Retroviral Vectors: Post Entry Events and Genomic Alterations

Abstract
1. Introduction
2. Trafficking to the Nucleus and the Retroviral Integration Reaction
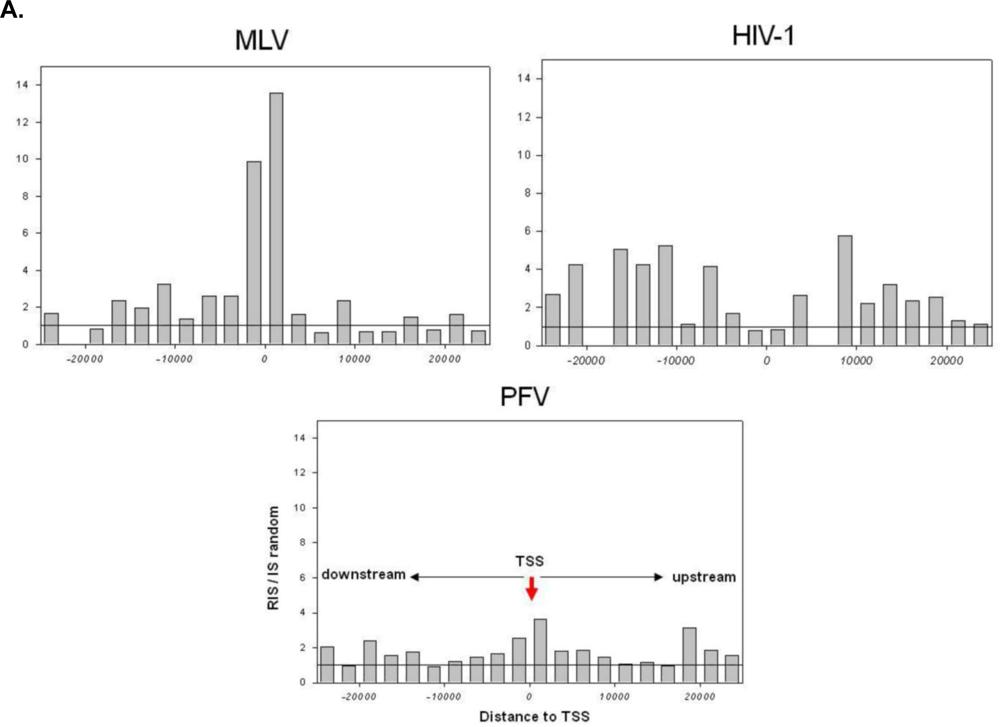
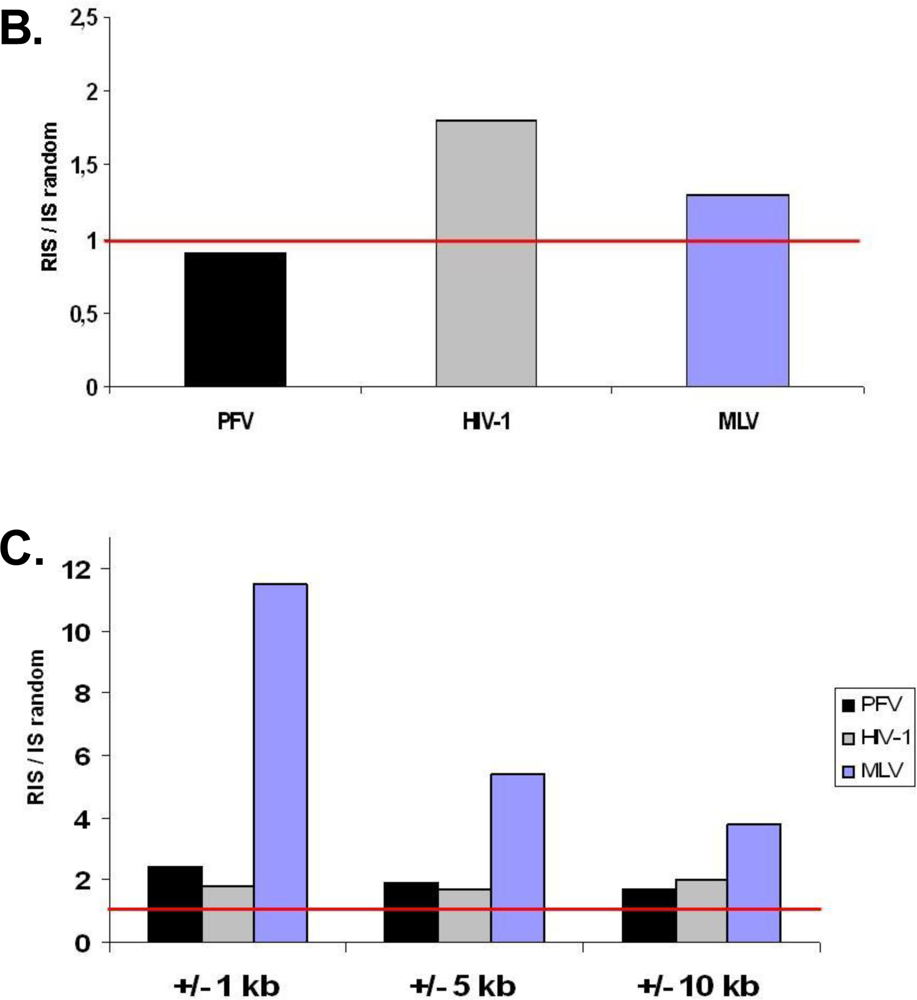
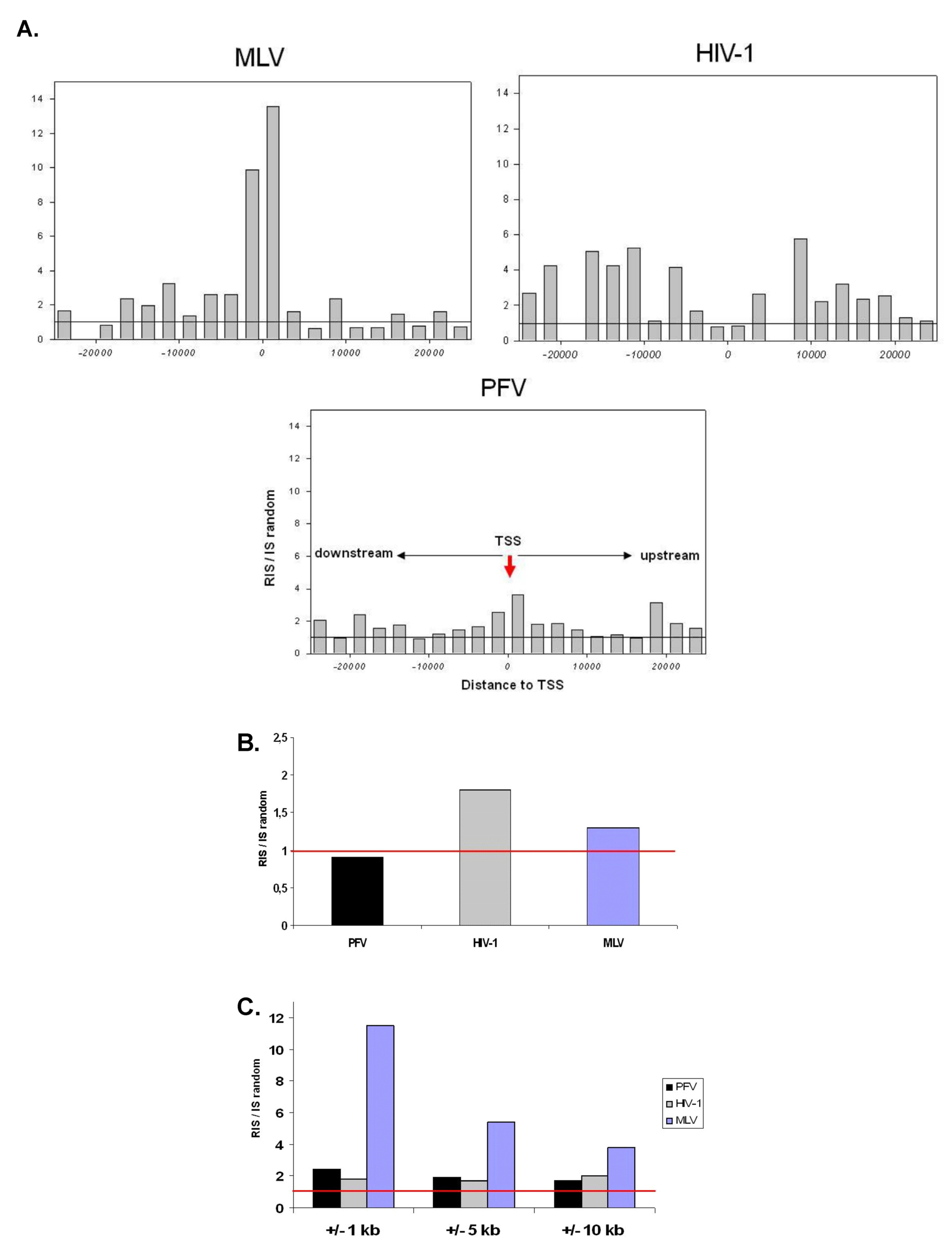
2.1. Distribution of Retroviral Integration Sites in the Cellular Genome in vitro and in vivo
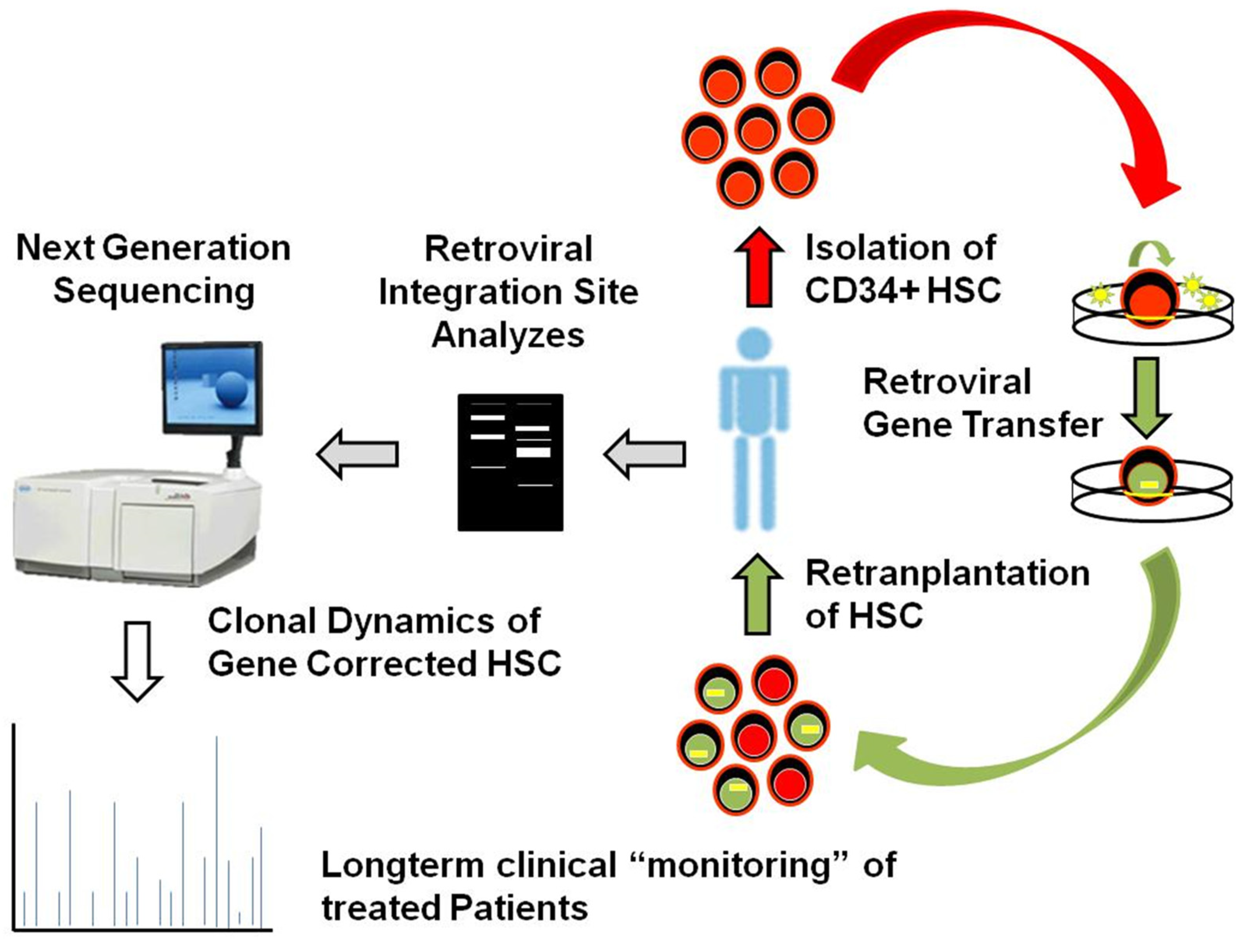
2.2. Next Generation Sequencing and Unbiased Retrieval of Vector Integration Sites
3. Side Effects in Clinical and Preclinical Gene Therapy Studies
New Strategies for Vector Biosafety in Gene Therapy
4. Conclusion and Future Perspective
Acknowledgments
References
- Vogt, P.K. Historical introduction to the general properties of retroviruses. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, E.H., Eds.; ColdSpring Harbor Laboratory Press: New York, NY, USA, 1997. [Google Scholar]
- Brown, P.O. Integration. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, E.H., Eds.; ColdSpring Harbor Laboratory Press: New York, NY, USA, 1997. [Google Scholar]
- Eglitis, M.A.; Kantoff, P.; Gilboa, E.; Anderson, W.F. Gene expression in mice after high efficiency retroviral-mediated gene transfer. Science 1985, 230, 1395–1398. [Google Scholar] [CrossRef]
- Dick, J.E.; Magli, M.C.; Huszar, D.; Phillips, R.A.; Bernstein, A. Introduction of a selectable gene into primitive stem cells capable of long-term reconstitution of the hemopoietic system of W/Wv mice. Cell 1985, 42, 71–79. [Google Scholar] [CrossRef]
- Williams, D.A.; Lemischka, I.R.; Nathan, D.G.; Mulligan, R.C. Introduction of new genetic material into pluripotent haematopoietic stem cells of the mouse. Nature 1984, 310, 476–80. [Google Scholar] [CrossRef] [PubMed]
- Mann, R.; Mulligan, R.C.; Baltimore, D. Construction of a retrovirus packaging mutant and its use to produce helper-free defective retrovirus. Cell 1983, 33, 153–159. [Google Scholar] [CrossRef]
- Miller, A.D. Development and applications of retroviral vectors. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, E.H., Eds.; ColdSpring Harbor Laboratory Press: New York, NY, USA, 1997. [Google Scholar]
- Anderson, W.F. Prospects for human gene therapy. Science 1984, 226, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Craigie, R. The road to chromatin - nuclear entry of retroviruses. Nat. Rev. Microbiol. 2007, 5, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Turlure, F.; Devroe, E.; Silver, P.A.; Engelman, A. Human cell proteins and human immunodeficiency virus DNA integration. Front. Biosci. 2004, 9, 3187–3208. [Google Scholar] [CrossRef]
- Edelstein, M.L.; Abedi, M.R.; Wixon, J. Gene therapy clinical trials worldwide to 2007—An update. J. Gene Med. 2007, 9, 833–842. [Google Scholar] [CrossRef]
- Verma, I.M.; Weitzman, M.D. Gene therapy: twenty-first century medicine. Annu. Rev. Biochem. 2005, 74, 711–738. [Google Scholar] [CrossRef]
- von Kalle, C.; Fehse, B.; Layh-Schmitt, G.; Schmidt, M.; Kelly, P.; Baum, C. Stem cell clonality and genotoxicity in hematopoietic cells: Gene activation side effects should be avoidable. Semin. Hematol. 2004, 41, 303–318. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Miller, D.G.; Jacobs, M.A.; Allen, J.M.; Kiem, H.P.; Kaul, R.; Russell, D.W. Foamy virus vector integration sites in normal human cells. Proc. Nat. Acad. Sci. USA 2006, 103, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Nowrouzi, A.; Dittrich, M.; Klanke, C.; Heinkelein, M.; Rammling, M.; Dandekar, T.; von Kalle, C.; Rethwilm, A. Genome-wide mapping of foamy virus vector integrations into a human cell line. J. Gen. Virol. 2006, 87, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Hacker, C.V.; Vink, C.A.; Wardell, T.W.; Lee, S.; Treasure, P.; Kingsman, S.M.; Mitrophanous, K.A.; Miskin, J.E. The integration profile of EIAV-based vectors. Mol. Ther. 2006, 14, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Narezkina, A.; Taganov, K.D.; Litwin, S.; Stoyanova, R.; Hayashi, J.; Seeger, C.; Skalka, A.M.; Katz, R.A. Genome-wide analyses of avian sarcoma virus integration sites. J. Virol. 2004, 78, 11656–11663. [Google Scholar] [CrossRef]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004, 2, E234. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M. Transcription start regions in the human genome are favored targets for MLV integration. Science 2003, 300, 1749–1751. [Google Scholar] [CrossRef]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Bushman, F.; Lewinski, M.; Ciuffi, A.; Barr, S.; Leipzig, J.; Hannenhalli, S.; Hoffmann, C. Genome-wide analysis of retroviral DNA integration. Nat. Rev. Microbiol. 2005, 3, 848–858. [Google Scholar] [CrossRef]
