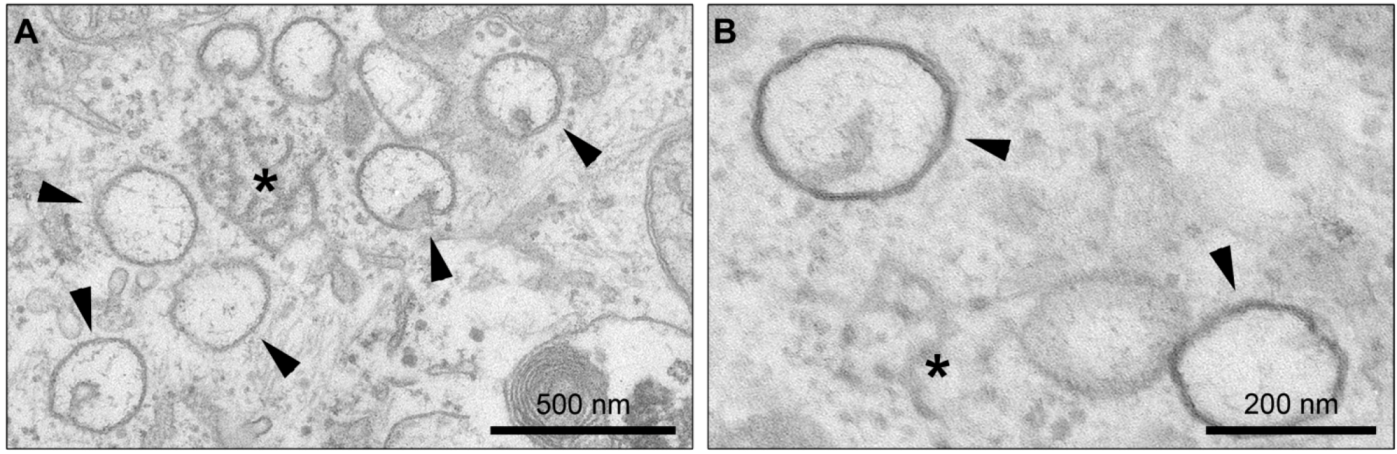
4. Membrane Rearrangements
4.1. Organelle-like Membranous Replicative Structures
4.2. Involvement of Cellular Pathways
4.3. Role of Viral Proteins
5. Dynamics of CoV Replicative Structures and Associated Proteins
5.1. Dynamics of the CoV Replicative Structures
5.2. Mobility of Replicative Structure-Associated Proteins
6. CoV Replicative Structures and RNA Synthesis
7. Future Directions
Acknowledgments
Conflict of Interest
References and Notes
- den Boon, J.A.; Ahlquist, P. Organelle-Like Membrane Compartmentalization of Positive-Strand RNA Virus Replication Factories. Annu. Rev. Microbiol. 2010, 64, 241–256. [Google Scholar] [CrossRef]
- den Boon, J.A.; Diaz, A.; Ahlquist, P. Cytoplasmic Viral Replication Complexes. Cell Host Microbe 2010, 8, 77–85. [Google Scholar] [CrossRef]
- Miller, S.; Krijnse-Locker, J. Modification of Intracellular Membrane Structures for Virus Replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef]
- Knoops, K.; Kikkert, M.; van den Worm, S.H.E.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-Coronavirus Replication is Supported by a Reticulovesicular Network of Modified Endoplasmic Reticulum. PLoS Biol. 2008, 6, 1957–1974. [Google Scholar]
- Goldsmith, C.S.; Tatti, K.M.; Ksiazek, T.G.; Rollin, P.E.; Comer, J.A.; Lee, W.W.; Rota, P.A.; Bankamp, B.; Bellini, W.J.; Zaki, S.R. Ultrastructural Characterization of SARS Coronavirus. Emerg. Infect. Dis. 2004, 10, 320–326. [Google Scholar]
- Drosten, C.; Günther, S.; Preiser, W.; van der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef]
- Guan, Y.; Zheng, B.J.; He, Y.Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Guan, Y.J.; et al. Isolation and Characterization of Viruses Related to the SARS Coronavirus from Animals in Southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are Natural Reservoirs of SARS-Like Coronaviruses. Science 2005, 310, 676–679. [Google Scholar]
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the Largest RNA Virus Genome. Virus Res. 2006, 117, 17–37. [Google Scholar] [CrossRef]
- Sawicki, S.G.; Sawicki, D.L.; Siddell, S.G. A Contemporary View of Coronavirus Transcription. J. Virol. 2007, 81, 20–29. [Google Scholar] [CrossRef]
- Sawicki, S.G.; Sawicki, D.L. Coronaviruses use Discontinuous Extension for Synthesis of Subgenome-Length Negative Strands. Adv. Exp. Med. Biol. 1995, 380, 499–506. [Google Scholar] [CrossRef]
- Zuniga, S.; Sola, I.; Alonso, S.; Enjuanes, L. Sequence Motifs Involved in the Regulation of Discontinuous Coronavirus Subgenomic RNA synthesis. J. Virol. 2004, 78, 980–994. [Google Scholar] [CrossRef]
- Sawicki, S.G.; Sawicki, D.L. Coronavirus Minus-Strand RNA Synthesis and Effect of Cycloheximide on Coronavirus RNA Synthesis. J. Virol. 1986, 57, 328–334. [Google Scholar]
- Sawicki, D.; Wang, T.; Sawicki, S. The RNA Structures Engaged in Replication and Transcription of the A59 Strain of Mouse Hepatitis Virus. J. Gen. Virol. 2001, 82, 385–396. [Google Scholar]
- Bredenbeek, P.J.; Pachuk, C.J.; Noten, A.F.; Charite, J.; Luytjes, W.; Weiss, S.R.; Spaan, W.J. The Primary Structure and Expression of the Second Open Reading Frame of the Polymerase Gene of the Coronavirus MHV-A59; a Highly Conserved Polymerase is Expressed by an Efficient Ribosomal Frameshifting Mechanism. Nucleic Acids Res. 1990, 18, 1825–1832. [Google Scholar]
- Brierley, I.; Digard, P.; Inglis, S.C. Characterization of an Efficient Coronavirus Ribosomal Frameshifting Signal: Requirement for an RNA Pseudoknot. Cell 1989, 57, 537–547. [Google Scholar] [CrossRef]
- Ziebuhr, J.; Snijder, E.J.; Gorbalenya, A.E. Virus-Encoded Proteinases and Proteolytic Processing in the Nidovirales. J. Gen. Virol. 2000, 81, 853–879. [Google Scholar]
- Deming, D.J.; Graham, R.L.; Denison, M.R.; Baric, R.S. Processing of Open Reading Frame 1a Replicase Proteins nsp7 to nsp10 in Murine Hepatitis Virus Strain A59 Replication. J. Virol. 2007, 81, 10280–10291. [Google Scholar]
- Sawicki, S.G.; Sawicki, D.L.; Younker, D.; Meyer, Y.; Thiel, V.; Stokes, H.; Siddell, S.G. Functional and Genetic Analysis of Coronavirus Replicase-Transcriptase Proteins. PLoS Pathog. 2005, 1, e39. [Google Scholar] [CrossRef]
- te Velthuis, A.J.; Arnold, J.J.; Cameron, C.E.; van den Worm, S.H.; Snijder, E.J. The RNA Polymerase Activity of SARS-Coronavirus nsp12 is Primer Dependent. Nucleic Acids Res. 2010, 38, 203–214. [Google Scholar]
