Foamy Virus Biology and Its Application for Vector Development


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
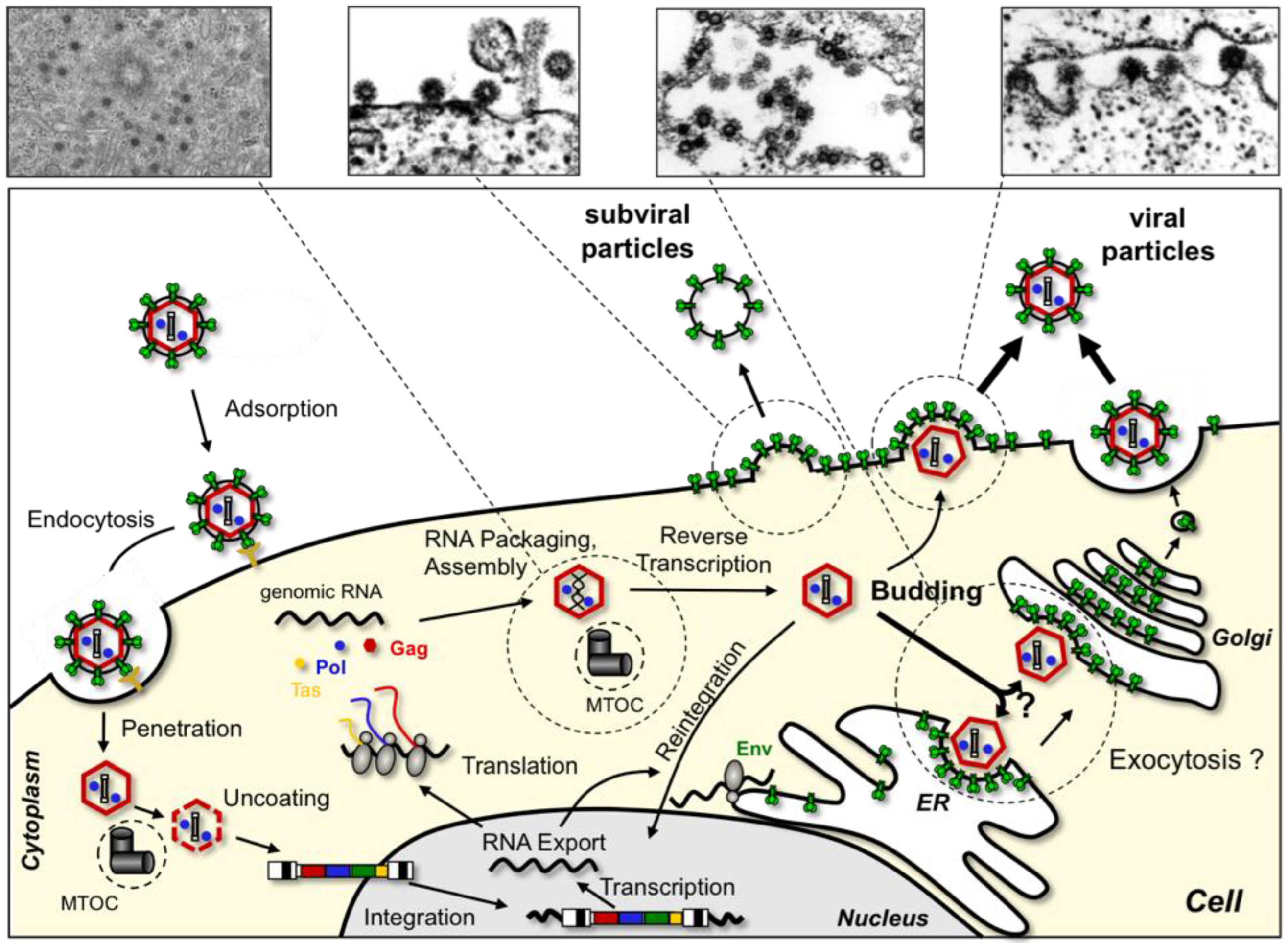
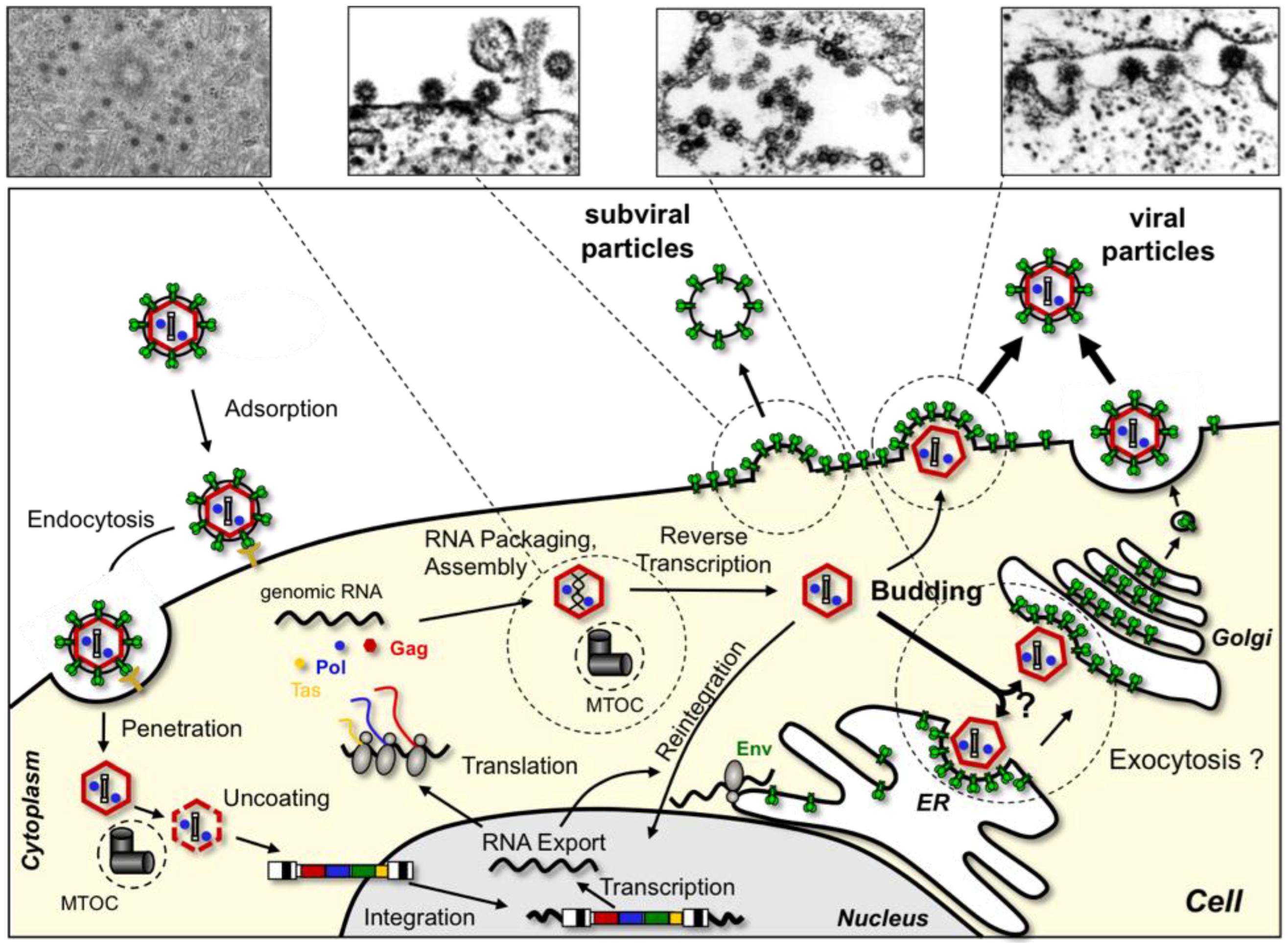
2. The FV Replication Cycle, an Overview
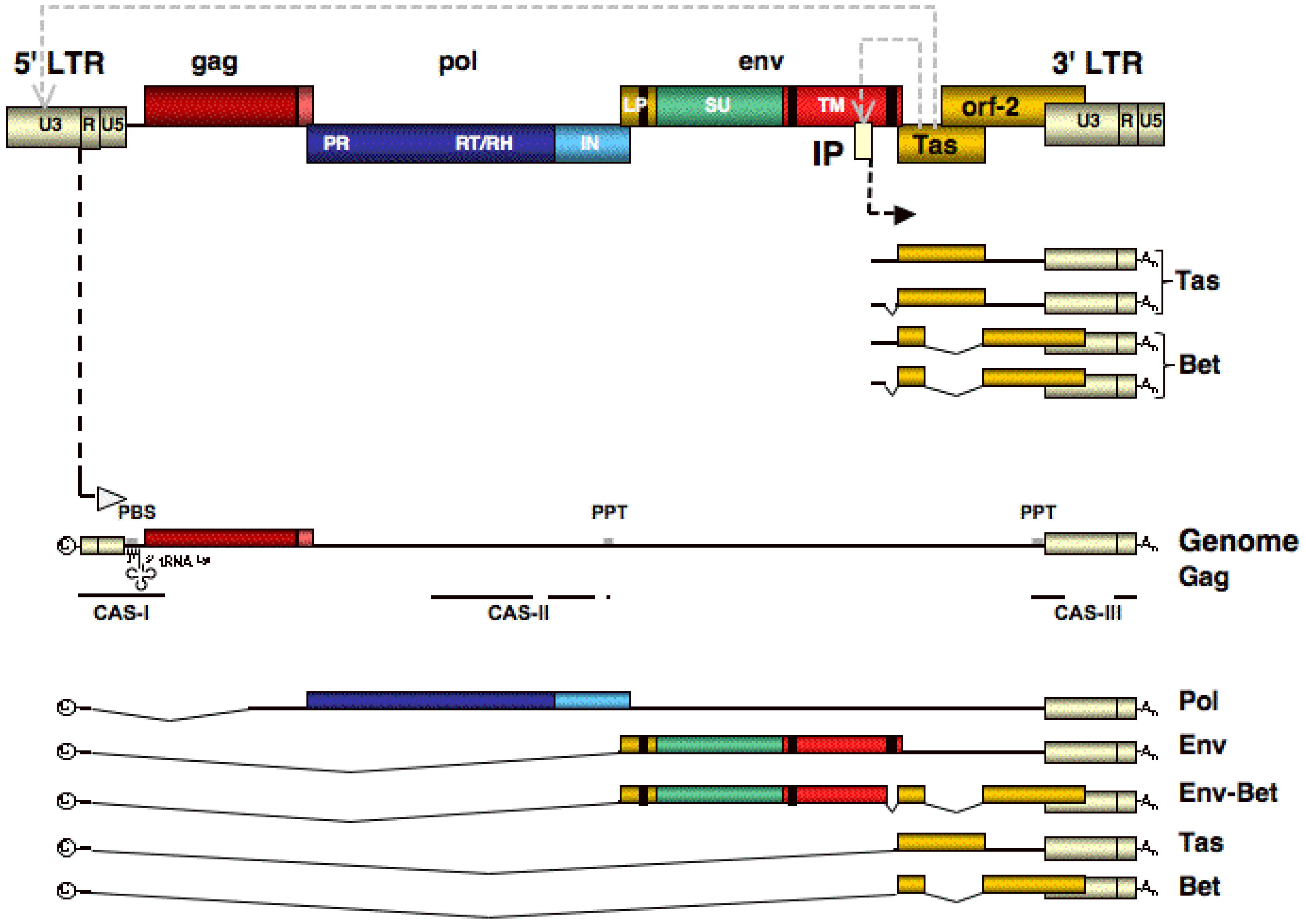
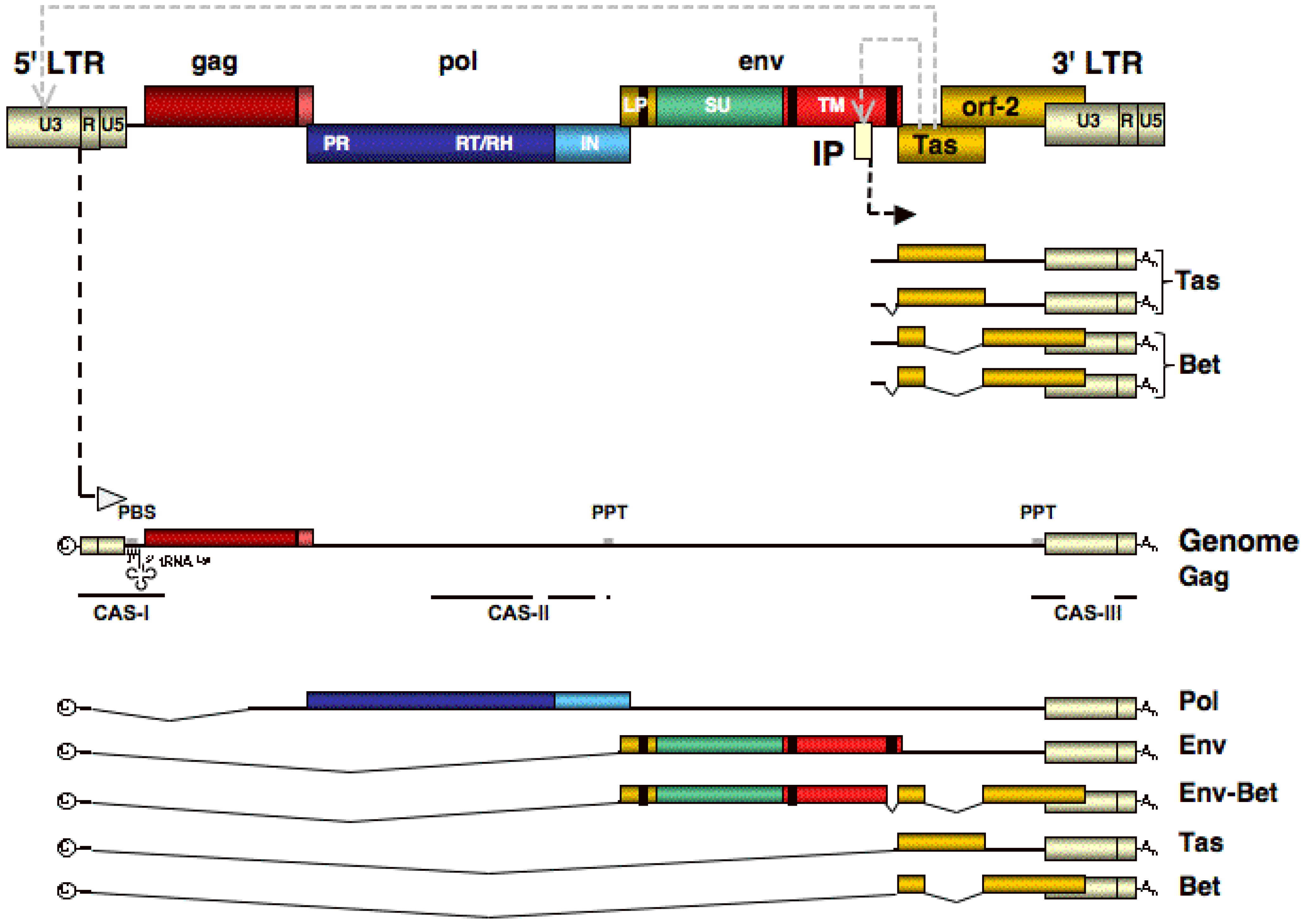
3. Genome Organization and Transcription
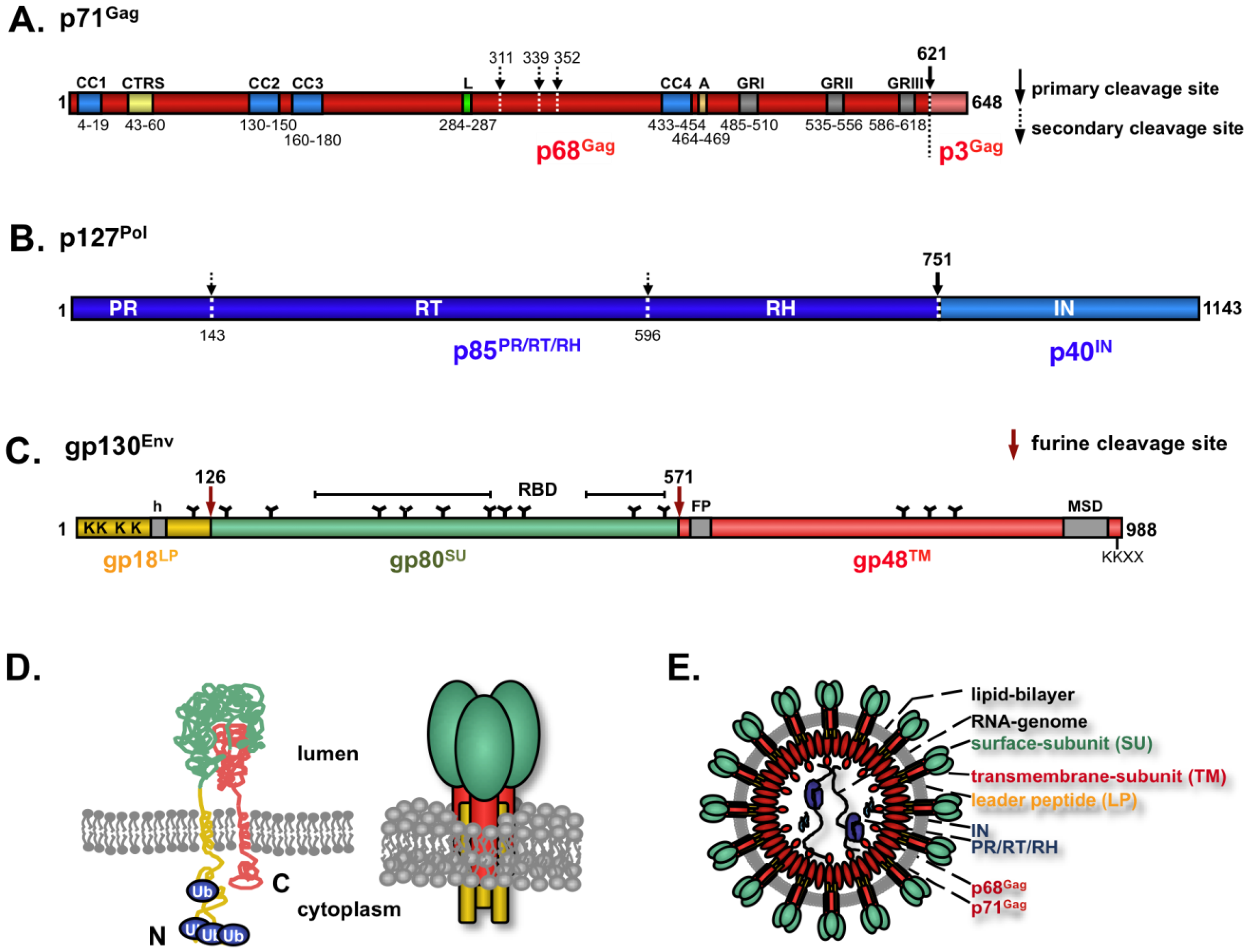
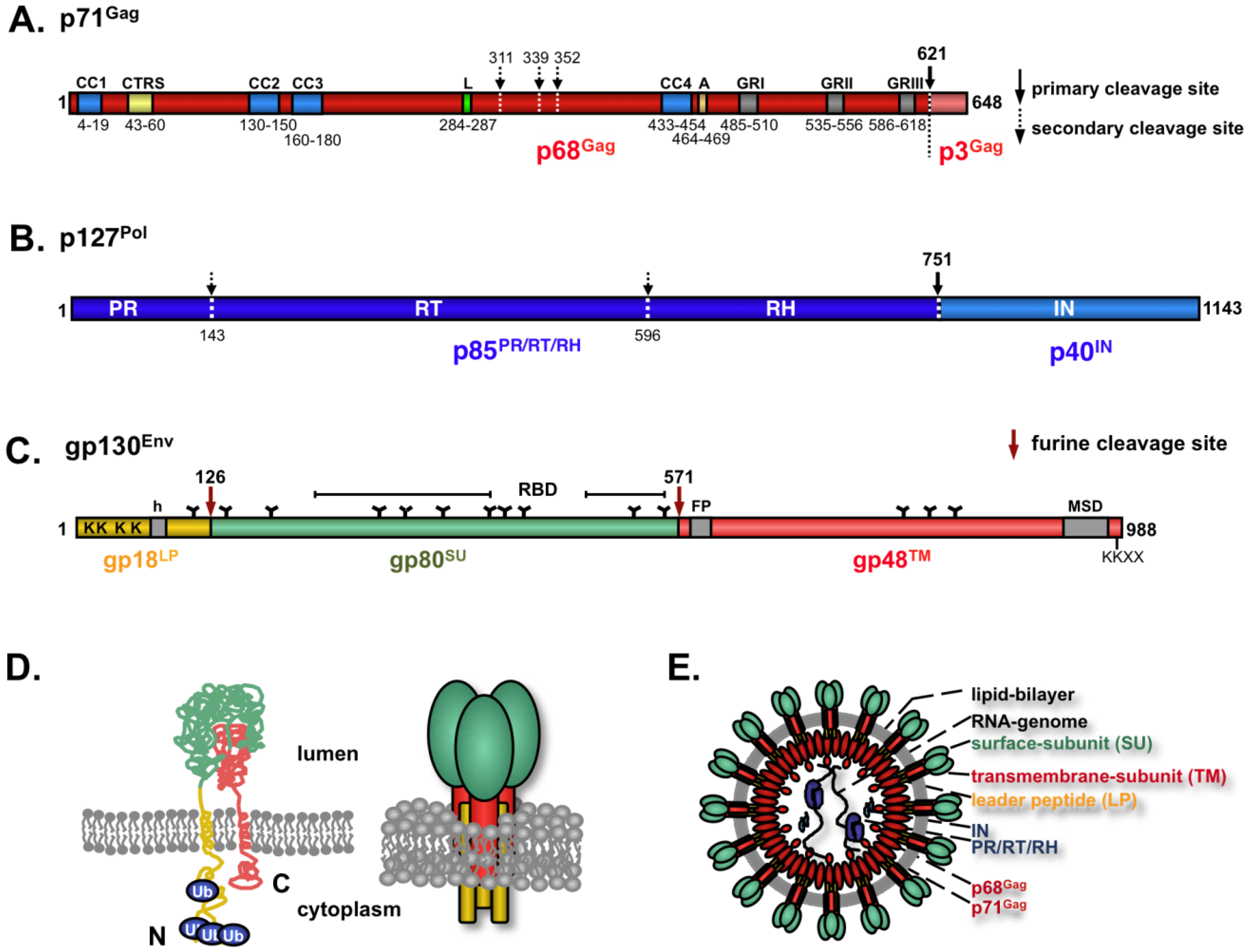
4. FV Structural Proteins
5. FV Gag
6. FV Pol
7. FV Env
8. FV Egress
9. FV Entry
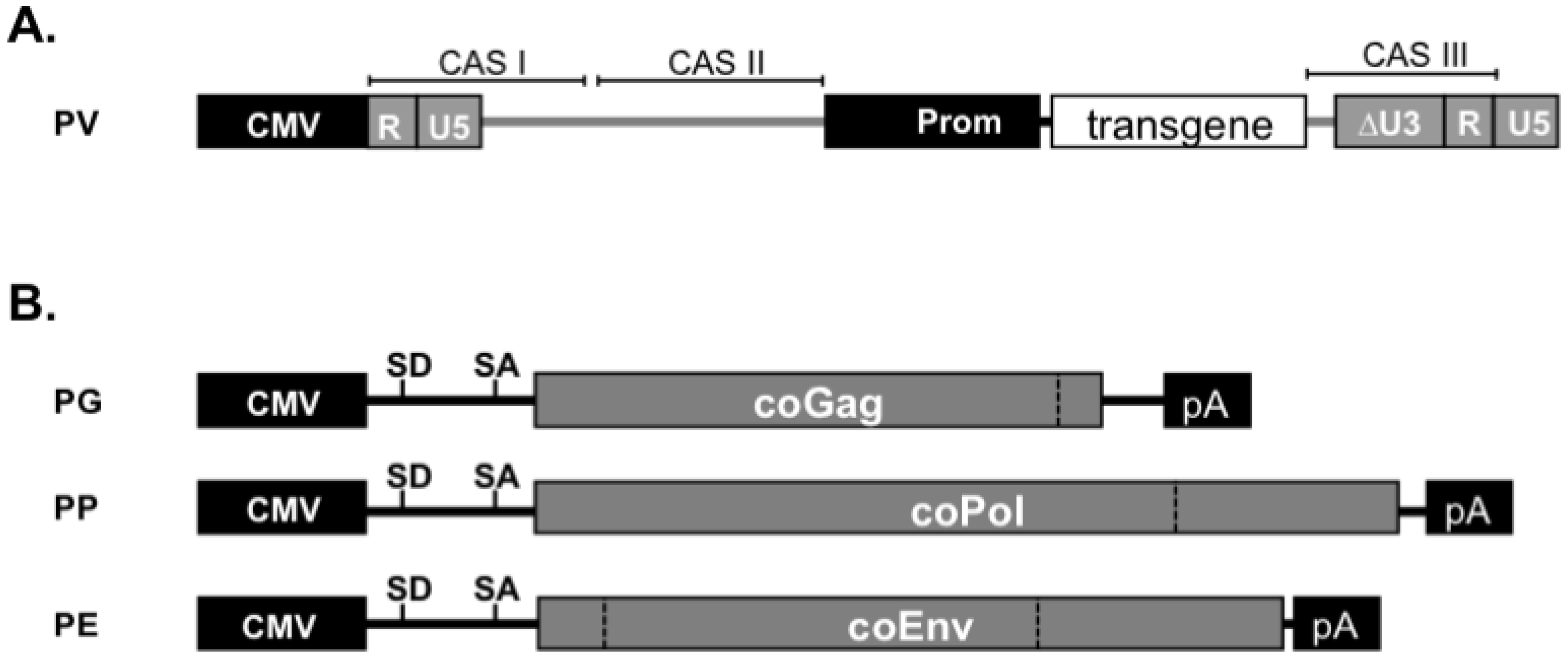
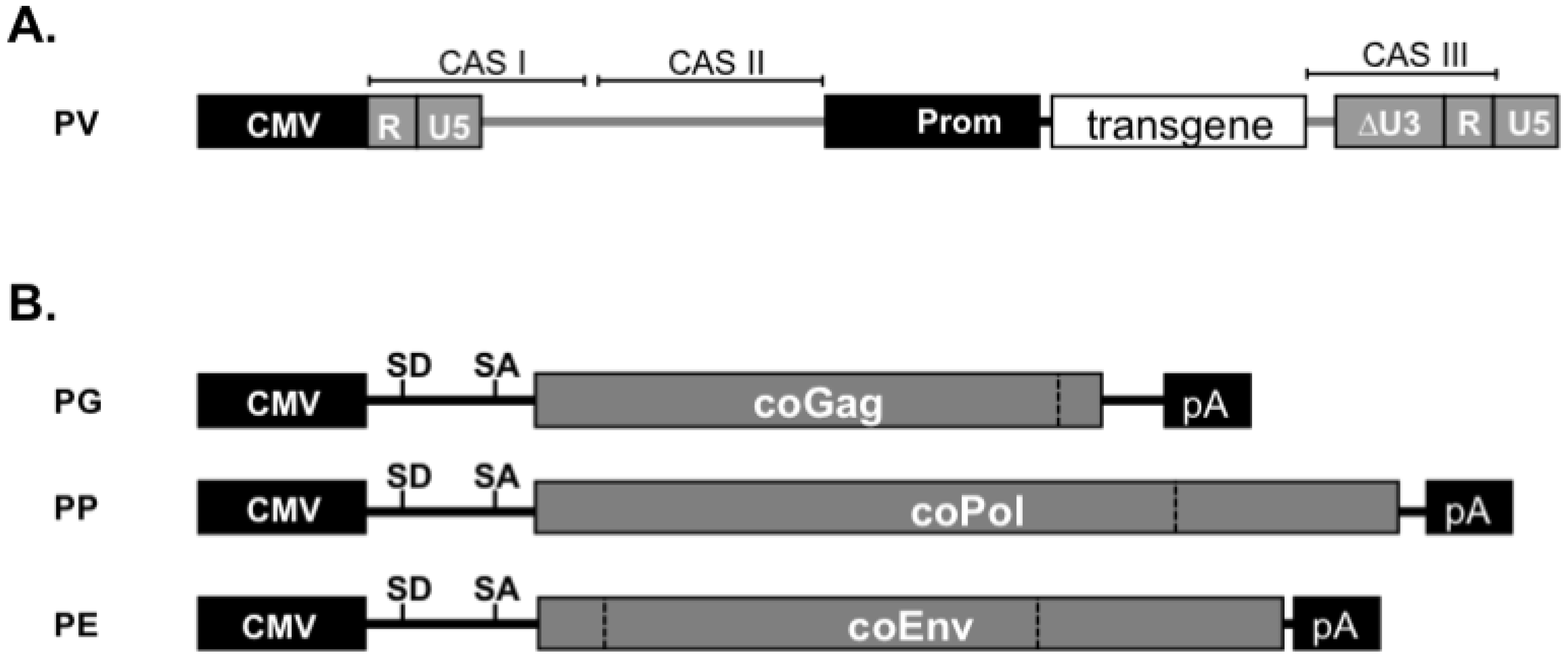
10. FV Vector Systems
11. Experimental FV Vector Applications
12. Pseudotyping of Orthoretroviral Vectors with FV Env
13. Safety of FV Vectors
14. Conclusions and Outlook
Acknowledgments
References and Notes
- Switzer, W.M.; Salemi, M.; Shanmugam, V.; Gao, F.; Cong, M.E.; Kuiken, C.; Bhullar, V.; Beer, B.E.; Vallet, D.; Gautier-Hion, A.; et al. Ancient co-speciation of simian foamy viruses and primates. Nature 2005, 434, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Saib, A. Non-primate foamy viruses. Curr. Top. Microbiol. Immunol. 2003, 277, 197–211. [Google Scholar] [PubMed]
- Switzer, W.M.; Bhullar, V.; Shanmugam, V.; Cong, M.E.; Parekh, B.; Lerche, N.W.; Yee, J.L.; Ely, J.J.; Boneva, R.; Chapman, L.E.; et al. Frequent simian foamy virus infection in persons occupationally exposed to nonhuman primates. J. Virol. 2004, 78, 2780–2789. [Google Scholar] [CrossRef] [PubMed]
- Heneine, W.; Schweizer, M.; Sandstrom, P.; Folks, T. Human infection with foamy viruses. Curr. Top. Microbiol. Immunol. 2003, 277, 181–196. [Google Scholar] [PubMed]
- Delelis, O.; Lehmann-Che, J.; Saib, A. Foamy viruses—A world apart. Curr. Opin. Microbiol. 2004, 7, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Linial, M.L.; Fan, H.; Hahn, B.; Löwer, R.; Neil, J.; Quackenbush, S.; Rethwilm, A.; Sonigo, P.; Stoye, J.P.; Tristem, M. Retroviridae. In Virus Taxonomy; Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., Ball, L.A., Eds.; Elsevier Academic Press: London, UK, 2005; pp. 421–440. [Google Scholar]
- Achong, B.G.; Mansell, P.W.A.; Epstein, M.A.; Clifford, P. An unusual virus in cultures from a human nasopharyngeal carcinoma. J. Natl. Cancer Inst. 1971, 46, 299–307. [Google Scholar] [PubMed]
- Herchenröder, O.; Renne, R.; Loncar, D.; Cobb, E.K.; Murthy, K.K.; Schneider, J.; Mergia, A.; Luciw, P.A. Isolation, cloning, and sequencing of simian foamy viruses from chimpanzees (SFVcpz): High homology to human foamy virus (HFV). Virology 1994, 201, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, M.; Turek, R.; Hahn, H.; Schliephake, A.; Netzer, K.O.; Eder, G.; Reinhardt, M.; Rethwilm, A.; Neumann-Haefelin, D. Markers of foamy virus infections in monkeys, apes, and accidentally infected humans: Appropriate testing fails to confirm suspected foamy virus prevalence in humans. AIDS Res. Hum. Retroviruses 1995, 11, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, M.; Turek, R.; Reinhardt, M.; Neumann-Haefelin, D. Absence of foamy virus DNA in Graves’ disease. AIDS Res. Hum. Retroviruses 1994, 10, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Epstein, M.A. Simian retroviral infectins in human beings. Lancet (Correspondence) 2004, 364, 137–140. [Google Scholar]
- Jones-Engel, L.; May, C.C.; Engel, G.A.; Steinkraus, K.A.; Schillaci, M.A.; Fuentes, A.; Rompis, A.; Chalise, M.K.; Aggimarangsee, N.; Feeroz, M.M.; et al. Diverse contexts of zoonotic transmission of simian foamy viruses in Asia. Emerg. Infect. Dis. 2008, 14, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Picard-Maureau, M.; Jarmy, G.; Berg, A.; Rethwilm, A.; Lindemann, D. Foamy virus envelope glycoprotein-mediated entry involves a pH-dependent fusion process. J. Virol. 2003, 77, 4722–4730. [Google Scholar] [CrossRef] [PubMed]
- Petit, C.; Giron, M.L.; Tobaly-Tapiero, J.; Bittoun, P.; Real, E.; Jacob, Y.; Tordo, N.; De The, H.; Saib, A. Targeting of incoming retroviral Gag to the centrosome involves a direct interaction with the dynein light chain 8. J. Cell Sci. 2003, 116, 3433–3442. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Che, J.; Renault, N.; Giron, M.L.; Roingeard, P.; Clave, E.; Tobaly-Tapiero, J.; Bittoun, P.; Toubert, A.; de The, H.; Saib, A. Centrosomal latency of incoming foamy viruses in resting cells. PLoS Pathog. 2007, 3, e74. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Che, J.; Giron, M.L.; Delelis, O.; Lochelt, M.; Bittoun, P.; Tobaly-Tapiero, J.; de The, H.; Saib, A. Protease-dependent uncoating of a complex retrovirus. J. Virol. 2005, 79, 9244–9253. [Google Scholar] [CrossRef] [PubMed]
