Improving Adenovirus Based Gene Transfer: Strategies to Accomplish Immune Evasion

{kind=link}
Abstract
:1. Introduction
2. Pre-emptive treatment of the host as a strategy to allow immune evasion by Ads
2.1. Suppression of innate immune responses induced by Ad mediated gene transfer
2.1.1. Global immunosuppression approach (innate)
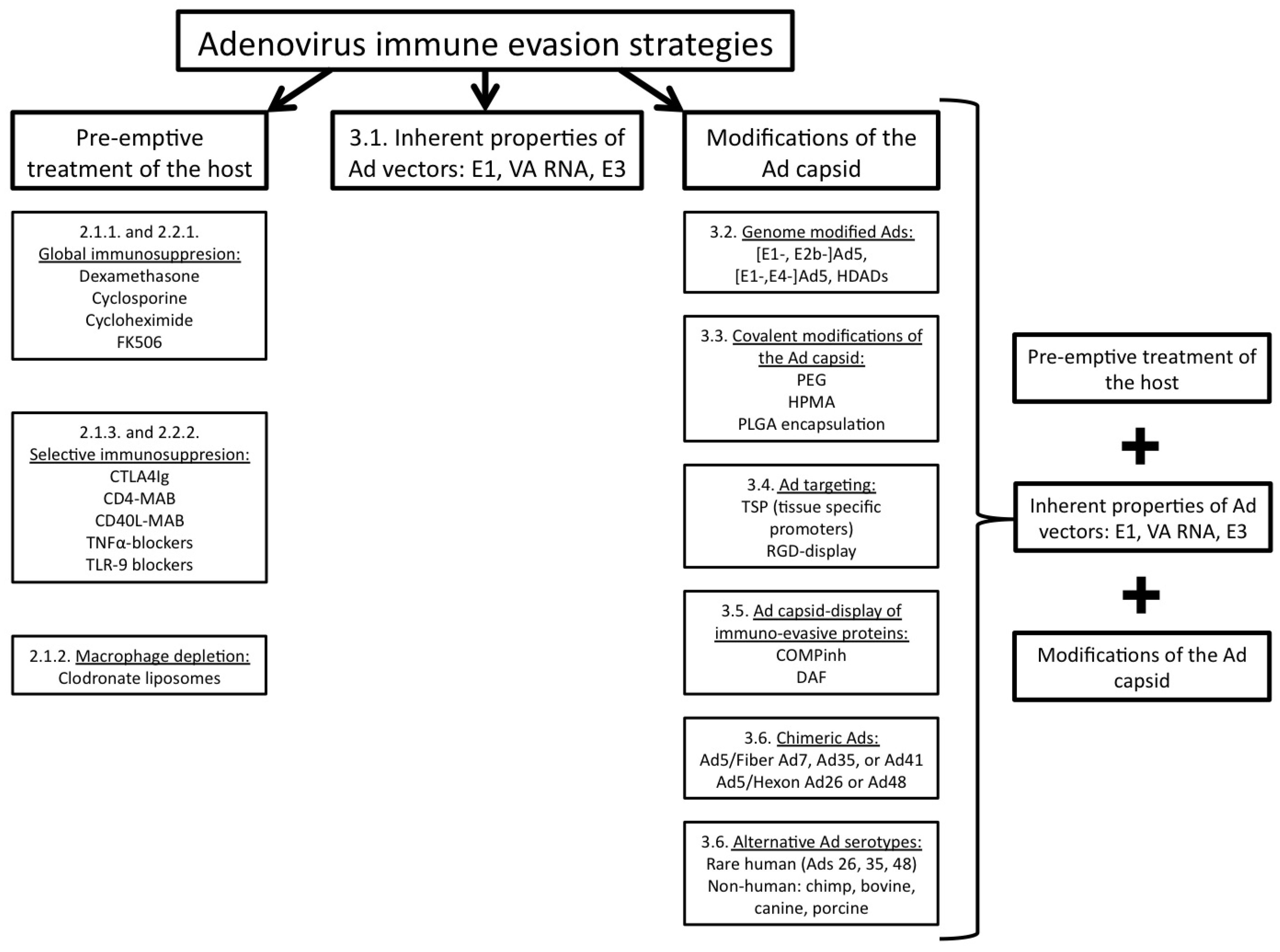
2.1.2. Macrophage depletion
2.1.3. Selective immunosuppression approach (innate)
2.2. Suppression of adaptive immune responses induced by Ad mediated gene transfer
2.2.1. Global immunosuppression approach (adaptive)
2.2.2. Selective immunosuppression approach (adaptive)
3. Modification of the Ad genome and/or capsid as a strategy to facilitate immune evasion by Ads
3.1. Inherent properties of Ad vectors
3.2. Genome modified Ad vectors
3.3. Covalent modifications of the Ad capsid
3.4. Ad targeting
3.5. Ad capsid-display of immuno-evasive proteins
3.6. Chimeric Ad vectors and Ad vectors, derived from alternative Ad serotypes
4. Conclusions and future directions
References and Notes
- Amalfitano, A. Utilization of adenovirus vectors for multiple gene transfer applications. Methods 2004, 33, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Bangari, D.S.; Mittal, S.K. Current strategies and future directions for eluding adenoviral vector immunity. Curr. Gene Ther. 2006, 6, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.C.; Kiang, A.; Everett, R.S.; Serra, D.; Yang, X.Y.; Clay, T.M.; Amalfitano, A. Adenovirus infection triggers a rapid, MyD88-regulated transcriptome response critical to acute-phase and adaptive immune responses in vivo. J. Virol. 2007, 81, 1796–1812. [Google Scholar] [CrossRef] [PubMed]
- Appledorn, D.M.; McBride, A.; Seregin, S.; Scott, J.M.; Schuldt, N.; Kiang, A.; Godbehere, S.; Amalfitano, A. Complex interactions with several arms of the complement system dictate innate and humoral immunity to adenoviral vectors. Gene Ther. 2008, 15, 1606–1617. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Appledorn, D.M.; Amalfitano, A. Adenovirus vector induced innate immune responses: impact upon efficacy and toxicity in gene therapy and vaccine applications. Virus Res. 2008, 132, 1–14. [Google Scholar] [CrossRef]
- Kiang, A.; Hartman, Z.C.; Everett, R.S.; Serra, D.; Jiang, H.; Frank, M.M.; Amalfitano, A. Multiple innate inflammatory responses induced after systemic adenovirus vector delivery depend on a functional complement system. Mol. Ther. 2006, 14, 588–598. [Google Scholar] [CrossRef]
- Seregin, S.S.; Aldhamen, Y.A.; Appledorn, D.M.; Schuldt, N.J.; McBride, A.J.; Bujold, M.; Godbehere, S.; Amalfitano, A. CR1/2 is an important suppressor of Adenovirus-induced innate immune responses and is required for induction of neutralizing antibodies. Gene Ther. 2009, 16, 1245–1259. [Google Scholar] [CrossRef]