- Nienhuis, A.W.; Dunbar, C.E.; Sorrentino, B.P. Genotoxicity of retroviral integration in hematopoietic cells. Mol. Ther. 2006, 13, 1031–1049. [Google Scholar] [CrossRef]
- Temin, H.M.; Mizutani, S. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature 1970, 226, 1211–1213. [Google Scholar] [CrossRef]
- Baltimore, D. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature 1970, 226, 1209–1211. [Google Scholar] [CrossRef] [PubMed]
- Temin, H.M. The DNA provirus hypothesis. Science 1976, 192, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Temin, H.M. The protovirus hypothesis: speculations on the significance of RNA-directed DNA synthesis for normal development and for carcinogenesis. J. Nat. Canc. Ins. 1971, 46, 3–7. [Google Scholar]
- Temin, H.M. Origin of retroviruses from cellular moveable genetic elements. Cell 1980, 21, 599–600. [Google Scholar] [CrossRef]
- Cohen, J.C.; Shank, P.R.; Morris, V.L.; Cardiff, R.; Varmus, H.E. Integration of the DNA of mouse mammary tumor virus in virus-infected normal and neoplastic tissue of the mouse. Cell 1979, 16, 333–345. [Google Scholar] [CrossRef]
- Steffen, D.; Weinberg, R.A. The integrated genome of murine leukemia virus. Cell 1978, 15, 1003–1010. [Google Scholar] [CrossRef]
- Shank, P.R.; Hughes, S.H.; Kung, H.J.; Majors, J.E.; Quintrell, N.; Guntaka, R.V.; Bishop, J.M.; Varmus, H.E. Mapping unintegrated avian sarcoma virus DNA: termini of linear DNA bear 300 nucleotides present once or twice in two species of circular DNA. Cell 1978, 15, 1383–1395. [Google Scholar] [CrossRef]
- Hughes, S.H.; Shank, P.R.; Spector, D.H.; Kung, H.J.; Bishop, J.M.; Varmus, H.E.; Vogt, P.K.; Breitman, M.L. Proviruses of avian sarcoma virus are terminally redundant, co-extensive with unintegrated linear DNA and integrated at many sites. Cell 1978, 15, 1397–1410. [Google Scholar] [CrossRef]
- Battula, N.; Temin, H.M. Sites of integration of infectious DNA of avian reticuloendotheliosis viruses in different avian cellular DNAs. Cell 1978, 13, 387–398. [Google Scholar] [CrossRef]
- Moebes, A.; Enssle, J.; Bieniasz, P.D.; Heinkelein, M.; Lindemann, D.; Bock, M.; McClure, M.O.; Rethwilm, A. Human foamy virus reverse transcription that occurs late in the viral replication cycle. J. Virol. 1997, 71, 7305–7311. [Google Scholar] [CrossRef]
- Rethwilm, A. The replication strategy of foamy viruses. Curr. Topics Microbiol. Immunol. 2003, 277, 1–26. [Google Scholar]
- Telesnitsky, A.; Goff, S.P. Reverse transkriptase and the generation of retroviral DNA. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, E.H., Eds.; ColdSpring Harbor Laboratory Press: New York, NY, USA, 1997. [Google Scholar]
- Bowerman, B.; Brown, P.O.; Bishop, J.M.; Varmus, H.E. A nucleoprotein complex mediates the integration of retroviral DNA. Gene. Develop. 1989, 3, 469–478. [Google Scholar] [CrossRef]
- Fujiwara, T.; Mizuuchi, K. Retroviral DNA integration: structure of an integration intermediate. Cell 1988, 54, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.O.; Bowerman, B.; Varmus, H.E.; Bishop, J.M. Correct integration of retroviral DNA in vitro. Cell 1987, 49, 347–356. [Google Scholar] [CrossRef]
- Petit, C.; Giron, M.L.; Tobaly-Tapiero, J.; Bittoun, P.; Real, E.; Jacob, Y.; Tordo, N.; De The, H.; Saib, A. Targeting of incoming retroviral Gag to the centrosome involves a direct interaction with the dynein light chain 8. J. Cell Sci. 2003, 116, 3433–3442. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Vodicka, M.A.; Lucero, G.; Svitkina, T.M.; Borisy, G.G.; Emerman, M.; Hope, T.J. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 2002, 159, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Saib, A.; Puvion-Dutilleul, F.; Schmid, M.; Peries, J.; de The, H. Nuclear targeting of incoming human foamy virus Gag proteins involves a centriolar step. J. Virol. 1997, 71, 1155–1161. [Google Scholar] [CrossRef]
- Roe, T.; Reynolds, T.C.; Yu, G.; Brown, P.O. Integration of murine leukemia virus DNA depends on mitosis. EMBO J. 1993, 12, 2099–2108. [Google Scholar] [CrossRef]
- Naldini, L.; Blomer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef]
- Bushman, F.D. Targeting survival: integration site selection by retroviruses and LTR-retrotransposons. Cell 2003, 115, 135–138. [Google Scholar] [CrossRef]