- Patton, G.S.; Erlwein, O.; McClure, M.O. Cell-cycle dependence of foamy virus vectors. J. Gen. Virol. 2004, 85, 2925–2930. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.; Russell, D.W. Cell cycle requirements for transduction by foamy virus vectors compared to those of oncovirus and lentivirus vectors. J. Virol. 2004, 78, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Tobaly-Tapiero, J.; Bittoun, P.; Lehmann-Che, J.; Delelis, O.; Giron, M.L.; de The, H.; Saib, A. Chromatin tethering of incoming foamy virus by the structural Gag protein. Traffic 2008, 9, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Löchelt, M. Foamy virus transactivation and gene expression. Curr. Top. Microbiol. Immunol. 2003, 277, 27–61. [Google Scholar] [PubMed]
- Bodem, J.; Schied, T.; Gabriel, R.; Rammling, M.; Rethwilm, A. Foamy virus nuclear RNA export is distinct from that of other retroviruses. J. Virol. 2011, 85, 2333–2341. [Google Scholar] [CrossRef] [PubMed]
- Renault, N.; Tobaly-Tapiero, J.; Paris, J.; Giron, M.L.; Coiffic, A.; Roingeard, P.; Saib, A. A nuclear export signal within the structural Gag protein is required for prototype foamy virus replication. Retrovirology 2011, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Goepfert, P.A.; Shaw, K.L.; Ritter, G.D., Jr.; Mulligan, M.J. A sorting motif localizes the foamy virus glycoprotein to the endoplasmic reticulum. J. Virol. 1997, 71, 778–784. [Google Scholar] [PubMed]
- Stanke, N.; Stange, A.; Lüftenegger, D.; Zentgraf, H.; Lindemann, D. Ubiquitination of the prototype foamy virus envelope glycoprotein leader peptide regulates subviral particle release. J. Virol. 2005, 79, 15074–15083. [Google Scholar] [CrossRef] [PubMed]
- Moebes, A.; Enssle, J.; Bieniasz, P.D.; Heinkelein, M.; Lindemann, D.; Bock, M.; McClure, M.O.; Rethwilm, A. Human foamy virus reverse transcription that occurs late in the viral replication cycle. J. Virol. 1997, 71, 7305–7311. [Google Scholar] [PubMed]
- Yu, S.F.; Sullivan, M.D.; Linial, M.L. Evidence that the human foamy virus genome is DNA. J. Virol. 1999, 73, 1565–1572. [Google Scholar] [PubMed]
- Roy, J.; Rudolph, W.; Juretzek, T.; Gartner, K.; Bock, M.; Herchenroder, O.; Lindemann, D.; Heinkelein, M.; Rethwilm, A. Feline foamy virus genome and replication strategy. J. Virol. 2003, 77, 11324–11331. [Google Scholar] [CrossRef] [PubMed]
- Heinkelein, M.; Pietschmann, T.; Jarmy, G.; Dressler, M.; Imrich, H.; Thurow, J.; Lindemann, D.; Bock, M.; Moebes, A.; Roy, J.; et al. Efficient intracellular retrotransposition of an exogenous primate retrovirus genome. EMBO J. 2000, 19, 3436–3445. [Google Scholar] [CrossRef] [PubMed]
- Heinkelein, M.; Rammling, M.; Juretzek, T.; Lindemann, D.; Rethwilm, A. Retrotransposition and cell-to-cell transfer of foamy viruses. J. Virol. 2003, 77, 11855–11858. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, D.N.; Linial, M.L. The roles of Pol and Env in the assembly pathway of human foamy virus. J. Virol. 1998, 72, 3658–3665. [Google Scholar] [PubMed]
- Fischer, N.; Heinkelein, M.; Lindemann, D.; Enssle, J.; Baum, C.; Werder, E.; Zentgraf, H.; Muller, J.G.; Rethwilm, A. Foamy virus particle formation. J. Virol. 1998, 72, 1610–1615. [Google Scholar] [PubMed]
- Patton, G.S.; Morris, S.A.; Chung, W.; Bieniasz, P.D.; McClure, M.O. Identification of domains in gag important for prototypic foamy virus egress. J. Virol. 2005, 79, 6392–6399. [Google Scholar] [CrossRef] [PubMed]
- Stange, A.; Mannigel, I.; Peters, K.; Heinkelein, M.; Stanke, N.; Cartellieri, M.; Gottlinger, H.; Rethwilm, A.; Zentgraf, H.; Lindemann, D. Characterization of prototype foamy virus gag late assembly domain motifs and their role in particle egress and infectivity. J. Virol. 2005, 79, 5466–5476. [Google Scholar] [CrossRef] [PubMed]
- Löchelt, M.; Muranyi, W.; Flügel, R.M. Human foamy virus genome possesses an internal, Bel-1-dependent and functional promoter. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 7317–7321. [Google Scholar] [CrossRef] [PubMed]
- Löchelt, M.; Flugel, R.M.; Aboud, M. The human foamy virus internal promoter directs the expression of the functional Bel 1 transactivator and Bet protein early after infection. J. Virol. 1994, 68, 638–645. [Google Scholar] [PubMed]