- Seregin, S.S.; Appledorn, D.M.; McBride, A.J.; Schuldt, N.J.; Aldhamen, Y.A.; Voss, T.; Wei, J.; Bujold, M.; Nance, W.; Godbehere, S.; Amalfitano, A. Transient pretreatment with glucocorticoid ablates innate toxicity of systemically delivered adenoviral vectors without reducing efficacy. Mol. Ther. 2009, 17, 685–696. [Google Scholar] [CrossRef]
- Raper, S.E.; Chirmule, N.; Lee, F.S.; Wivel, N.A.; Bagg, A.; Gao, G.P.; Wilson, J.M.; Batshaw, M.L. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003, 80, 148–158. [Google Scholar] [CrossRef]
- Makower, D.; Rozenblit, A.; Kaufman, H.; Edelman, M.; Lane, M.E.; Zwiebel, J.; Haynes, H.; Wadler, S. Phase II clinical trial of intralesional administration of the oncolytic adenovirus ONYX-015 in patients with hepatobiliary tumors with correlative p53 studies. Clin. Cancer Res. 2003, 9, 693–702. [Google Scholar]
- Grines, C.L.; Watkins, M.W.; Mahmarian, J.J.; Iskandrian, A.E.; Rade, J.J.; Marrott, P.; Pratt, C.; Kleiman, N. A randomized, double-blind, placebo-controlled trial of Ad5FGF-4 gene therapy and its effect on myocardial perfusion in patients with stable angina. J. Am. Coll. Cardiol. 2003, 42, 1339–1347. [Google Scholar] [CrossRef]
- Atencio, I.A.; Warren, R.; Venook, A.P.; Kemeny, M.M.; Staley, C.; Fraker, D.L.; Horwitz, J.; Rybak, M.; Freeman, S.; Indelicato, S.; Grace, M.; et al. A phase I clinical trial of intra-arterial adenovirus p53 (SCH 58500) gene therapy for colorectal tumors metastatic to the liver. Mol. Ther. 2001, 3 (Suppl. 5), part 2 of 2 parts. [Google Scholar]
- Muruve, D.A.; Barnes, M.J.; Stillman, I.E.; Libermann, T.A. Adenoviral gene therapy leads to rapid induction of multiple chemokines and acute neutrophil-dependent hepatic injury in vivo. Hum. Gene Ther. 1999, 10, 965–976. [Google Scholar] [CrossRef]
- Lieber, A.; He, C.Y.; Meuse, L.; Schowalter, D.; Kirillova, I.; Winther, B.; Kay, M.A. The role of Kupffer cell activation and viral gene expression in early liver toxicity after infusion of recombinant adenovirus vectors. J. Virol. 1997, 71, 8798–8807. [Google Scholar] [CrossRef]
- Zhang, Y.; Chirmule, N.; Gao, G.P.; Qian, R.; Croyle, M.; Joshi, B.; Tazelaar, J.; Wilson, J.M. Acute cytokine response to systemic adenoviral vectors in mice is mediated by dendritic cells and macrophages. Mol. Ther. 2001, 3, 697–707. [Google Scholar] [CrossRef]
- Seregin, S.S.; Amalfitano, A. Overcoming pre-existing adenovirus immunity by genetic engineering of adenovirus-based vectors. Expert Opin. Biol. Ther. 2009, 9, 1521–1531. [Google Scholar] [CrossRef]
- Wolins, N.; Lozier, J.; Eggerman, T.L.; Jones, E.; Aguilar-Cordova, E.; Vostal, J.G. Intravenous administration of replication-incompetent adenovirus to rhesus monkeys induces thrombocytopenia by increasing in vivo platelet clearance. Br. J. Haematol. 2003, 123, 903–905. [Google Scholar] [CrossRef]