- Imbert, I.; Guillemot, J.C.; Bourhis, J.M.; Bussetta, C.; Coutard, B.; Egloff, M.P.; Ferron, F.; Gorbalenya, A.E.; Canard, B. A Second, Non-Canonical RNA-Dependent RNA Polymerase in SARS Coronavirus. EMBO J. 2006, 25, 4933–4942. [Google Scholar]
- Zhai, Y.; Sun, F.; Li, X.; Pang, H.; Xu, X.; Bartlam, M.; Rao, Z. Insights into SARS-CoV Transcription and Replication from the Structure of the nsp7-nsp8 Hexadecamer. Nat. Struct. Mol. Biol. 2005, 12, 980–986. [Google Scholar]
- Xiao, Y.; Ma, Q.; Restle, T.; Shang, W.; Svergun, D.I.; Ponnusamy, R.; Sczakiel, G.; Hilgenfeld, R. Nonstructural Proteins 7 and 8 of Feline Coronavirus Form a 2:1 Heterotrimer that Exhibits Primer-Independent RNA Polymerase Activity. J. Virol. 2012, 86, 4444–4454. [Google Scholar] [CrossRef]
- te Velthuis, A.J.; van den Worm, S.H.; Snijder, E.J. The SARS-Coronavirus nsp7+nsp8 Complex is a Unique Multimeric RNA Polymerase Capable of both De Novo Initiation and Primer Extension. Nucleic Acids Res. 2012, 40, 1737–1747. [Google Scholar] [CrossRef]
- Seybert, A.; Posthuma, C.C.; van Dinten, L.C.; Snijder, E.J.; Gorbalenya, A.E.; Ziebuhr, J. A Complex Zinc Finger Controls the Enzymatic Activities of Nidovirus Helicases. J. Virol. 2005, 79, 696–704. [Google Scholar] [CrossRef]
- Seybert, A.; Hegyi, A.; Siddell, S.G.; Ziebuhr, J. The Human Coronavirus 229E Superfamily 1 Helicase has RNA and DNA Duplex-Unwinding Activities with 5'-to-3' Polarity. RNA 2000, 6, 1056–1068. [Google Scholar] [CrossRef]
- Tanner, J.A.; Watt, R.M.; Chai, Y.B.; Lu, L.Y.; Lin, M.C.; Peiris, J.S.; Poon, L.L.; Kung, H.F.; Huang, J.D. The Severe Acute Respiratory Syndrome (SARS) Coronavirus NTPase/helicase Belongs to a Distinct Class of 5' to 3' Viral Helicases. J. Biol. Chem. 2003, 278, 39578–39582. [Google Scholar]
- Ivanov, K.A.; Thiel, V.; Dobbe, J.C.; van der Meer, Y.; Snijder, E.J.; Ziebuhr, J. Multiple Enzymatic Activities Associated with Severe Acute Respiratory Syndrome Coronavirus Helicase. J. Virol. 2004, 78, 5619–5632. [Google Scholar] [CrossRef]
- Ivanov, K.A.; Ziebuhr, J. Human Coronavirus 229E Nonstructural Protein 13: Characterization of Duplex-Unwinding, Nucleoside Triphosphatase, and RNA 5'-Triphosphatase Activities. J. Virol. 2004, 78, 7833–7838. [Google Scholar] [CrossRef]
- Chen, Y.; Cai, H.; Pan, J.; Xiang, N.; Tien, P.; Ahola, T.; Guo, D. Functional Screen Reveals SARS Coronavirus Nonstructural Protein nsp14 as a Novel Cap N7 Methyltransferase. Proc. Natl. Acad. Sci. USA 2009, 106, 3484–3489. [Google Scholar]
- Decroly, E.; Imbert, I.; Coutard, B.; Bouvet, M.; Selisko, B.; Alvarez, K.; Gorbalenya, A.E.; Snijder, E.J.; Canard, B. Coronavirus Nonstructural Protein 16 is a Cap-0 Binding Enzyme Possessing (Nucleoside-2'O)-Methyltransferase Activity. J. Virol. 2008, 82, 8071–8084. [Google Scholar] [CrossRef]
- Bouvet, M.; Debarnot, C.; Imbert, I.; Selisko, B.; Snijder, E.J.; Canard, B.; Decroly, E. In Vitro Reconstitution of SARS-Coronavirus mRNA Cap Methylation. PLoS Pathog. 2010, 6, e1000863. [Google Scholar]
- Lugari, A.; Betzi, S.; Decroly, E.; Bonnaud, E.; Hermant, A.; Guillemot, J.C.; Debarnot, C.; Borg, J.P.; Bouvet, M.; Canard, B. et al. Molecular Mapping of the RNA Cap 2'-O-Methyltransferase Activation Interface between Severe Acute Respiratory Syndrome Coronavirus nsp10 and nsp16. J. Biol. Chem. 2010, 285, 33230–33241. [Google Scholar]
- Minskaia, E.; Hertzig, T.; Gorbalenya, A.E.; Campanacci, V.; Cambillau, C.; Canard, B.; Ziebuhr, J. Discovery of an RNA Virus 3'→5' Exoribonuclease that is Critically Involved in Coronavirus RNA Synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 5108–5113. [Google Scholar]
- Ivanov, K.A.; Hertzig, T.; Rozanov, M.; Bayer, S.; Thiel, V.; Gorbalenya, A.E.; Ziebuhr, J. Major Genetic Marker of Nidoviruses Encodes a Replicative Endoribonuclease. Proc. Natl. Acad. Sci. USA 2004, 101, 12694–12699. [Google Scholar]
- Bhardwaj, K.; Sun, J.; Holzenburg, A.; Guarino, L.A.; Kao, C.C. RNA Recognition and Cleavage by the SARS Coronavirus Endoribonuclease. J. Mol. Biol. 2006, 361, 243–256. [Google Scholar]
- Eckerle, L.D.; Becker, M.M.; Halpin, R.A.; Li, K.; Venter, E.; Lu, X.; Scherbakova, S.; Graham, R.L.; Baric, R.S.; Stockwell, T.B.; et al. Infidelity of SARS-CoV Nsp14-Exonuclease Mutant Virus Replication is Revealed by Complete Genome Sequencing. PLoS Pathog. 2010, 6, e1000896. [Google Scholar] [CrossRef]
- Denison, M.R.; Graham, R.L.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses: An RNA Proofreading Machine Regulates Replication Fidelity and Diversity. RNA Biol. 2011, 8, 270–279. [Google Scholar] [CrossRef]