- Löchelt, M.; Yu, S.F.; Linial, M.L.; Flügel, R.M. The human foamy virus internal promoter is required for efficient gene expression and infectivity. Virology 1995, 206, 601–610. [Google Scholar] [CrossRef]
- Rethwilm, A.; Erlwein, O.; Baunach, G.; Maurer, B.; ter Meulen, V. The transcriptional transactivator of human foamy virus maps to the bel 1 genomic region. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Partin, K.M.; Löchelt, M.; Bannert, H.; Flügel, R.M.; Cullen, B.R. Characterization of the transcriptional trans activator of human foamy retrovirus. J. Virol. 1991, 65, 2589–2594. [Google Scholar] [PubMed]
- Kang, Y.; Blair, W.S.; Cullen, B.R. Identification and functional characterization of a high-affinity Bel-1 DNA binding site located in the human foamy virus internal promoter. J. Virol. 1998, 72, 504–511. [Google Scholar] [PubMed]
- He, F.; Blair, W.S.; Fukushima, J.; Cullen, B.R. The human foamy virus Bel-1 transcription factor is a sequence-specific DNA binding protein. J. Virol. 1996, 70, 3902–3908. [Google Scholar] [PubMed]
- Wang, J.; Tan, J.; Guo, H.; Zhang, Q.; Jia, R.; Xu, X.; Geng, Y.; Qiao, W. Bovine foamy virus transactivator BTas interacts with cellular RelB to enhance viral transcription. J. Virol. 2010, 84, 11888–11897. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, J.; Tan, J.; Zhang, X.; Guo, H.; Zhang, Q.; Guo, T.; Geng, Y.; Qiao, W. BFV activates the NF-kappaB pathway through its transactivator (BTas) to enhance viral transcription. Virology 2010, 400, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Hao, P.; Jia, R.; Yang, W.; Liu, R.; Wang, J.; Xi, Z.; Geng, Y.; Qiao, W. Identification and functional characterization of BTas transactivator as a DNA-binding protein. Virology 2010, 405, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Baunach, G.; Maurer, B.; Hahn, H.; Kranz, M.; Rethwilm, A. Functional analysis of human foamy virus accessory reading frames. J. Virol. 1993, 67, 5411–5418. [Google Scholar] [PubMed]
- Muranyi, W.; Flugel, R.M. Analysis of splicing patterns of human spumaretrovirus by polymerase chain reaction reveals complex RNA structures. J. Virol. 1991, 65, 727–735. [Google Scholar] [PubMed]
- Schmidt, M.; Rethwilm, A. Replicating foamy virus-based vectors directing high level expression of foreign genes. Virology 1995, 210, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Löchelt, M.; Romen, F.; Bastone, P.; Muckenfuss, H.; Kirchner, N.; Kim, Y.B.; Truyen, U.; Rosler, U.; Battenberg, M.; Saib, A.; et al. The antiretroviral activity of APOBEC3 is inhibited by the foamy virus accessory Bet protein. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 7982–7987. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.A.; Wiegand, H.L.; Moore, M.D.; Schafer, A.; McClure, M.O.; Cullen, B.R. Foamy virus Bet proteins function as novel inhibitors of the APOBEC3 family of innate antiretroviral defense factors. J. Virol. 2005, 79, 8724–8731. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, M.; Schmidt, S.; Marino, D.; Russell, R.A.; Stauch, B.; Hofmann, H.; Kopietz, F.; Kloke, B.P.; Zielonka, J.; Strover, H.; et al. Species-specific inhibition of APOBEC3C by the prototype foamy virus protein bet. J. Biol. Chem. 2009, 284, 5819–5826. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. Using retroviruses to study the nuclear export of mRNA. Results Probl. Cell Differ. 2002, 35, 151–168. [Google Scholar] [PubMed]
- Cullen, B.R. Nuclear RNA export. J. Cell Sci. 2003, 116, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Gruter, P.; Tabernero, C.; von Kobbe, C.; Schmitt, C.; Saavedra, C.; Bachi, A.; Wilm, M.; Felber, B.K.; Izaurralde, E. TAP, the human homolog of Mex67p, mediates CTE-dependent RNA export from the nucleus. Mol. Cell 1998, 1, 649–659. [Google Scholar] [CrossRef]
- Zolotukhin, A.S.; Michalowski, D.; Smulevitch, S.; Felber, B.K. Retroviral constitutive transport element evolved from cellular TAP(NXF1)-binding sequences. J. Virol. 2001, 75, 5567–5575. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.F.; Baldwin, D.N.; Gwynn, S.R.; Yendapalli, S.; Linial, M.L. Human foamy virus replication: a pathway distinct from that of retroviruses and hepadnaviruses. Science 1996, 271, 1579–1582. [Google Scholar] [CrossRef] [PubMed]
- Bodem, J.; Lochelt, M.; Winkler, I.; Flower, R.P.; Delius, H.; Flügel, R.M. Characterization of the spliced pol transcript of feline foamy virus: the splice acceptor site of the pol transcript is located in gag of foamy viruses. J. Virol. 1996, 70, 9024–9027. [Google Scholar] [PubMed]