- Schiedner, G.; Bloch, W.; Hertel, S.; Johnston, M.; Molojavyi, A.; Dries, V.; Varga, G.; Van Rooijen, N.; Kochanek, S. A hemodynamic response to intravenous adenovirus vector particles is caused by systemic Kupffer cell-mediated activation of endothelial cells. Hum. Gene Ther. 2003, 14, 1631–1641. [Google Scholar] [CrossRef]
- Muruve, D.A.; Petrilli, V.; Zaiss, A.K.; White, L.R.; Clark, S.A.; Ross, P.J.; Parks, R.J.; Tschopp, J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008, 452, 103–107. [Google Scholar] [CrossRef]
- Varnavski, A.N.; Calcedo, R.; Bove, M.; Gao, G.; Wilson, J.M. Evaluation of toxicity from high-dose systemic administration of recombinant adenovirus vector in vector-naive and pre-immunized mice. Gene Ther. 2005, 12, 427–436. [Google Scholar] [CrossRef]
- Jiang, H.; Wang, Z.; Serra, D.; Frank, M.M.; Amalfitano, A. Recombinant adenovirus vectors activate the alternative complement pathway, leading to the binding of human complement protein C3 independent of anti-ad antibodies. Mol. Ther. 2004, 10, 1140–1142. [Google Scholar] [CrossRef] [PubMed]
- Cichon, G.; Boeckh-Herwig, S.; Schmidt, H.H.; Wehnes, E.; Muller, T.; Pring-Akerblom, P.; Burger, R. Complement activation by recombinant adenoviruses. Gene Ther. 2001, 8, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Appledorn, D.M.; Kiang, A.; McBride, A.; Jiang, H.; Seregin, S.; Scott, J.M.; Stringer, R.; Kousa, Y.; Hoban, M.; Frank, M.M.; Amalfitano, A. Wild-type adenoviruses from groups A-F evoke unique innate immune responses, of which HAd3 and SAd23 are partially complement dependent. Gene Ther. 2008, 15, 885–901. [Google Scholar] [CrossRef]
- Ding, E.; Hu, H.; Hodges, B.L.; Migone, F.; Serra, D.; Xu, F.; Chen, Y.T.; Amalfitano, A. Efficacy of gene therapy for a prototypical lysosomal storage disease (GSD-II) is critically dependent on vector dose, transgene promoter, and the tissues targeted for vector transduction. Mol. Ther. 2002, 5, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Schowalter, D.B.; Himeda, C.L.; Winther, B.L.; Wilson, C.B.; Kay, M.A. Implication of interfering antibody formation and apoptosis as two different mechanisms leading to variable duration of adenovirus-mediated transgene expression in immune-competent mice. J. Virol. 1999, 73, 4755–4766. [Google Scholar] [CrossRef]
- Amalfitano, A.; Parks, R.J. Separating fact from fiction: assessing the potential of modified adenovirus vectors for use in human gene therapy. Curr. Gene Ther. 2002, 2, 111–133. [Google Scholar] [CrossRef]