- Bouvet, M.; Imbert, I.; Subissi, L.; Gluais, L.; Canard, B.; Decroly, E. RNA 3'-End Mismatch Excision by the Severe Acute Respiratory Syndrome Coronavirus Nonstructural Protein nsp10/nsp14 Exoribonuclease Complex. Proc. Natl. Acad. Sci. USA 2012, 109, 9372–9377. [Google Scholar]
- Harcourt, B.H.; Jukneliene, D.; Kanjanahaluethai, A.; Bechill, J.; Severson, K.M.; Smith, C.M.; Rota, P.A.; Baker, S.C. Identification of Severe Acute Respiratory Syndrome Coronavirus Replicase Products and Characterization of Papain-Like Protease Activity. J. Virol. 2004, 78, 13600–13612. [Google Scholar] [CrossRef]
- Oostra, M.; Hagemeijer, M.C.; van Gent, M.; Bekker, C.P.; te Lintelo, E.G.; Rottier, P.J.; de Haan, C.A. Topology and Membrane Anchoring of the Coronavirus Replication Complex: Not all Hydrophobic Domains of nsp3 and nsp6 are Membrane Spanning. J. Virol. 2008, 82, 12392–12405. [Google Scholar] [CrossRef]
- Oostra, M.; te Lintelo, E.G.; Deijs, M.; Verheije, M.H.; Rottier, P.J.; de Haan, C.A. Localization and Membrane Topology of Coronavirus Nonstructural Protein 4: Involvement of the Early Secretory Pathway in Replication. J. Virol. 2007, 81, 12323–12336. [Google Scholar] [CrossRef]
- Kanjanahaluethai, A.; Chen, Z.; Jukneliene, D.; Baker, S.C. Membrane Topology of Murine Coronavirus Replicase Nonstructural Protein 3. Virology 2007, 361, 391–401. [Google Scholar] [CrossRef]
- Baliji, S.; Cammer, S.A.; Sobral, B.; Baker, S.C. Detection of Nonstructural Protein 6 in Murine Coronavirus-Infected Cells and Analysis of the Transmembrane Topology by using Bioinformatics and Molecular Approaches. J. Virol. 2009, 83, 6957–6962. [Google Scholar] [CrossRef]
- Kanjanahaluethai, A.; Baker, S.C. Identification of Mouse Hepatitis Virus Papain-like Proteinase 2 Activity. J. Virol. 2000, 74, 7911–7921. [Google Scholar]
- Gosert, R.; Kanjanahaluethai, A.; Egger, D.; Bienz, K.; Baker, S.C. RNA Replication of Mouse Hepatitis Virus Takes Place at Double-Membrane Vesicles. J. Virol. 2002, 76, 3697–3708. [Google Scholar]
- Sutton, G.; Fry, E.; Carter, L.; Sainsbury, S.; Walter, T.; Nettleship, J.; Berrow, N.; Owens, R.; Gilbert, R.; Davidson, A.; et al. The nsp9 Replicase Protein of SARS-Coronavirus, Structure and Functional Insights. Structure 2004, 12, 341–353. [Google Scholar]
- Su, D.; Lou, Z.; Sun, F.; Zhai, Y.; Yang, H.; Zhang, R.; Joachimiak, A.; Zhang, X.C.; Bartlam, M.; Rao, Z. Dodecamer Structure of Severe Acute Respiratory Syndrome Coronavirus Nonstructural Protein nsp10. J. Virol. 2006, 80, 7902–7908. [Google Scholar] [CrossRef]
- Zust, R.; Cervantes-Barragan, L.; Kuri, T.; Blakqori, G.; Weber, F.; Ludewig, B.; Thiel, V. Coronavirus Non-Structural Protein 1 is a Major Pathogenicity Factor: Implications for the Rational Design of Coronavirus Vaccines. PLoS Pathog. 2007, 3, e109. [Google Scholar]
- Wang, G.; Chen, G.; Zheng, D.; Cheng, G.; Tang, H. PLP2 of Mouse Hepatitis Virus A59 (MHV-A59) Targets TBK1 to Negatively Regulate Cellular Type I Interferon Signaling Pathway. PLoS One 2011, 6, e17192. [Google Scholar]
- Wathelet, M.G.; Orr, M.; Frieman, M.B.; Baric, R.S. Severe Acute Respiratory Syndrome Coronavirus Evades Antiviral Signaling: Role of nsp1 and Rational Design of an Attenuated Strain. J. Virol. 2007, 81, 11620–11633. [Google Scholar] [CrossRef]
- Narayanan, K.; Huang, C.; Lokugamage, K.; Kamitani, W.; Ikegami, T.; Tseng, C.T.; Makino, S. Severe Acute Respiratory Syndrome Coronavirus nsp1 Suppresses Host Gene Expression, Including that of Type I Interferon, in Infected Cells. J. Virol. 2008, 82, 4471–4479. [Google Scholar]
- Kuri, T.; Eriksson, K.K.; Putics, A.; Zust, R.; Snijder, E.J.; Davidson, A.D.; Siddell, S.G.; Thiel, V.; Ziebuhr, J.; Weber, F. The ADP-Ribose-1"-Monophosphatase Domains of SARS-Coronavirus and Human Coronavirus 229E Mediate Resistance to Antiviral Interferon Responses. J. Gen. Virol. 2011, 92, 1899–1905. [Google Scholar]
- Kamitani, W.; Narayanan, K.; Huang, C.; Lokugamage, K.; Ikegami, T.; Ito, N.; Kubo, H.; Makino, S. Severe Acute Respiratory Syndrome Coronavirus nsp1 Protein Suppresses Host Gene Expression by Promoting Host mRNA Degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 12885–12890. [Google Scholar]
- Kamitani, W.; Huang, C.; Narayanan, K.; Lokugamage, K.G.; Makino, S. A Two-Pronged Strategy to Suppress Host Protein Synthesis by SARS Coronavirus Nsp1 Protein. Nat. Struct. Mol. Biol. 2009, 16, 1134–1140. [Google Scholar]