- Jordan, I.; Enssle, J.; Guttler, E.; Mauer, B.; Rethwilm, A. Expression of human foamy virus reverse transcriptase involves a spliced pol mRNA. Virology 1996, 224, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Enssle, J.; Jordan, I.; Mauer, B.; Rethwilm, A. Foamy virus reverse transcriptase is expressed independently from the Gag protein. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 4137–4141. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.G.; Kuppers, D.; Horn, M.; Roy, J.; May, C.; Linial, M.L. A premature termination codon mutation at the C terminus of foamy virus Gag downregulates the levels of spliced pol mRNA. J. Virol. 2008, 82, 1656–1664. [Google Scholar] [CrossRef] [PubMed]
- Swiersy, A.; Wiek, C.; Reh, J.; Zentgraf, H.; Lindemann, D. Orthoretroviral-like Prototype Foamy Virus Gag-Pol Expression is Compatible with Viral Replication. Retrovirology 2011. submitted for publication. [Google Scholar] [CrossRef] [PubMed]
- Heinkelein, M.; Leurs, C.; Rammling, M.; Peters, K.; Hanenberg, H.; Rethwilm, A. Pregenomic RNA is required for efficient incorporation of pol polyprotein into foamy virus capsids. J. Virol. 2002, 76, 10069–10073. [Google Scholar] [CrossRef] [PubMed]
- Wiktorowicz, T.; Peters, K.; Armbruster, N.; Steinert, A.F.; Rethwilm, A. Generation of an improved foamy virus vector by dissection of cis-acting sequences. J. Gen. Virol. 2009, 90, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.; Wiktorowicz, T.; Heinkelein, M.; Rethwilm, A. RNA and protein requirements for incorporation of the pol protein into foamy virus particles. J. Virol. 2005, 79, 7005–7013. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.G.; Linial, M.L. The C terminus of foamy retrovirus Gag contains determinants for encapsidation of Pol protein into virions. J. Virol. 2008, 82, 10803–10810. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; Linial, M.L. Role of the foamy virus pol cleavage site in viral replication. J. Virol. 2007, 81, 4956–4962. [Google Scholar] [CrossRef] [PubMed]
- Heinkelein, M.; Dressler, M.; Jarmy, G.; Rammling, M.; Imrich, H.; Thurow, J.; Lindemann, D.; Rethwilm, A. Improved primate foamy virus vectors and packaging constructs. J. Virol. 2002, 76, 3774–3783. [Google Scholar] [CrossRef] [PubMed]
- Heinkelein, M.; Thurow, J.; Dressler, M.; Imrich, H.; Neumann-Haefelin, D.; McClure, M.O.; Rethwilm, A. Complex effects of deletions in the 5' untranslated region of primate foamy virus on viral gene expression and RNA packaging. J. Virol. 2000, 74, 3141–3148. [Google Scholar] [CrossRef] [PubMed]
- Flügel, R.M.; Pfrepper, K.I. Proteolytic processing of foamy virus Gag and Pol proteins. Curr. Top. Microbiol. Immunol. 2003, 277, 63–88. [Google Scholar] [PubMed]
- Cartellieri, M.; Rudolph, W.; Herchenröder, O.; Lindemann, D.; Rethwilm, A. Determination of the relative amounts of Gag and Pol proteins in foamy virus particles. Retrovirology 2005, 2, 44. [Google Scholar] [CrossRef] [PubMed]
- Enssle, J.; Fischer, N.; Moebes, A.; Mauer, B.; Smola, U.; Rethwilm, A. Carboxy-terminal cleavage of the human foamy virus Gag precursor molecule is an essential step in the viral life cycle. J. Virol. 1997, 71, 7312–7317. [Google Scholar] [PubMed]
- Zemba, M.; Wilk, T.; Rutten, T.; Wagner, A.; Flügel, R.M.; Löchelt, M. The carboxy-terminal p3Gag domain of the human foamy virus Gag precursor is required for efficient virus infectivity. Virology 1998, 247, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Giron, M.L.; Colas, S.; Wybier, J.; Rozain, F.; Emanoil-Ravier, R. Expression and maturation of human foamy virus Gag precursor polypeptides. J. Virol. 1997, 71, 1635–1639. [Google Scholar] [PubMed]
- Matthes, D.; Wiktorowicz, T.; Zahn, J.; Bodem, J.; Stanke, N.; Lindemann, D.; Rethwilm, A. Basic Residues in the Foamy Virus Gag Protein. J. Virol. 2011, 85, 3986–3995. [Google Scholar] [CrossRef] [PubMed]
- Tobaly-Tapiero, J.; Bittoun, P.; Giron, M.L.; Neves, M.; Koken, M.; Saib, A.; de The, H. Human foamy virus capsid formation requires an interaction domain in the N terminus of Gag. J. Virol. 2001, 75, 4367–4375. [Google Scholar] [CrossRef] [PubMed]
- Eastman, S.W.; Linial, M.L. Identification of a conserved residue of foamy virus Gag required for intracellular capsid assembly. J. Virol. 2001, 75, 6857–6864. [Google Scholar] [CrossRef] [PubMed]