- Craigie, R.; Fujiwara, T.; Bushman, F. The IN protein of Moloney murine leukemia virus processes the viral DNA ends and accomplishes their integration in vitro. Cell 1990, 62, 829–837. [Google Scholar] [CrossRef]
- Hare, S.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 2010, 464, 232–236. [Google Scholar] [CrossRef]
- Maertens, G.N.; Hare, S.; Cherepanov, P. The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature 2010, 468, 326–329. [Google Scholar] [CrossRef]
- Katzman, M.; Mack, J.P.; Skalka, A.M.; Leis, J. A covalent complex between retroviral integrase and nicked substrate DNA. Proc. Natl. Acad. Sci. USA 1991, 88, 4695–1699. [Google Scholar] [CrossRef]
- Engelman, A.; Mizuuchi, K.; Craigie, R. HIV-1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell 1991, 67, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.D.; Fujiwara, T.; Craigie, R. Retroviral DNA integration directed by HIV integration protein in vitro. Science 1990, 249, 1555–1558. [Google Scholar] [CrossRef] [PubMed]
- Yoder, K.E.; Bushman, F.D. Repair of gaps in retroviral DNA integration intermediates. J. Virol. 2000, 74, 11191–1200. [Google Scholar] [CrossRef] [PubMed]
- Holman, A.G.; Coffin, J.M. Symmetrical base preferences surrounding HIV-1, avian sarcoma/leukosis virus, and murine leukemia virus integration sites. Proc. Natl. Acad. Sci. USA 2005, 102, 6103–6107. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M.; Munroe, D.J. Weak palindromic consensus sequences are a common feature found at the integration target sites of many retroviruses. J. Virol. 2005, 79, 5211–5214. [Google Scholar] [CrossRef]
- Pryciak, P.M.; Sil, A.; Varmus, H.E. Retroviral integration into minichromosomes in vitro. EMBO J. 1992, 11, 291–303. [Google Scholar] [CrossRef]
- Pryciak, P.M.; Varmus, H.E. Nucleosomes, DNA-binding proteins, and DNA sequence modulate retroviral integration target site selection. Cell 1992, 69, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.P.; Varmus, H.E. DNA bending creates favored sites for retroviral integration: an explanation for preferred insertion sites in nucleosomes. EMBO J. 1994, 13, 4704–4714. [Google Scholar] [CrossRef]
- Pruss, D.; Reeves, R.; Bushman, F.D.; Wolffe, A.P. The influence of DNA and nucleosome structure on integration events directed by HIV integrase. J. Biol. Chem. 1994, 269, 25031–25041. [Google Scholar] [CrossRef] [PubMed]
- Scherdin, U.; Rhodes, K.; Breindl, M. Transcriptionally active genome regions are preferred targets for retrovirus integration. J. Virol. 1990, 64, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Mooslehner, K.; Karls, U.; Harbers, K. Retroviral integration sites in transgenic Mov mice frequently map in the vicinity of transcribed DNA regions. J. Virol. 1990, 64, 3056–3058. [Google Scholar] [CrossRef] [PubMed]
- Rohdewohld, H.; Weiher, H.; Reik, W.; Jaenisch, R.; Breindl, M. Retrovirus integration and chromatin structure: Moloney murine leukemia proviral integration sites map near DNase I-hypersensitive sites. J. Virol. 1987, 61, 336–343. [Google Scholar] [CrossRef]
- Vijaya, S.; Steffen, D.L.; Robinson, H.L. Acceptor sites for retroviral integrations map near DNase I-hypersensitive sites in chromatin. J. Virol. 1986, 60, 683–692. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Le Deist, F.; Carlier, F.; Bouneaud, C.; Hue, C.; De Villartay, J.P.; Thrasher, A.J.; Wulffraat, N.; Sorensen, R.; Dupuis-Girod, S.; et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N. Engl. J. Med. 2002, 346, 1185–1193. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.L.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 2008, 118, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Hematti, P.; Hong, B.K.; Ferguson, C.; Adler, R.; Hanawa, H.; Sellers, S.; Holt, I.E.; Eckfeldt, C.E.; Sharma, Y.; Schmidt, M.; et al. Distinct genomic integration of MLV and SIV vectors in primate hematopoietic stem and progenitor cells. PLoS Biol. 2004, 2, e423. [Google Scholar] [CrossRef] [PubMed]
- Themis, M.; Waddington, S.N.; Schmidt, M.; von Kalle, C.; Wang, Y.; Al-Allaf, F.; Gregory, L.G.; Nivsarkar, M.; Themis, M.; Holder, M.V.; et al. Oncogenesis following delivery of a nonprimate lentiviral gene therapy vector to fetal and neonatal mice. Mol. Ther. 2005, 12, 763–771. [Google Scholar] [CrossRef]
- Lewinski, M.K.; Yamashita, M.; Emerman, M.; Ciuffi, A.; Marshall, H.; Crawford, G.; Collins, F.; Shinn, P.; Leipzig, J.; Hannenhalli, S. Retroviral DNA integration: Viral and cellular determinants of target-site selection. PLoS Pathog. 2006, 2, e60. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Cherepanov, P. The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS Pathog. 2008, 4, e1000046. [Google Scholar] [CrossRef] [PubMed]