- Chen, C.J.; Sugiyama, K.; Kubo, H.; Huang, C.; Makino, S. Murine Coronavirus Nonstructural Protein p28 Arrests Cell Cycle in G0/G1 Phase. J. Virol. 2004, 78, 10410–10419. [Google Scholar]
- Gadlage, M.J.; Graham, R.L.; Denison, M.R. Murine Coronaviruses Encoding nsp2 at Different Genomic Loci have Altered Replication, Protein Expression, and Localization. J. Virol. 2008, 82, 11964–11969. [Google Scholar] [CrossRef]
- Graham, R.L.; Sims, A.C.; Brockway, S.M.; Baric, R.S.; Denison, M.R. The nsp2 Replicase Proteins of Murine Hepatitis Virus and Severe Acute Respiratory Syndrome Coronavirus are Dispensable for Viral Replication. J. Virol. 2005, 79, 13399–13411. [Google Scholar]
- Perlman, S.; Netland, J. Coronaviruses Post-SARS: Update on Replication and Pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef]
- Ziebuhr, J. The Coronavirus Replicase: Insights into a Sophisticated Enzyme Machinery. Adv. Exp. Med. Biol. 2006, 581, 3–11. [Google Scholar] [CrossRef]
- Ulasli, M.; Verheije, M.H.; de Haan, C.A.; Reggiori, F. Qualitative and Quantitative Ultrastructural Analysis of the Membrane Rearrangements Induced by Coronavirus. Cell. Microbiol. 2010, 12, 844–861. [Google Scholar] [CrossRef]
- Snijder, E.J.; van der Meer, Y.; Zevenhoven-Dobbe, J.; Onderwater, J.J.; van der Meulen, J.; Koerten, H.K.; Mommaas, A.M. Ultrastructure and Origin of Membrane Vesicles Associated with the Severe Acute Respiratory Syndrome Coronavirus Replication Complex. J. Virol. 2006, 80, 5927–5940. [Google Scholar] [CrossRef]
- David-Ferreira, J.F.; Manaker, R.A. An Electron Microscope Study of the Development of a Mouse Hepatitis Virus in Tissue Culture Cells. J. Cell. Biol. 1965, 24, 57–78. [Google Scholar] [CrossRef]
- Ng, M.L.; Tan, S.H.; See, E.E.; Ooi, E.E.; Ling, A.E. Proliferative Growth of SARS Coronavirus in Vero E6 Cells. J. Gen. Virol. 2003, 84, 3291–3303. [Google Scholar]
- Knoops, K.; Swett-Tapia, C.; van den Worm, S.H.E.; te Velthuis, A.J.W.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J.; Kikkert, M. Integrity of the Early Secretory Pathway Promotes, but is Not Required for, Severe Acute Respiratory Syndrome Coronavirus RNA Synthesis and Virus-Induced Remodeling of Endoplasmic Reticulum Membranes. J. Virol. 2010, 84, 833–846. [Google Scholar]
- Verheije, M.H.; Raaben, M.; Mari, M.; Te Lintelo, E.G.; Reggiori, F.; van Kuppeveld, F.J.; Rottier, P.J.; de Haan, C.A. Mouse Hepatitis Coronavirus RNA Replication Depends on GBF1-Mediated ARF1 Activation. PLoS Pathog. 2008, 4, e1000088. [Google Scholar] [CrossRef]
- Prentice, E.; McAuliffe, J.; Lu, X.; Subbarao, K.; Denison, M.R. Identification and Characterization of Severe Acute Respiratory Syndrome Coronavirus Replicase Proteins. J. Virol. 2004, 78, 9977–9986. [Google Scholar] [CrossRef]
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Calì, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; de Haan, C.A.M.; Molinari, M. Coronaviruses Hijack the LC3-I-Positive EDEMosomes, ER-Derived Vesicles Exporting Short-Lived ERAD Regulators, for Replication. Cell Host Microbe 2010, 7, 500–508. [Google Scholar] [CrossRef]
- Shi, S.T.; Schiller, J.J.; Kanjanahaluethai, A.; Baker, S.C.; Oh, J.W.; Lai, M.M. Colocalization and Membrane Association of Murine Hepatitis Virus Gene 1 Products and De Novo-Synthesized Viral RNA in Infected Cells. J. Virol. 1999, 73, 5957–5969. [Google Scholar]
- van der Meer, Y.; Snijder, E.J.; Dobbe, J.C.; Schleich, S.; Denison, M.R.; Spaan, W.J.; Locker, J.K. Localization of Mouse Hepatitis Virus Nonstructural Proteins and RNA Synthesis Indicates a Role for Late Endosomes in Viral Replication. J. Virol. 1999, 73, 7641–7657. [Google Scholar]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus Replication Complex Formation Utilizes Components of Cellular Autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar]
- Zhao, Z.; Thackray, L.B.; Miller, B.C.; Lynn, T.M.; Becker, M.M.; Ward, E.; Mizushima, N.N.; Denison, M.R.; Virgin, H.W., 4th. Coronavirus Replication does Not Require the Autophagy Gene ATG5. Autophagy 2007, 3, 581–585. [Google Scholar]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus nsp6 Proteins Generate Autophagosomes from the Endoplasmic Reticulum via an Omegasome Intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef]
- de Haan, C.A.M.; Reggiori, F.; Molinari, M. Autophagy-Independent LC3 Function in Vesicular Traffic. Autophagy 2010, 6, 994–996. [Google Scholar] [CrossRef]