- Cartellieri, M.; Herchenröder, O.; Rudolph, W.; Heinkelein, M.; Lindemann, D.; Zentgraf, H.; Rethwilm, A. N-terminal Gag domain required for foamy virus particle assembly and export. J. Virol. 2005, 79, 12464–12476. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.F.; Eastman, S.W.; Linial, M.L. Foamy virus capsid assembly occurs at a pericentriolar region through a cytoplasmic targeting/retention signal in Gag. Traffic 2006, 7, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.S.; Hunter, E. A single amino acid substitution within the matrix protein of a type D retrovirus converts its morphogenesis to that of a type C retrovirus. Cell 1990, 63, 77–86. [Google Scholar] [CrossRef]
- Mannigel, I.; Stange, A.; Zentgraf, H.; Lindemann, D. Correct capsid assembly mediated by a conserved YXXLGL motif in prototype foamy virus Gag is essential for infectivity and reverse transcription of the viral genome. J. Virol. 2007, 81, 3317–3326. [Google Scholar] [CrossRef] [PubMed]
- Swanstrom, R.; Wills, J.W. Synthesis, Assembly, and Processing of Viral Proteins. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, H.E., Eds.; Cold Spring Harbour Laboratory Press: Cold Spring Harbour, NY, USA, 1997. [Google Scholar]
- Schliephake, A.W.; Rethwilm, A. Nuclear localization of foamy virus Gag precursor protein. J. Virol. 1994, 68, 4946–4954. [Google Scholar] [PubMed]
- Müllers, E.; Uhlig, T.; Stirnnagel, K.; Fiebig, U.; Zentgraf, H.; Lindemann, D. Novel functions of Prototype Foamy Virus Gag GR boxes in reverse transcription and particle morphogenesis. J. Virol. 2011, 85, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.F.; Edelmann, K.; Strong, R.K.; Moebes, A.; Rethwilm, A.; Linial, M.L. The carboxyl terminus of the human foamy virus Gag protein contains separable nucleic acid binding and nuclear transport domains. J. Virol. 1996, 70, 8255–8262. [Google Scholar] [PubMed]
- Bodem, J.; Zemba, M.; Flügel, R.M. Nuclear localization of the functional Bel 1 transactivator but not of the gag proteins of the feline foamy virus. Virology 1998, 251, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Zhadina, M.; McClure, M.O.; Johnson, M.C.; Bieniasz, P.D. Ubiquitin-dependent virus particle budding without viral protein ubiquitination. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 20031–20036. [Google Scholar] [CrossRef] [PubMed]
- Imrich, H.; Heinkelein, M.; Herchenröder, O.; Rethwilm, A. Primate foamy virus Pol proteins are imported into the nucleus. J. Gen. Virol. 2000, 81, 2941–2947. [Google Scholar] [CrossRef] [PubMed]
- An, D.G.; Hyun, U.; Shin, C.G. Characterization of nuclear localization signals of the prototype foamy virus integrase. J. Gen. Virol. 2008, 89, 1680–1684. [Google Scholar] [CrossRef] [PubMed]
- Hartl, M.J.; Mayr, F.; Rethwilm, A.; Wohrl, B.M. Biophysical and enzymatic properties of the simian and prototype foamy virus reverse transcriptases. Retrovirology 2010, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Rinke, C.S.; Boyer, P.L.; Sullivan, M.D.; Hughes, S.H.; Linial, M.L. Mutation of the catalytic domain of the foamy virus reverse transcriptase leads to loss of processivity and infectivity. J. Virol. 2002, 76, 7560–7570. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.L.; Stenbak, C.R.; Clark, P.K.; Linial, M.L.; Hughes, S.H. Characterization of the polymerase and RNase H activities of human foamy virus reverse transcriptase. J. Virol. 2004, 78, 6112–6121. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.L.; Stenbak, C.R.; Hoberman, D.; Linial, M.L.; Hughes, S.H. In vitro fidelity of the prototype primate foamy virus (PFV) RT compared to HIV-1 RT. Virology 2007, 367, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, K.; Wiktorowicz, T.; Park, J.; Mergia, A.; Rethwilm, A.; Scheller, C. Accuracy estimation of foamy virus genome copying. Retrovirology 2009, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Hartl, M.J.; Schweimer, K.; Reger, M.H.; Schwarzinger, S.; Bodem, J.; Rosch, P.; Wohrl, B.M. Formation of transient dimers by a retroviral protease. Biochem J 2010, 427, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Hartl, M.J.; Wohrl, B.M.; Rosch, P.; Schweimer, K. The solution structure of the simian foamy virus protease reveals a monomeric protein. J. Mol. Biol. 2008, 381, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.G.; Roy, J.; Jackson, D.; Clark, P.; Boyer, P.L.; Hughes, S.H.; Linial, M. Foamy retrovirus integrase contains a Pol dimerization domain required for protease activation. J. Virol. 2011, 85, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Hartl, M.J.; Bodem, J.; Jochheim, F.; Rethwilm, A.; Rösch, P.; Wöhrl, B.M. Regulation of foamy virus protease activity by viral RNA—A novel and unique mechanism among retroviruses. J. Virol. 