- Ding, E.Y.; Hodges, B.L.; Hu, H.; McVie-Wylie, A.J.; Serra, D.; Migone, F.K.; Pressley, D.; Chen, Y.T.; Amalfitano, A. Long-term efficacy after [E1-, polymerase-] adenovirus-mediated transfer of human acid-alpha-glucosidase gene into glycogen storage disease type II knockout mice. Hum. Gene Ther. 2001, 12, 955–965. [Google Scholar] [CrossRef]
- Palmer, D.J.; Ng, P. Helper-dependent adenoviral vectors for gene therapy. Hum. Gene Ther. 2005, 16, 1–16. [Google Scholar] [CrossRef]
- Svensson, E.C.; Black, H.B.; Dugger, D.L.; Tripathy, S.K.; Goldwasser, E.; Hao, Z.; Chu, L.; Leiden, J.M. Long-term erythropoietin expression in rodents and non-human primates following intramuscular injection of a replication-defective adenoviral vector. Hum. Gene Ther. 1997, 8, 1797–1806. [Google Scholar] [CrossRef]
- Tripathy, S.K.; Goldwasser, E.; Lu, M.M.; Barr, E.; Leiden, J.M. Stable delivery of physiologic levels of recombinant erythropoietin to the systemic circulation by intramuscular injection of replication-defective adenovirus. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 11557–11561. [Google Scholar] [CrossRef]
- Tripathy, S.K.; Black, H.B.; Goldwasser, E.; Leiden, J.M. Immune responses to transgene-encoded proteins limit the stability of gene expression after injection of replication-defective adenovirus vectors. Nat. Med. 1996, 2, 545–550. [Google Scholar] [CrossRef]
- Kiang, A.; Hartman, Z.C.; Liao, S.; Xu, F.; Serra, D.; Palmer, D.J.; Ng, P.; Amalfitano, A. Fully deleted adenovirus persistently expressing GAA accomplishes long-term skeletal muscle glycogen correction in tolerant and nontolerant GSD-II mice. Mol. Ther. 2006, 13, 127–134. [Google Scholar] [CrossRef]
- Rivera, V.M.; Gao, G.P.; Grant, R.L.; Schnell, M.A.; Zoltick, P.W.; Rozamus, L.W.; Clackson, T.; Wilson, J.M. Long-term pharmacologically regulated expression of erythropoietin in primates following AAV-mediated gene transfer. Blood 2005, 105, 1424–1430. [Google Scholar] [CrossRef]
- Sarukhan, A.; Camugli, S.; Gjata, B.; von Boehmer, H.; Danos, O.; Jooss, K. Successful interference with cellular immune responses to immunogenic proteins encoded by recombinant viral vectors. J. Virol. 2001, 75, 269–277. [Google Scholar] [CrossRef]
- Yang, Y.; Nunes, F.A.; Berencsi, K.; Furth, E.E.; Gonczol, E.; Wilson, J.M. Cellular immunity to viral antigens limits E1-deleted adenoviruses for gene therapy. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 4407–4411. [Google Scholar] [CrossRef]
- Abbink, P.; Lemckert, A.A.; Ewald, B.A.; Lynch, D.M.; Denholtz, M.; Smits, S.; Holterman, L.; Damen, I.; Vogels, R.; Thorner, A.R.; O’Brien, K.L.; Carville, A.; Mansfield, K.G.; Goudsmit, J.; Havenga, M.J.; Barouch, D.H. Comparative seroprevalence and immunogenicity of six rare serotype recombinant adenovirus vaccine vectors from subgroups B and D. J. Virol. 2007, 81, 4654–4663. [Google Scholar] [CrossRef]