- Stertz, S.; Reichelt, M.; Spiegel, M.; Kuri, T.; Martinez-Sobrido, L.; Garcia-Sastre, A.; Weber, F.; Kochs, G. The Intracellular Sites of Early Replication and Budding of SARS-Coronavirus. Virology 2007, 361, 304–315. [Google Scholar]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In Vivo Analysis of Autophagy in Response to Nutrient Starvation using Transgenic Mice Expressing a Fluorescent Autophagosome Marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar]
- Cali, T.; Galli, C.; Olivari, S.; Molinari, M. Segregation and Rapid Turnover of EDEM1 by an Autophagy-Like Mechanism Modulates Standard ERAD and Folding Activities. Biochem. Biophys. Res. Commun. 2008, 371, 405–410. [Google Scholar] [CrossRef]
- Cali, T.; Vanoni, O.; Molinari, M. The Endoplasmic Reticulum Crossroads for Newly Synthesized Polypeptide Chains. Prog. Mol. Biol. Transl. Sci. 2008, 83, 135–179. [Google Scholar] [CrossRef]
- Bernasconi, R.; Galli, C.; Noack, J.; Bianchi, S.; de Haan, C.A.; Reggiori, F.; Molinari, M. Role of the SEL1L:LC3-I Complex as an ERAD Tuning Receptor in the Mammalian ER. Mol. Cell. 2012, 46, 809–819. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Ulasli, M.; Vonk, A.; Reggiori, F.; Rottier, P.J.M.; de Haan, C.A.M. Mobility and Interactions of the Coronavirus Nonstructural Protein 4. J. Virol. 2011, 85, 4572–4577. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; de Haan, C.A.M. Utrecht University: Utrecht, the Netherlands, 2012; Unpublished work.
- Gadlage, M.J.; Sparks, J.S.; Beachboard, D.C.; Cox, R.G.; Doyle, J.D.; Stobart, C.C.; Denison, M.R. Murine Hepatitis Virus Nonstructural Protein 4 Regulates Virus-Induced Membrane Modifications and Replication Complex Function. J. Virol. 2010, 84, 280–290. [Google Scholar] [CrossRef]
- Sparks, J.S.; Lu, X.; Denison, M.R. Genetic Analysis of Murine Hepatitis Virus nsp4 in Virus Replication. J. Virol. 2007, 81, 12554–12563. [Google Scholar] [CrossRef]
- Snijder, E.J.; van Tol, H.; Roos, N.; Pedersen, K.W. Non-Structural Proteins 2 and 3 Interact to Modify Host Cell Membranes during the Formation of the Arterivirus Replication Complex. J. Gen. Virol. 2001, 82, 985–994. [Google Scholar]
- Pedersen, K.W.; van der Meer, Y.; Roos, N.; Snijder, E.J. Open Reading Frame 1a-Encoded Subunits of the Arterivirus Replicase Induce Endoplasmic Reticulum-Derived Double-Membrane Vesicles which Carry the Viral Replication Complex. J. Virol. 1999, 73, 2016–2026. [Google Scholar]
- Posthuma, C.C.; Pedersen, K.W.; Lu, Z.; Joosten, R.G.; Roos, N.; Zevenhoven-Dobbe, J.C.; Snijder, E.J. Formation of the Arterivirus replication/transcription Complex: A Key Role for Nonstructural Protein 3 in the Remodeling of Intracellular Membranes. J. Virol. 2008, 82, 4480–4491. [Google Scholar]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Buhler, S.; Bartenschlager, R. The Non-Structural Protein 4A of Dengue Virus is an Integral Membrane Protein Inducing Membrane Alterations in a 2K-Regulated Manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar] [CrossRef]
- Roosendaal, J.; Westaway, E.G.; Khromykh, A.; Mackenzie, J.M. Regulated Cleavages at the West Nile Virus NS4A-2K-NS4B Junctions Play a Major Role in Rearranging Cytoplasmic Membranes and Golgi Trafficking of the NS4A Protein. J. Virol. 2006, 80, 4623–4632. [Google Scholar]
- Egger, D.; Wolk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of Hepatitis C Virus Proteins Induces Distinct Membrane Alterations Including a Candidate Viral Replication Complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar] [CrossRef]
- den Boon, J.A.; Chen, J.; Ahlquist, P. Identification of Sequences in Brome Mosaic Virus Replicase Protein 1a that Mediate Association with Endoplasmic Reticulum Membranes. J. Virol. 2001, 75, 12370–12381. [Google Scholar]
- Echeverri, A.C.; Dasgupta, A. Amino Terminal Regions of Poliovirus 2C Protein Mediate Membrane Binding. Virology 1995, 208, 540–553. [Google Scholar] [CrossRef]
- Lundin, M.; Monne, M.; Widell, A.; von Heijne, G.; Persson, M.A.A. Topology of the Membrane-Associated Hepatitis C Virus Protein NS4B. J. Virol. 2003, 77, 5428–5438. [Google Scholar] [CrossRef]
- Miller, D.J.; Ahlquist, P. Flock House Virus RNA Polymerase is a Transmembrane Protein with Amino-Terminal Sequences Sufficient for Mitochondrial Localization and Membrane Insertion. J. Virol. 2002, 76, 9856–9867. [Google Scholar] [CrossRef]