- Vandekerckhove, L.; Christ, F.; Van Maele, B.; De Rijck, J.; Gijsbers, R.; Van den Haute, C.; Witvrouw, M.; Debyser, Z. Transient and stable knockdown of the integrase cofactor LEDGF/p75 reveals its role in the replication cycle of human immunodeficiency virus. J. Virol. 2006, 80, 1886–1896. [Google Scholar] [CrossRef]
- Llano, M.; Delgado, S.; Vanegas, M.; Poeschla, E.M. Lens epithelium-derived growth factor/p75 prevents proteasomal degradation of HIV-1 integrase. J. Biol. Chem. 2004, 279, 55570–55577. [Google Scholar] [CrossRef]
- Busschots, K.; Vercammen, J.; Emiliani, S.; Benarous, R.; Engelborghs, Y.; Christ, F.; Debyser, Z. The interaction of LEDGF/p75 with integrase is lentivirus-specific and promotes DNA binding. J. Biol. Chem. 2005, 280, 17841–17847. [Google Scholar] [CrossRef]
- Emiliani, S.; Mousnier, A.; Busschots, K.; Maroun, M.; Van Maele, B.; Tempe, D.; Vandekerckhove, L.; Moisant, F.; Ben-Slama, L.; Witvrouw, M. Integrase mutants defective for interaction with LEDGF/p75 are impaired in chromosome tethering and HIV-1 replication. J. Biol. Chem. 2005, 280, 25517–25523. [Google Scholar] [CrossRef]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar] [CrossRef] [PubMed]
- Gijsbers, R.; Ronen, K.; Vets, S.; Malani, N.; De Rijck, J.; McNeely, M.; Bushman, F.D.; Debyser, Z. LEDGF hybrids efficiently retarget lentiviral integration into heterochromatin. Mol. Ther. 2010, 18, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Marshall, H.M.; Ronen, K.; Berry, C.; Llano, M.; Sutherland, H.; Saenz, D.; Bickmore, W.; Poeschla, E.; Bushman, F.D. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS ONE 2007, 2, e1340. [Google Scholar] [CrossRef] [PubMed]
- Shun, M.C.; Raghavendra, N.K.; Vandegraaff, N.; Daigle, J.E.; Hughes, S.; Kellam, P.; Cherepanov, P.; Engelman, A. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Gene. Develop. 2007, 21, 1767–1778. [Google Scholar] [CrossRef]
- Bartholomae, C.C.; Arens, A.; Balaggan, K.S.; Yanez-Munoz, R.J.; Montini, E.; Howe, S.J.; Paruzynski, A.; Korn, B.; Appelt, J.U.; Macneil, A. Lentiviral Vector Integration Profiles Differ in Rodent Postmitotic Tissues. Mol. Ther. 2011, 19, 703–710. [Google Scholar] [CrossRef]
- Hendrix, J.; Gijsbers, R.; De Rijck, J.; Voet, A.; Hotta, J.; McNeely, M.; Hofkens, J.; Debyser, Z.; Engelborghs, Y. The transcriptional co-activator LEDGF/p75 displays a dynamic scan-and-lock mechanism for chromatin tethering. Nucl. Acid. Res. 2011, 39, 1310–1325. [Google Scholar] [CrossRef]
- Silvers, R.M.; Smith, J.A.; Schowalter, M.; Litwin, S.; Liang, Z.; Geary, K.; Daniel, R. Modification of integration site preferences of an HIV-1-based vector by expression of a novel synthetic protein. Hum. Gene. Ther. 2010, 21, 337–349. [Google Scholar] [CrossRef]
- Meehan, A.M.; Poeschla, E.M. Chromatin tethering and retroviral integration: recent discoveries and parallels with DNA viruses. Biochim. Biophys. Acta 2010, 1799, 182–191. [Google Scholar] [CrossRef]
- Ferris, A.L.; Wu, X.; Hughes, C.M.; Stewart, C.; Smith, S.J.; Milne, T.A.; Wang, G.G.; Shun, M.C.; Allis, C.D.; Engelman, A.; Hughes, S.H. Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration. Proc. Natl. Acad. Sci. USA 2010, 107, 3135–3140. [Google Scholar] [CrossRef]
- Meekings, K.N.; Leipzig, J.; Bushman, F.D.; Taylor, G.P.; Bangham, C.R. HTLV-1 integration into transcriptionally active genomic regions is associated with proviral expression and with HAM/TSP. PLoS Pathog. 2008, 4, e1000027. [Google Scholar] [CrossRef]
- Derse, D.; Crise, B.; Li, Y.; Princler, G.; Lum, N.; Stewart, C.; McGrath, C.F.; Hughes, S.H.; Munroe, D.J.; Wu, X. Human T-cell leukemia virus type 1 integration target sites in the human genome: comparison with those of other retroviruses. J. Virol. 2007, 81, 6731–6741. [Google Scholar] [CrossRef] [PubMed]
- Maxfield, L.F.; Fraize, C.D.; Coffin, J.M. Relationship between retroviral DNA-integration-site selection and host cell transcription. Proc. Nat. Acad. Sci. USA 2005, 102, 1436–1441. [Google Scholar] [CrossRef]