2011, 85, 4462–4469. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 2010, 464, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P. Integrase iluminated. EMBO Rep. 2010, 11, 328. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Maertens, G.N.; Hare, S. Structural insights into the retroviral DNA integration apperatus. Curr Opin Struct Bio. 2011, 21, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Vos, A.M.; Clayton, R.R.; Thuring, J.W.; Cummings, M.D.; Cherepanov, P. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20057–20062. [Google Scholar] [CrossRef] [PubMed]
- Maertens, G.N.; Hare, S.; Cherepanov, P. The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature 2010, 468, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, L.; Li, X.; Naraharisetty, H.L.; Hare, S.; Cherepanov, P.; Engelman, A. Structure-based modeling of the functional HIV-1 intasome and its inhibition. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 15910–15915. [Google Scholar] [CrossRef] [PubMed]
- Wilk, T.; Geiselhart, V.; Frech, M.; Fuller, S.D.; Flügel, R.M.; Löchelt, M. Specific interaction of a novel foamy virus Env leader protein with the N-terminal Gag domain. J. Virol. 2001, 75, 7995–8007. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, D.; Pietschmann, T.; Picard-Maureau, M.; Berg, A.; Heinkelein, M.; Thurow, J.; Knaus, P.; Zentgraf, H.; Rethwilm, A. A particle-associated glycoprotein signal peptide essential for virus maturation and infectivity. J. Virol. 2001, 75, 5762–5771. [Google Scholar] [CrossRef] [PubMed]
- Duda, A.; Stange, A.; Luftenegger, D.; Stanke, N.; Westphal, D.; Pietschmann, T.; Eastman, S.W.; Linial, M.L.; Rethwilm, A.; Lindemann, D. Prototype foamy virus envelope glycoprotein leader peptide processing is mediated by a furin-like cellular protease, but cleavage is not essential for viral infectivity. J. Virol. 2004, 78, 13865–13870. [Google Scholar] [CrossRef] [PubMed]
- Geiselhart, V.; Bastone, P.; Kempf, T.; Schnolzer, M.; Löchelt, M. Furin-mediated cleavage of the feline foamy virus Env leader protein. J. Virol. 2004, 78, 13573–13581. [Google Scholar] [CrossRef] [PubMed]
- Luftenegger, D.; Picard-Maureau, M.; Stanke, N.; Rethwilm, A.; Lindemann, D. Analysis and function of prototype foamy virus envelope N glycosylation. J. Virol. 2005, 79, 7664–7672. [Google Scholar] [CrossRef] [PubMed]
- Geiselhart, V.; Schwantes, A.; Bastone, P.; Frech, M.; Löchelt, M. Features of the Env leader protein and the N-terminal Gag domain of feline foamy virus important for virus morphogenesis. Virology 2003, 310, 235–244. [Google Scholar] [CrossRef]
- Goepfert, P.A.; Shaw, K.; Wang, G.; Bansal, A.; Edwards, B.H.; Mulligan, M.J. An endoplasmic reticulum retrieval signal partitions human foamy virus maturation to intracytoplasmic membranes. J. Virol. 1999, 73, 7210–7217. [Google Scholar] [PubMed]
- Stange, A.; Lüftenegger, D.; Reh, J.; Weissenhorn, W.; Lindemann, D. Subviral particle release determinants of prototype foamy virus. J. Virol. 2008, 82, 9858–9869. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, T.; Heinkelein, M.; Heldmann, M.; Zentgraf, H.; Rethwilm, A.; Lindemann, D. Foamy virus capsids require the cognate envelope protein for particle export. J. Virol. 1999, 73, 2613–2621. [Google Scholar] [PubMed]
- Hill, C.L.; Bieniasz, P.D.; McClure, M.O. Properties of human foamy virus relevant to its development as a vector for gene therapy. J. Gen. Virol. 1999, 80, 2003–2009. [Google Scholar] [CrossRef] [PubMed]
- Stirnnagel, K.; Lüftenegger, D.; Stange, A.; Swiersy, A.; Mullers, E.; Reh, J.; Stanke, N.; Grosse, A.; Chiantia, S.; Keller, H.; et al. Analysis of prototype foamy virus particle-host cell interaction with autofluorescent retroviral particles. Retrovirology 2010, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Goepfert, P.A.; Wang, G.; Mulligan, M.J. Identification of an ER retrieval signal in a retroviral glycoprotein. Cell 1995, 82, 543–544. [Google Scholar] [CrossRef]
- Winkler, I.; Bodem, J.; Haas, L.; Zemba, M.; Delius, H.; Flower, R.; Flügel, R.M.; Löchelt, M. Characterization of the genome of feline foamy virus and its proteins shows distinct features different from those of primate spumaviruses. J. Virol. 1997, 71, 6727–6741. [Google Scholar] [PubMed]