- Miller, S.; Sparacio, S.; Bartenschlager, R. Subcellular Localization and Membrane Topology of the Dengue Virus Type 2 Non-Structural Protein 4B. J. Biol. Chem. 2006, 281, 8854–8863. [Google Scholar]
- O'Reilly, E.K.; Wang, Z.; French, R.; Kao, C.C. Interactions between the Structural Domains of the RNA Replication Proteins of Plant-Infecting RNA Viruses. J. Virol. 1998, 72, 7160–7169. [Google Scholar]
- O'Reilly, E.; Paul, J.; Kao, C. Analysis of the Interaction of Viral RNA Replication Proteins by using the Yeast Two-Hybrid Assay. J. Virol. 1997, 71, 7526–7532. [Google Scholar]
- Paul, D.; Romero-Brey, I.; Gouttenoire, J.; Stoitsova, S.; Krijnse-Locker, J.; Moradpour, D.; Bartenschlager, R. NS4B Self-Interaction through Conserved C-Terminal Elements is Required for the Establishment of Functional Hepatitis C Virus Replication Complexes. J. Virol. 2011, 85, 6963–6976. [Google Scholar]
- Mackenzie, J.M.; Khromykh, A.A.; Jones, M.K.; Westaway, E.G. Subcellular Localization and some Biochemical Properties of the Flavivirus Kunjin Nonstructural Proteins NS2A and NS4A. Virology 1998, 245, 203–215. [Google Scholar] [CrossRef]
- McMahon, H.T.; Gallop, J.L. Membrane Curvature and Mechanisms of Dynamic Cell Membrane Remodelling. Nature 2005, 438, 590–596. [Google Scholar] [CrossRef]
- Shibata, Y.; Hu, J.; Kozlov, M.M.; Rapoport, T.A. Mechanisms Shaping the Membranes of Cellular Organelles. Annu. Rev. Cell Dev. Biol. 2009, 25, 329–354. [Google Scholar] [CrossRef]
- Zimmerberg, J.; Kozlov, M.M. How Proteins Produce Cellular Membrane Curvature. Nat. Rev. Mol. Cell Biol. 2006, 7, 9–19. [Google Scholar] [CrossRef]
- Jones, D.M.; Gretton, S.N.; McLauchlan, J.; Targett-Adams, P. Mobility Analysis of an NS5A–GFP Fusion Protein in Cells Actively Replicating Hepatitis C Virus Subgenomic RNA. J. Gen. Virol. 2007, 88, 470–475. [Google Scholar]
- Gretton, S.N.; Taylor, A.I.; McLauchlan, J. Mobility of the Hepatitis C Virus NS4B Protein on the Endoplasmic Reticulum Membrane and Membrane-Associated Foci. J. Gen. Virol. 2005, 86, 1415–1421. [Google Scholar]
- Cui, Z.; Zhang, Z.; Zhang, X.; Wen, J.; Zhou, Y.; Xie, W. Visualizing the Dynamic Behavior of Poliovirus Plus-Strand RNA in Living Host Cells. Nucleic Acids Res. 2005, 33, 3245–3252. [Google Scholar] [CrossRef]
- Kujala, P.; Ikaheimonen, A.; Ehsani, N.; Vihinen, H.; Auvinen, P.; Kaariainen, L. Biogenesis of the Semliki Forest Virus RNA Replication Complex. J. Virol. 2001, 75, 3873–3884. [Google Scholar]
- Spuul, P.; Balistreri, G.; Kaariainen, L.; Ahola, T. Phosphatidylinositol 3-Kinase-, Actin-, and Microtubule-Dependent Transport of Semliki Forest Virus Replication Complexes from the Plasma Membrane to Modified Lysosomes. J. Virol. 2010, 84, 7543–7557. [Google Scholar] [CrossRef]
- Wolk, B.; Buchele, B.; Moradpour, D.; Rice, C.M. A Dynamic View of Hepatitis C Virus Replication Complexes. J. Virol. 2008, 82, 10519–10531. [Google Scholar] [CrossRef]
- Egger, D.; Bienz, K. Intracellular Location and Translocation of Silent and Active Poliovirus Replication Complexes. J. Gen. Virol. 2005, 86, 707–718. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Verheije, M.H.; Ulasli, M.; Shaltiël, I.A.; de Vries, L.A.; Reggiori, F.; Rottier, P.J.M.; de Haan, C.A.M. Dynamics of Coronavirus Replication-Transcription Complexes. J. Virol. 2010, 84, 2134–2149. [Google Scholar]
- Ma, S.; Chisholm, R.L. Cytoplasmic Dynein-Associated Structures Move Bidirectionally in Vivo. J. Cell Sci. 2002, 115, 1453–1460. [Google Scholar]
- Bost, A.G.; Venable, D.; Liu, L.; Heinz, B.A. Cytoskeletal Requirements for Hepatitis C Virus (HCV) RNA Synthesis in the HCV Replicon Cell Culture System. J. Virol. 2003, 77, 4401–4408. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Vonk, A.M.; Monastyrska, I.; Rottier, P.J.; de Haan, C.A. Visualizing Coronavirus RNA Synthesis in Time by using Click Chemistry. J. Virol. 2012, 86, 5808–5816. [Google Scholar]
- Imbert, I.; Snijder, E.J.; Dimitrova, M.; Guillemot, J.C.; Lecine, P.; Canard, B. The SARS-Coronavirus PLnc Domain of nsp3 as a replication/transcription Scaffolding Protein. Virus Res. 2008, 133, 136–148. [Google Scholar]
- Pan, J.; Peng, X.; Gao, Y.; Li, Z.; Lu, X.; Chen, Y.; Ishaq, M.; Liu, D.; DeDiego, M.L.; Enjuanes, L.; et al. Genome-Wide Analysis of Protein-Protein Interactions and Involvement of Viral Proteins in SARS-CoV Replication. PLoS One 2008, 3, e3299. [Google Scholar]
- von Brunn, A.; Teepe, C.; Simpson, J.C.; Pepperkok, R.; Friedel, C.C.; Zimmer, R.; Roberts, R.; Baric, R.; Haas, J. Analysis of Intraviral Protein-Protein Interactions of the SARS Coronavirus ORFeome. PLoS One 2007, 2, e459. [Google Scholar]