- Lewinski, M.K.; Bisgrove, D.; Shinn, P.; Chen, H.; Hoffmann, C.; Hannenhalli, S.; Verdin, E.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Genome-wide analysis of chromosomal features repressing human immunodeficiency virus transcription. J. Virol. 2005, 79, 6610–6619. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Cuesta, I.; Daniel, R.; Cirillo, L.A.; Katz, R.A.; Zaret, K.S.; Skalka, A.M. Integrase-specific enhancement and suppression of retroviral DNA integration by compacted chromatin structure in vitro. J. Virol. 2004, 78, 5848–5855. [Google Scholar] [CrossRef] [PubMed]
- Felice, B.; Cattoglio, C.; Cittaro, D.; Testa, A.; Miccio, A.; Ferrari, G.; Luzi, L.; Recchia, A.; Mavilio, F. Transcription factor binding sites are genetic determinants of retroviral integration in the human genome. PLoS ONE 2009, 4, e4571. [Google Scholar] [CrossRef] [PubMed]
- Biasco, L.; Ambrosi, A.; Pellin, D.; Bartholomae, C.; Brigida, I.; Roncarolo, M.G.; Di Serio, C.; von Kalle, C.; Schmidt, M.; Aiuti, A. Integration profile of retroviral vector in gene therapy treated patients is cell-specific according to gene expression and chromatin conformation of target cell. EMBO Mol. Med. 2011, 3, 89–101. [Google Scholar] [CrossRef]
- Baum, C. Parachuting in the epigenome: the biology of gene vector insertion profiles in the context of clinical trials. EMBO Mol. Med. 2011, 3, 75–77. [Google Scholar] [CrossRef]
- Baum, C.; Dullmann, J.; Li, Z.; Fehse, B.; Meyer, J.; Williams, D.A.; von Kalle, C. Side effects of retroviral gene transfer into hematopoietic stem cells. Blood 2003, 101, 2099–2114. [Google Scholar] [CrossRef]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Invest. 2009, 119, 964–975. [Google Scholar] [CrossRef]
- Maruggi, G.; Porcellini, S.; Facchini, G.; Perna, S.K.; Cattoglio, C.; Sartori, D.; Ambrosi, A.; Schambach, A.; Baum, C.; Bonini, C.; et al. Transcriptional enhancers induce insertional gene deregulation independently from the vector type and design. Mol. Ther. 2009, 17, 851–856. [Google Scholar] [CrossRef]
- Modlich, U.; Bohne, J.; Schmidt, M.; von Kalle, C.; Knoss, S.; Schambach, A.; Baum, C. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood 2006, 108, 2545–2553. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.H.; Turner, G.; Trubetskoy, A.; Verhoeven, E.; Wientjens, E.; Hulsman, D.; Russell, R.; DePinho, R.A.; Lenz, J.; van Lohuizen, M. Genome-wide retroviral insertional tagging of genes involved in cancer in Cdkn2a-deficient mice. Nat. Genet. 2002, 32, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Ponzoni, M.; Bartholomae, C.; Sergi, L.S.; Benedicenti, F.; Ambrosi, A.; Di Serio, C. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 2006, 24, 687–696. [Google Scholar] [CrossRef]
- Bauer, T.R. Jr.; Allen, J.M.; Hai, M.; Tuschong, L.M.; Khan, I.F.; Olson, E.M.; Adler, R.L.; Burkholder, T.H.; Gu, Y.C.; Russell, D.W. Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors. Nat. Med. 2008, 14, 93–97. [Google Scholar] [CrossRef]
- Li, J.; Shen, H.; Himmel, K.L.; Dupuy, A.J.; Largaespada, D.A.; Nakamura, T.; Shaughnessy, J.D.Jr.; Jenkins, N.A.; Copeland, N.G. Leukaemia disease genes: large-scale cloning and pathway predictions. Nat. Genet. 1999, 23, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; Steigerwald, S.D.; Mueller, P.R.; Wold, B.; Riggs, A.D. Genomic sequencing and methylation analysis by ligation mediated PCR. Science 1989, 246, 810–813. [Google Scholar] [CrossRef]
- Mueller, P.R.; Wold, B. In vivo footprinting of a muscle specific enhancer by ligation mediated PCR. Science 1989, 246, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.; Keerikatte, V. Novel use of polymerase chain reaction to amplify cellular DNA adjacent to an integrated provirus. J. Virol. 1989, 63, 1924–1928. [Google Scholar] [CrossRef]
- Schmidt, M.; Schwarzwaelder, K.; Bartholomae, C.; Zaoui, K.; Ball, C.; Pilz, I.; Braun, S.; Glimm, H.; von Kalle, C. High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR). Nat. Methods 2007, 4, 1051–1057. [Google Scholar] [CrossRef]
- Schmidt, M.; Hoffmann, G.; Wissler, M.; Lemke, N.; Mussig, A.; Glimm, H.; Williams, D.A.; Ragg, S.; Hesemann, C.U.; von Kalle, C. Detection and direct genomic sequencing of multiple rare unknown flanking DNA in highly complex samples. Hum. Gene. Ther. 2001, 12, 743–749. [Google Scholar] [CrossRef]