- Tobaly-Tapiero, J.; Bittoun, P.; Neves, M.; Guillemin, M.C.; Lecellier, C.H.; Puvion-Dutilleul, F.; Gicquel, B.; Zientara, S.; Giron, M.L.; de The, H.; et al. Isolation and characterization of an equine foamy virus. J. Virol. 2000, 74, 4064–4073. [Google Scholar] [CrossRef] [PubMed]
- Zamborlini, A.; Renault, N.; Saib, A.; Delelis, O. Early reverse transcription is essential for productive foamy virus infection. PLoS ONE 2010, 5, e11023. [Google Scholar] [CrossRef] [PubMed]
- Delelis, O.; Saib, A.; Sonigo, P. Biphasic DNA synthesis in spumaviruses. J. Virol. 2003, 77, 8141–8146. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Worobey, M.; Li, Y.; Keele, B.F.; Bibollet-Ruche, F.; Guo, Y.; Goepfert, P.A.; Santiago, M.L.; Ndjango, J.B.; Neel, C.; et al. Molecular ecology and natural history of simian foamy virus infection in wild-living chimpanzees. PLoS Pathog. 2008, 4, e1000097. [Google Scholar] [CrossRef] [PubMed]
- Stirnnagel, K.; Dupont, A.; Schupp, D.; Perrotton, F.; Müllers, E.; Lindemann, D.; Lamb, D.C. Insights into uptake and fusion strategies exploited by a non-conventional retrovirus. Technische Universität Dresden: Dresden, Germany, Unpublished work. 2011. [Google Scholar]
- Russell, D.W.; Miller, A.D. Foamy virus vectors. J. Virol. 1996, 70, 217–222. [Google Scholar] [PubMed]
- Lo, Y.T.; Tian, T.; Nadeau, P.E.; Park, J.; Mergia, A. The foamy virus genome remains unintegrated in the nuclei of G1/S phase-arrested cells, and integrase is critical for preintegration complex transport into the nucleus. J. Virol. 2010, 84, 2832–2842. [Google Scholar] [CrossRef] [PubMed]
- Bieniasz, P.D.; Weiss, R.A.; McClure, M.O. Cell cycle dependence of foamy retrovirus infection. J. Virol. 1995, 69, 7295–7299. [Google Scholar] [PubMed]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Maetzig, T.; Galla, M.; Baum, C.; Schambach, A. Gammaretroviral Vectors: Biology, Technology and Application. Viruses 2011, in press. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M. Transcription start regions in the human genome are favored targets for MLV integration. Science 2003, 300, 1749–1751. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.D.; Miller, D.G.; Jacobs, M.A.; Allen, J.M.; Kiem, H.P.; Kaul, R.; Russell, D.W. Foamy virus vector integration sites in normal human cells. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Schröder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integrationin the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Nowrouzi, A.; Dittrich, M.; Klanke, C.; Heinkelein, M.; Rammling, M.; Dandekar, T.; von Kalle, C.; Rethwilm, A. Genome-wide mapping of foamy virus vector integrations into a human cell line. J Gen Virol. 2006, 87, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Juretzek, T.; Holm, T.; Gärtner, K.; Kanzler, S.; Lindemann, D.; Herchenröder, O.; Picard-Maureau, M.; Rammling, M.; Heinkelein, M.; Rethwilm, A. Foamy virus integration. J. Virol. 2004, 78, 2472–2477. [Google Scholar] [CrossRef] [PubMed]
- Enssle, J.; Moebes, A.; Heinkelein, M.; Panhuysen, M.; Mauer, B.; Schweizer, M.; Neumann-Haefelin, D.; Rethwilm, A. An active foamy virus integrase is required for virus replication. J. Gen. Virol. 1999, 80, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Heinkelein, M.; Schmidt, M.; Fischer, N.; Moebes, A.; Lindemann, D.; Enssle, J.; Rethwilm, A. Characterization of a cis-acting sequence in the Pol region required to transfer human foamy virus vectors. J. Virol. 1998, 72, 6307–6314. [Google Scholar] [PubMed]
- Erlwein, O.; Bieniasz, P.D.; McClure, M.O. Sequences in pol are required for transfer of human foamy virus-based vectors. J. Virol. 1998, 72, 5510–5516. [Google Scholar] [PubMed]
- Wu, M.; Chari, S.; Yanchis, T.; Mergia, A. cis-Acting sequences required for simian foamy virus type 1 vectors. J. Virol. 1998, 72, 3451–3454. [Google Scholar] [PubMed]
- Trobridge, G.; Josephson, N.; Vassilopoulos, G.; Mac, J.; Russell, D.W. Improved foamy virus vectors with minimal viral sequences. Mol. Ther. 2002, 6, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Maurer, B.; Bannert, H.; Darai, G.; Flügel, R.M. Analysis of the primary structure of the long terminal repeat and the gag and the pol genes of the human spumaretrovirs. J. Virol. 1988, 62, 1590–1597. [Google Scholar] [PubMed]
- Lindemann, D.; Technische Universität Dresden, Dresden, Germany. Unpublished work. 2011.