- Bost, A.G.; Carnahan, R.H.; Lu, X.T.; Denison, M.R. Four Proteins Processed from the Replicase Gene Polyprotein of Mouse Hepatitis Virus Colocalize in the Cell Periphery and Adjacent to Sites of Virion Assembly. J. Virol. 2000, 74, 3379–3387. [Google Scholar]
- Denison, M.R.; Spaan, W.J.; van der Meer, Y.; Gibson, C.A.; Sims, A.C.; Prentice, E.; Lu, X.T. The Putative Helicase of the Coronavirus Mouse Hepatitis Virus is Processed from the Replicase Gene Polyprotein and Localizes in Complexes that are Active in Viral RNA Synthesis. J. Virol. 1999, 73, 6862–6871. [Google Scholar]
- Hurst, K.R.; Ye, R.; Goebel, S.J., Javaraman; Masters, P.S. An Interaction between the Nucleocapsid Protein and a Component of the Replicase-Transcriptase Complex is Crucial for the Infectivity of Coronavirus Genomic RNA. J. Virol. 2010, 84, 10276–10288. [Google Scholar]
- Verheije, M.H.; Hagemeijer, M.C.; Ulasli, M.; Reggiori, F.; Rottier, P.J.M.; Masters, P.S.; de Haan, C.A.M. The Coronavirus Nucleocapsid Protein is Dynamically Associated with the Replication-Transcription Complexes. J. Virol. 2010, 84, 11575–11579. [Google Scholar] [CrossRef]
- Almazan, F.; Galan, C.; Enjuanes, L. The Nucleoprotein is Required for Efficient Coronavirus Genome Replication. J. Virol. 2004, 78, 12683–12688. [Google Scholar] [CrossRef]
- Compton, S.R.; Rogers, D.B.; Holmes, K.V.; Fertsch, D.; Remenick, J.; McGowan, J.J. In Vitro Replication of Mouse Hepatitis Virus Strain A59. J. Virol. 1987, 61, 1814–1820. [Google Scholar]
- Schelle, B.; Karl, N.; Ludewig, B.; Siddell, S.G.; Thiel, V. Selective Replication of Coronavirus Genomes that Express Nucleocapsid Protein. J. Virol. 2005, 79, 6620–6630. [Google Scholar] [CrossRef]
- de Haan, C.A.; Rottier, P.J. Molecular Interactions in the Assembly of Coronaviruses. Adv. Virus Res. 2005, 64, 165–230. [Google Scholar] [CrossRef]
- van Hemert, M.J.; van den Worm, S.H.; Knoops, K.; Mommaas, A.M.; Gorbalenya, A.E.; Snijder, E.J. SARS-Coronavirus replication/transcription Complexes are Membrane-Protected and Need a Host Factor for Activity in Vitro. PLoS Pathog. 2008, 4, e1000054. [Google Scholar]
- Schwartz, M.; Chen, J.; Janda, M.; Sullivan, M.; den Boon, J.; Ahlquist, P. A Positive-Strand RNA Virus Replication Complex Parallels Form and Function of Retrovirus Capsids. Mol. Cell 2002, 9, 505–514. [Google Scholar] [CrossRef]
- Kopek, B.G.; Perkins, G.; Miller, D.J.; Ellisman, M.H.; Ahlquist, P. Three-Dimensional Analysis of a Viral RNA Replication Complex Reveals a Virus-Induced Mini-Organelle. PLoS Biol. 2007, 5, e220. [Google Scholar]
- Westaway, E.G.; Khromykh, A.A.; Mackenzie, J.M. Nascent Flavivirus RNA Colocalizedin Situwith Double-Stranded RNA in Stable Replication Complexes. Virology 1999, 258, 108–117. [Google Scholar] [CrossRef]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The Endoplasmic Reticulum Provides the Membrane Platform for Biogenesis of the Flavivirus Replication Complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and Three-Dimensional Architecture of the Dengue Virus Replication and Assembly Sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef]
- Sola, I.; Mateos-Gomez, P.A.; Almazan, F.; Zuniga, S.; Enjuanes, L. RNA-RNA and RNA-Protein Interactions in Coronavirus Replication and Transcription. RNA Biol. 2011, 8, 237–248. [Google Scholar] [CrossRef]
- Diaz, A.; Wang, X.; Ahlquist, P. Membrane-Shaping Host Reticulon Proteins Play Crucial Roles in Viral RNA Replication Compartment Formation and Function. Proc. Natl. Acad. Sci. USA 2010, 107, 16291–16296. [Google Scholar] [CrossRef]
- Hu, J.; Shibata, Y.; Zhu, P.P.; Voss, C.; Rismanchi, N.; Prinz, W.A.; Rapoport, T.A.; Blackstone, C. A Class of Dynamin-Like GTPases Involved in the Generation of the Tubular ER Network. Cell 2009, 138, 549–561. [Google Scholar]
- Hu, J.; Shibata, Y.; Voss, C.; Shemesh, T.; Li, Z.; Coughlin, M.; Kozlov, M.M.; Rapoport, T.A.; Prinz, W.A. Membrane Proteins of the Endoplasmic Reticulum Induce High-Curvature Tubules. Science 2008, 319, 1247–1250. [Google Scholar] [CrossRef]
- Voeltz, G.K.; Prinz, W.A.; Shibata, Y.; Rist, J.M.; Rapoport, T.A. A Class of Membrane Proteins Shaping the Tubular Endoplasmic Reticulum. Cell 2006, 124, 573–586. [Google Scholar]