- Schmidt, M.; Carbonaro, D.A.; Speckmann, C.; Wissler, M.; Bohnsack, J.; Elder, M.; Aronow, B.J.; Nolta, J.A.; Kohn, D.B.; von Kalle, C. Clonality analysis after retroviral-mediated gene transfer to CD34+ cells from the cord blood of ADA-deficient SCID neonates. Nat. Med. 2003, 9, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, R.; Eckenberg, R.; Paruzynski, A.; Bartholomae, C.C.; Nowrouzi, A.; Arens, A.; Howe, S.J.; Recchia, A.; Cattoglio, C.; Wang, W.; et al. Comprehensive genomic access to vector integration in clinical gene therapy. Nat. Med. 2009, 15, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Paruzynski, A.; Arens, A.; Gabriel, R.; Bartholomae, C.C.; Scholz, S.; Wang, W.; Wolf, S.; Glimm, H.; Schmidt, M.; von Kalle, C. Genome-wide high-throughput integrome analyses by nrLAM-PCR and next-generation sequencing. Nat. Protocol. 2010, 5, 1379–1395. [Google Scholar] [CrossRef] [PubMed]
- Warlich, E.; Kuehle, J.; Cantz, T.; Brugman, M.H.; Maetzig, T.; Galla, M.; Filipczyk, A.A.; Halle, S.; Klump, H.; Scholer, H.R.; et al. Lentiviral Vector Design and Imaging Approaches to Visualize the Early Stages of Cellular Reprogramming. Mol. Ther. 2011, 19, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Schwarzwaelder, K.; Bartholomae, C.C.; Glimm, H.; von Kalle, C. Detection of retroviral integration sites by linear amplification-mediated PCR and tracking of individual integration clones in different samples. Meth. Mol. Biol. 2009, 506, 363–372. [Google Scholar]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef]
- Schwarzwaelder, K.; Howe, S.J.; Schmidt, M.; Brugman, M.H.; Deichmann, A.; Glimm, H.; Schmidt, S.; Prinz, C.; Wissler, M.; King, D.J.; et al. Gammaretrovirus-mediated correction of SCID-X1 is associated with skewed vector integration site distribution in vivo. J. Clin. Invest. 2007, 117, 2241–2249. [Google Scholar] [CrossRef]
- Deichmann, A.; Hacein-Bey-Abina, S.; Schmidt, M.; Garrigue, A.; Brugman, M.H.; Hu, J.; Glimm, H.; Gyapay, G.; Prum, B.; Fraser, C.C.; et al. Vector integration is nonrandom and clustered and influences the fate of lymphopoiesis in SCID-X1 gene therapy. J. Clin. Invest. 2007, 117, 2225–2232. [Google Scholar] [CrossRef]
- Ott, M.G.; Schmidt, M.; Schwarzwaelder, K.; Stein, S.; Siler, U.; Koehl, U.; Glimm, H.; Kuhlcke, K.; Schilz, A.; Kunkel, H.; et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006, 12, 401–409. [Google Scholar] [CrossRef]
- Boztug, K.; Schmidt, M.; Schwarzer, A.; Banerjee, P.P.; Diez, I.A.; Dewey, R.A.; Bohm, M.; Nowrouzi, A.; Ball, C.R.; Glimm, H.; et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N. Engl. J. Med. 2010, 363, 1918–1927. [Google Scholar] [CrossRef]
- Wang, G.P.; Garrigue, A.; Ciuffi, A.; Ronen, K.; Leipzig, J.; Berry, C.; Lagresle-Peyrou, C.; Benjelloun, F.; Hacein-Bey-Abina, S.; Fischer, A.; et al. DNA bar coding and pyrosequencing to analyze adverse events in therapeutic gene transfer. Nucl. Acid. Res. 2008, 36, e49. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.P.; Berry, C.C.; Malani, N.; Leboulch, P.; Fischer, A.; Hacein-Bey-Abina, S.; Cavazzana-Calvo, M.; Bushman, F.D. Dynamics of gene-modified progenitor cells analyzed by tracking retroviral integration sites in a human SCID-X1 gene therapy trial. Blood 2010, 115, 4356–4366. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.; Ott, M.G.; Schultze-Strasser, S.; Jauch, A.; Burwinkel, B.; Kinner, A.; Schmidt, M.; Kramer, A.; Schwable, J.; Glimm, H.; et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010, 16, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Payne, G.S.; Bishop, J.M.; Varmus, H.E. Multiple arrangements of viral DNA and an activated host oncogene in bursal lymphomas. Nature 1982, 295, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Payne, G.S.; Courtneidge, S.A.; Crittenden, L.B.; Fadly, A.M.; Bishop, J.M.; Varmus, H.E. Analysis of avian leukosis virus DNA and RNA in bursal tumours: viral gene expression is not required for maintenance of the tumor state. Cell 1981, 23, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Neel, B.G.; Hayward, W.S.; Robinson, H.L.; Fang, J.; Astrin, S.M. Avian leukosis virus-induced tumors have common proviral integration sites and synthesize discrete new RNAs: oncogenesis by promoter insertion. Cell 1981, 23, 323–334. [Google Scholar] [CrossRef]
- Hayward, W.S.; Neel, B.G.; Astrin, S.M. Activation of a cellular onc gene by promoter insertion in ALV-induced lymphoid leukosis. Nature 1981, 290, 475–480. [Google Scholar] [CrossRef]
- Kool, J.; Berns, A. High-throughput insertional mutagenesis screens in mice to identify oncogenic networks. Nat. Rev. Canc. 2009, 9, 389–399. [Google Scholar] [CrossRef]
- Rosenberg, N.; Jolicoeur, P. Retroviral pathogenesis. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, E.H., Eds.; ColdSpring Harbor Laboratory Press: New York, NY, USA, 1997; pp. S475–S586. [Google Scholar]