- Rothenaigner, I.; Kramer, S.; Meggendorfer, M.; Rethwilm, A.; Brack-Werner, R. Transduction of human neural progenitor cells with foamy virus vectors for differentiation-dependent gene expression. Gen. Ther. 2009, 16, 349–358. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gharwan, H.; Hirata, R.K.; Wang, P.; Richard, R.E.; Wang, L.; Olson, E.; Allen, J.; Ware, C.B.; Russell, D.W. Transduction of human embryonic stem cells by foamy virus vectors. Mol. Ther. 2007, 15, 1827–1833. [Google Scholar] [CrossRef] [PubMed]
- Leurs, C.; Jansen, M.; Pollok, K.E.; Heinkelein, M.; Schmidt, M.; Wissler, M.; Lindemann, D.; Von Kalle, C.; Rethwilm, A.; Williams, D.A.; et al. Comparison of three retroviral vector systems for transduction of nonobese diabetic/severe combined immunodeficiency mice repopulating human CD34+ cord blood cells. Hum. Gene. Ther. 2003, 14, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Kiem, H.P.; Allen, J.; Trobridge, G.; Olson, E.; Keyser, K.; Peterson, L.; Russell, D.W. Foamy virus-mediated gene transfer to canine repopulating cells. Blood 2007, 109, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Josephson, N.C.; Trobridge, G.; Russell, D.W. Transduction of long-term and mobilized peripheral blood-derived NOD/SCID repopulating cells by foamy virus vectors. Hum. Gene. Ther. 2004, 15, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Hirata, R.K.; Miller, A.D.; Andrews, R.G.; Russell, D.W. Transduction of hematopoietic cells by foamy virus vectors. Blood 1996, 88, 3654–3661. [Google Scholar] [PubMed]
- Si, Y.; Pulliam, A.C.; Linka, Y.; Ciccone, S.; Leurs, C.; Yuan, J.; Eckermann, O.; Fruehauf, S.; Mooney, S.; Hanenberg, H.; et al. Overnight transduction with foamyviral vectors restores the long-term repopulating activity of Fancc-/- stem cells. Blood 2008, 112, 4458–4465. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.D.; Allen, J.M.; Peterson, L.; Ironside, C.G.; Russell, D.; Kiem, H.P. Foamy and Lentiviral Vectors Transduce Canine Long-term Repopulating Cells at Similar Efficiency. Hum. Gene. Ther. 2009, 20, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.R.; Allen, J.M.; Hai, M.; Tuschong, L.M.; Khan, I.F.; Olson, E.M.; Adler, R.L.; Burkholder, T.H.; Gu, Y.C.; Russell, D.; et al. Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors. Nat. Med. 2008, 14, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Wurm, M.; Schambach, A.; Lindemann, D.; Hanenberg, H.; Standker, L.; Forssmann, W.G.; Blasczyk, R.; Horn, P.A. The influence of semen-derived enhancer of virus infection on the efficiency of retroviral gene transfer. J. Gene Med. 2010, 12, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, D.; Bock, M.; Schweizer, M.; Rethwilm, A. Efficient pseudotyping of murine leukemia virus particles with chimeric human foamy virus envelope proteins. J. Virol. 1997, 71, 4815–4820. [Google Scholar] [PubMed]
- Trobridge, G.; Vassilopoulos, G.; Josephson, N.; Russell, D.W. Gene transfer with foamy virus vectors. Methods Enzymol. 2002, 346, 628–648. [Google Scholar] [CrossRef] [PubMed]
- Kühlke, K.; EUFETS GmbH, Idar-Oberstein, Germany. Unpublished work. 2011.
- Linial, M. Why aren’t foamy viruses pathogenic? Trends Microbiol. 2000, 8, 284–289. [Google Scholar] [CrossRef]
- German, A.C.; Harbour, D.A.; Helps, C.R.; Gruffydd-Jones, T.J. Is feline foamy virus really apathogenic? Vet. Immunol. Immunopathol. 2008, 123, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Ohmine, K.; Li, Y.; Bauer, T.R.; Hickstein, D.D.; Russell, D.W. Tracking of Specific Integrant Clones in Dogs Treated with Foamy Virus Vectors. Hum. Gene. Ther. 2011, 22, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Hendrie, P.C.; Huo, Y.; Stolitenko, R.B.; Russell, D.W. A rapid and quantitative assay for measuring neighboring gene activation by vector proviruses. Mol. Ther. 2008, 16, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Deyle, D.R.; Li, Y.; Olson, E.M.; Russell, D.W. Nonintegrating foamy virus vectors. J. Virol. 2010, 84, 9341–9349. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lindemann, D.; Rethwilm, A. Foamy Virus Biology and Its Application for Vector Development. Viruses 2011, 3, 561-585. https://doi.org/10.3390/v3050561
Lindemann D, Rethwilm A. Foamy Virus Biology and Its Application for Vector Development. Viruses. 2011; 3(5):561-585. https://doi.org/10.3390/v3050561
Chicago/Turabian StyleLindemann, Dirk, and Axel Rethwilm. 2011. "Foamy Virus Biology and Its Application for Vector Development" Viruses 3, no. 5: 561-585. https://doi.org/10.3390/v3050561
APA StyleLindemann, D., & Rethwilm, A. (2011). Foamy Virus Biology and Its Application for Vector Development. Viruses, 3(5), 561-585. https://doi.org/10.3390/v3050561