- Klopfenstein, D.R.; Klumperman, J.; Lustig, A.; Kammerer, R.A.; Oorschot, V.; Hauri, H. Subdomain-Specific Localization of Climp-63 (P63) in the Endoplasmic Reticulum is Mediated by its Luminal α-Helical Segment. J. Cell Biol. 2001, 153, 1287–1300. [Google Scholar] [CrossRef]
- Farsad, K.; Camilli, P.D. Mechanisms of Membrane Deformation. Curr. Opin. Cell Biol. 2003, 15, 372–381. [Google Scholar] [CrossRef]
- Shibata, Y.; Shemesh, T.; Prinz, W.A.; Palazzo, A.F.; Kozlov, M.M.; Rapoport, T.A. Mechanisms Determining the Morphology of the Peripheral ER. Cell 2010, 143, 774–788. [Google Scholar] [CrossRef]
- Hsu, N.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral Reorganization of the Secretory Pathway Generates Distinct Organelles for RNA Replication. Cell 2010, 141, 799–811. [Google Scholar]
- Reiss, S.; Rebhan, I.; Backes, P.; Romero-Brey, I.; Erfle, H.; Matula, P.; Kaderali, L.; Poenisch, M.; Blankenburg, H.; Hiet, M.; et al. Recruitment and Activation of a Lipid Kinase by Hepatitis C Virus NS5A is Essential for Integrity of the Membranous Replication Compartment. Cell Host Microbe 2011, 9, 32–45. [Google Scholar] [CrossRef]
- Guinea, R.; Carrasco, L. Phospholipid Biosynthesis and Poliovirus Genome Replication, Two Coupled Phenomena. EMBO J. 1990, 9, 2011–2016. [Google Scholar]
- Heaton, N.S.; Perera, R.; Berger, K.L.; Khadka, S.; LaCount, D.J.; Kuhn, R.J.; Randall, G. Dengue Virus Nonstructural Protein 3 Redistributes Fatty Acid Synthase to Sites of Viral Replication and Increases Cellular Fatty Acid Synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 17345–17350. [Google Scholar]
- Perez, L.; Guinea, R.; Carrasco, L. Synthesis of Semliki Forest Virus RNA Requires Continuous Lipid Synthesis. Virology 1991, 183, 74–82. [Google Scholar]
- Perera, R.; Riley, C.; Isaac, G.; Hopf-Jannasch, A.S.; Moore, R.J.; Weitz, K.W.; Pasa-Tolic, L.; Metz, T.O.; Adamec, J.; Kuhn, R.J. Dengue Virus Infection Perturbs Lipid Homeostasis in Infected Mosquito Cells. PLoS Pathog. 2012, 8, e1002584. [Google Scholar] [CrossRef]
- Lippincott-Schwartz, J.; Altan-Bonnet, N.; Patterson, G.H. Photobleaching and Photoactivation: Following Protein Dynamics in Living Cells. Nat. Cell. Biol. Suppl., S7–S14.
- Lippincott-Schwartz, J.; Patterson, G.H. Development and use of Fluorescent Protein Markers in Living Cells. Science 2003, 300, 87–91. [Google Scholar] [CrossRef]
- Fusco, D.; Accornero, N.; Lavoie, B.; Shenoy, S.M.; Blanchard, J.; Singer, R.H.; Bertrand, E. Single mRNA Molecules Demonstrate Probabilistic Movement in Living Mammalian Cells. Curr. Biol. 2003, 13, 161–167. [Google Scholar] [CrossRef]
- Daigle, N.; Ellenberg, J. LambdaN-GFP: An RNA Reporter System for Live-Cell Imaging. Nat. Methods 2007, 4, 633–636. [Google Scholar] [CrossRef]
- Tyagi, S.; Kramer, F.R. Molecular Beacons: Probes that Fluoresce upon Hybridization. Nat. Biotechnol. 1996, 14, 303–308. [Google Scholar] [CrossRef]
- Baskin, J.M.; Prescher, J.A.; Laughlin, S.T.; Agard, N.J.; Chang, P.V.; Miller, I.A.; Lo, A.; Codelli, J.A.; Bertozzi, C.R. Copper-Free Click Chemistry for Dynamic in Vivo Imaging. Proc. Natl. Acad. Sci. USA 2007, 104, 16793–16797. [Google Scholar]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-Diffraction-Limit Imaging by Stochastic Optical Reconstruction Microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar] [CrossRef]
- Hell, S.W.; Wichmann, J. Breaking the Diffraction Resolution Limit by Stimulated Emission: Stimulated-Emission-Depletion Fluorescence Microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar]
- van Rijnsoever, C.; Oorschot, V.; Klumperman, J. Correlative Light-Electron Microscopy (CLEM) Combining Live-Cell Imaging and Immunolabeling of Ultrathin Cryosections. Nat. Methods 2008, 5, 973–980. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hagemeijer, M.C.; Rottier, P.J.M.; Haan, C.A.M.d. Biogenesis and Dynamics of the Coronavirus Replicative Structures. Viruses 2012, 4, 3245-3269. https://doi.org/10.3390/v4113245
Hagemeijer MC, Rottier PJM, Haan CAMd. Biogenesis and Dynamics of the Coronavirus Replicative Structures. Viruses. 2012; 4(11):3245-3269. https://doi.org/10.3390/v4113245
Chicago/Turabian StyleHagemeijer, Marne C., Peter J.M. Rottier, and Cornelis A.M. de Haan. 2012. "Biogenesis and Dynamics of the Coronavirus Replicative Structures" Viruses 4, no. 11: 3245-3269. https://doi.org/10.3390/v4113245
APA StyleHagemeijer, M. C., Rottier, P. J. M., & Haan, C. A. M. d. (2012). Biogenesis and Dynamics of the Coronavirus Replicative Structures. Viruses, 4(11), 3245-3269. https://doi.org/10.3390/v4113245