- Moolten, F.L.; Cupples, L.A. A model for predicting the risk of cancer consequent to retroviral gene therapy. Hum. Gene. Ther. 1992, 3, 479–486. [Google Scholar] [CrossRef]
- Li, Z.; Dullmann, J.; Schiedlmeier, B.; Schmidt, M.; von Kalle, C.; Meyer, J.; Forster, M.; Stocking, C.; Wahlers, A.; Frank, O.; Ostertag, W.; et al. Murine leukemia induced by retroviral gene marking. Science 2002, 296, 497. [Google Scholar] [CrossRef] [PubMed]
- Modlich, U.; Kustikova, O.S.; Schmidt, M.; Rudolph, C.; Meyer, J.; Li, Z.; Kamino, K.; von Neuhoff, N.; Schlegelberger, B.; Kuehlcke, K.; et al. Leukemias following retroviral transfer of multidrug resistance 1 (MDR1) are driven by combinatorial insertional mutagenesis. Blood 2005, 105, 4235–4246. [Google Scholar] [CrossRef]
- Calmels, B.; Ferguson, C.; Laukkanen, M.O.; Adler, R.; Faulhaber, M.; Kim, H.J.; Sellers, S.; Hematti, P.; Schmidt, M.; von Kalle, C.; et al. Recurrent retroviral vector integration at the Mds1/Evi1 locus in nonhuman primate hematopoietic cells. Blood 2005, 106, 2530–2533. [Google Scholar] [CrossRef] [PubMed]
- Kustikova, O.; Fehse, B.; Modlich, U.; Yang, M.; Dullmann, J.; Kamino, K.; von Neuhoff, N.; Schlegelberger, B.; Li, Z.; Baum, C. Clonal dominance of hematopoietic stem cells triggered by retroviral gene marking. Science 2005, 308, 1171–1174. [Google Scholar] [CrossRef] [PubMed]
- Seggewiss, R.; Pittaluga, S.; Adler, R.L.; Guenaga, F.J.; Ferguson, C.; Pilz, I.H.; Ryu, B.; Sorrentino, B.P.; Young, W.S.; Donahue, R.E.; et al. Acute myeloid leukemia is associated with retroviral gene transfer to hematopoietic progenitor cells in a rhesus macaque. Blood 2006, 107, 3865–3867. [Google Scholar] [CrossRef]
- Aiuti, A.; Cattaneo, F.; Galimberti, S.; Benninghoff, U.; Cassani, B.; Callegaro, L.; Scaramuzza, S.; Andolfi, G.; Mirolo, M.; Brigida, I.; et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 2009, 360, 447–458. [Google Scholar] [CrossRef]
- Woods, N.B.; Bottero, V.; Schmidt, M.; von Kalle, C.; Verma, I.M. Gene therapy: Therapeutic gene causing lymphoma. Nature 2006, 440, 1123. [Google Scholar] [CrossRef]
- Thrasher, A.J.; Gaspar, H.B.; Baum, C.; Modlich, U.; Schambach, A.; Candotti, F.; Otsu, M.; Sorrentino, B.; Scobie, L.; Cameron, E.; et al. Gene therapy: X-SCID transgene leukaemogenicity. Nature 2006, 443, E5–E6; discussion E6–E7. [Google Scholar] [CrossRef]
- Dave, U.P.; Akagi, K.; Tripathi, R.; Cleveland, S.M.; Thompson, M.A.; Yi, M.; Stephens, R.; Downing, J.R.; Jenkins, N.A.; Copeland, N.G. Murine leukemias with retroviral insertions at Lmo2 are predictive of the leukemias induced in SCID-X1 patients following retroviral gene therapy. PLoS Genet. 2009, 5, e1000491. [Google Scholar] [CrossRef]
- Cartier, N.; Aubourg, P. Hematopoietic stem cell transplantation and hematopoietic stem cell gene therapy in X-linked adrenoleukodystrophy. Brain Pathol. 2010, 20, 857–862. [Google Scholar] [CrossRef]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Ponzoni, M.; Bartholomae, C.; Sergi Sergi, L.; Benedicenti, F.; Ambrosi, A.; Di Serio, C.; et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 2006, 24, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Modlich, U.; Navarro, S.; Zychlinski, D.; Maetzig, T.; Knoess, S.; Brugman, M.H.; Schambach, A.; Charrier, S.; Galy, A.; Thrasher, A.J.; et al. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol. Ther. 2009, 17, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Daya, S.; Berns, K.I. Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W. AAV vectors, insertional mutagenesis, and cancer. Mol. Ther. 2007, 15, 1740–1743. [Google Scholar] [CrossRef]
- Donsante, A.; Miller, D.G.; Li, Y.; Vogler, C.; Brunt, E.M.; Russell, D.W.; Sands, M.S. AAV vector integration sites in mouse hepatocellular carcinoma. Science 2007, 317, 477. [Google Scholar] [CrossRef] [PubMed]
- Wanisch, K.; Yanez-Munoz, R.J. Integration-deficient lentiviral vectors: A slow coming of age. Mol. Ther. 2009, 17, 1316–1332. [Google Scholar] [CrossRef]
- Yanez-Munoz, R.J.; Balaggan, K.S.; MacNeil, A.; Howe, S.J.; Schmidt, M.; Smith, A.J.; Buch, P.; MacLaren, R.E.; Anderson, P.N.; Barker, S.E.; et al. Effective gene therapy with nonintegrating lentiviral vectors. Nat. Med. 2006, 12, 348–353. [Google Scholar] [CrossRef]
- Bushman, F. Targeting retroviral integration? Mol. Ther. 2002, 6, 570–571. [Google Scholar] [CrossRef]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef]
- Ivics, Z.; Izsvak, Z. The expanding universe of transposon technologies for gene and cell engineering. Mob. DNA 2010, 1, 25. [Google Scholar] [CrossRef]
- Copeland, N.G.; Jenkins, N.A. Harnessing transposons for cancer gene discovery. Nat. Rev. Canc. 2010, 10, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S.; Blau, H.M. Nuclear reprogramming to a pluripotent state by three approaches. Nature 2010, 465, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Sudhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef]
- Szabo, E.; Rampalli, S.; Risueno, R.M.; Schnerch, A.; Mitchell, R.; Fiebig-Comyn, A.; Levadoux-Martin, M.; Bhatia, M. Direct conversion of human fibroblasts to multilineage blood progenitors. Nature 2010, 468, 521–526. [Google Scholar] [CrossRef]
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef]
- Papapetrou, E.P.; Lee, G.; Malani, N.; Setty, M.; Riviere, I.; Tirunagari, L.M.; Kadota, K.; Roth, S.L.; Giardina, P.; Viale, A.; et al. Genomic safe harbors permit high beta-globin transgene expression in thalassemia induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 73–78. [Google Scholar] [CrossRef]
- Yamanaka, S. Strategies and new developments in the generation of patient-specific pluripotent stem cells. Cell Stem. Cell 2007, 1, 39–49. [Google Scholar] [CrossRef]
- Ellis, J.; Baum, C.; Benvenisty, N.; Mostoslavsky, G.; Okano, H.; Stanford, W.L.; Porteus, M.; Sadelain, M. Benefits of utilizing gene-modified iPSCs for clinical applications. Cell Stem. Cell 2010, 7, 429–430. [Google Scholar] [CrossRef]
- Suzuki, T.; Shen, H.; Akagi, K.; Morse, H.C.; Malley, J.D.; Naiman, D.Q.; Jenkins, N.A.; Copeland, N.G. New genes involved in cancer identified by retroviral tagging. Nat. Genet. 2002, 32, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Akagi, K.; Suzuki, T.; Stephens, R.M.; Jenkins, N.A.; Copeland, N.G. RTCGD: Retroviral tagged cancer gene database. Nucl. Acid. Res. 2004, 32, D523–D527. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.K.; Allaei, R.; Silverstein, K.A.; Staggs, R.A.; Sarver, A.L.; Bergemann, T.L.; Gupta, M.; O’Sullivan, M.G.; Matise, I.; Dupuy, A.J.; et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science 2009, 323, 1747–1750. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, A.J.; Rogers, L.M.; Kim, J.; Nannapaneni, K.; Starr, T.K.; Liu, P.; Largaespada, D.A.; Scheetz, T.E.; Jenkins, N.A.; Copeland, N.G. A modified sleeping beauty transposon system that can be used to model a wide variety of human cancers in mice. Canc. Res. 2009, 69, 8150–8156. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Spence, S.E.; Jenkins, N.A.; Copeland, N.G. Cooperating cancer-gene identification through oncogenic-retrovirus-induced insertional mutagenesis. Blood 2005, 106, 2498–2505. [Google Scholar] [CrossRef]
- Du, Y.; Jenkins, N.A.; Copeland, N.G. Insertional mutagenesis identifies genes that promote the immortalization of primary bone marrow progenitor cells. Blood 2005, 106, 3932–3939. [Google Scholar] [CrossRef]
- Copeland, N.G.; Jenkins, N.A. Retroviral integration in murine myeloid tumors to identify Evi-1, a novel locus encoding a zinc-finger protein. Adv. Canc. Res. 1990, 54, 141–157. [Google Scholar]
- Neff, T.; Shotkoski, F.; Stamatoyannopoulos, G. Stem cell gene therapy, position effects and chromatin insulators. Stem Cells 1997, 15, 265–271. [Google Scholar]
- Valenzuela, L.; Kamakaka, R.T. Chromatin insulators. Annu. Rev. Genet. 2006, 40, 107–138. [Google Scholar] [CrossRef]
- Arumugam, P.I.; Higashimoto, T.; Urbinati, F.; Modlich, U.; Nestheide, S.; Xia, P.; Fox, C.; Corsinotti, A.; Baum, C.; Malik, P. Genotoxic potential of lineage-specific lentivirus vectors carrying the beta-globin locus control region. Mol. Ther. 2009, 17, 1929–1937. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2011 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nowrouzi, A.; Glimm, H.; Von Kalle, C.; Schmidt, M. Retroviral Vectors: Post Entry Events and Genomic Alterations. Viruses 2011, 3, 429-455. https://doi.org/10.3390/v3050429
Nowrouzi A, Glimm H, Von Kalle C, Schmidt M. Retroviral Vectors: Post Entry Events and Genomic Alterations. Viruses. 2011; 3(5):429-455. https://doi.org/10.3390/v3050429
Chicago/Turabian StyleNowrouzi, Ali, Hanno Glimm, Christof Von Kalle, and Manfred Schmidt. 2011. "Retroviral Vectors: Post Entry Events and Genomic Alterations" Viruses 3, no. 5: 429-455. https://doi.org/10.3390/v3050429
APA StyleNowrouzi, A., Glimm, H., Von Kalle, C., & Schmidt, M. (2011). Retroviral Vectors: Post Entry Events and Genomic Alterations. Viruses, 3(5), 429-455. https://doi.org/10.3390/v3050429