- Sumida, S.M.; Truitt, D.M.; Kishko, M.G.; Arthur, J.C.; Jackson, S.S.; Gorgone, D.A.; Lifton, M.A.; Koudstaal, W.; Pau, M.G.; Kostense, S.; Havenga, M.J.; Goudsmit, J.; Letvin, N.L.; Barouch, D.H. Neutralizing antibodies and CD8+ T lymphocytes both contribute to immunity to adenovirus serotype 5 vaccine vectors. J. Virol. 2004, 78, 2666–2673. [Google Scholar] [CrossRef]
- Tang, J.; Olive, M.; Pulmanausahakul, R.; Schnell, M.; Flomenberg, N.; Eisenlohr, L.; Flomenberg, P. Human CD8+ cytotoxic T cell responses to adenovirus capsid proteins. Virology 2006, 350, 312–322. [Google Scholar] [CrossRef]
- Mingozzi, F.; High, K.A. Immune responses to AAV in clinical trials. Curr. Gene Ther. 2007, 7, 316–324. [Google Scholar] [CrossRef]
- Zaiss, A.K.; Muruve, D.A. Immune responses to adeno-associated virus vectors. Curr. Gene Ther. 2005, 5, 323–331. [Google Scholar] [CrossRef]
- Barry, M.E.; Pinto-Gonzalez, D.; Orson, F.M.; McKenzie, G.J.; Petry, G.R.; Barry, M.A. Role of endogenous endonucleases and tissue site in transfection and CpG-mediated immune activation after naked DNA injection. Hum. Gene Ther. 1999, 10, 2461–2480. [Google Scholar] [CrossRef] [PubMed]
- Hensley, S.E.; Amalfitano, A. Toll-like receptors impact on safety and efficacy of gene transfer vectors. Mol. Ther. 2007, 15, 1417–1422. [Google Scholar] [CrossRef] [PubMed]
- Follenzi, A.; Santambrogio, L.; Annoni, A. Immune responses to lentiviral vectors. Curr. Gene Ther. 2007, 7, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, S.; Sarkari, B. Comparative efficacy of dexamethasone versus hydrocortisone in severe acute pediatric asthma. Iran. J. Allergy Asthma Immunol. 2007, 6, 159–160. [Google Scholar] [PubMed]
- Otake, K.; Ennist, D.L.; Harrod, K.; Trapnell, B.C. Nonspecific inflammation inhibits adenovirus-mediated pulmonary gene transfer and expression independent of specific acquired immune responses. Hum. Gene Ther. 1998, 9, 2207–2222. [Google Scholar] [CrossRef]
- Bohn, M.C.; Choi-Lundberg, D.L.; Davidson, B.L.; Leranth, C.; Kozlowski, D.A.; Smith, J.C.; O’Banion, M.K.; Redmond, D.E., Jr. Adenovirus-mediated transgene expression in nonhuman primate brain. Hum. Gene Ther. 1999, 10, 1175–1184. [Google Scholar] [CrossRef]
- National Gene Vector Laboratories: Toxicology database. http://www.ngvl.org/include/tox (accessed on 2 September 2010).
- Worgall, S.; Wolff, G.; Falck-Pedersen, E.; Crystal, R.G. Innate immune mechanisms dominate elimination of adenoviral vectors following in vivo administration. Hum. Gene Ther. 1997, 8, 37–44. [Google Scholar] [CrossRef]
- Worgall, S.; Leopold, P.L.; Wolff, G.; Ferris, B.; Van Roijen, N.; Crystal, R.G. Role of alveolar macrophages in rapid elimination of adenovirus vectors administered to the epithelial surface of the respiratory tract. Hum. Gene Ther. 1997, 8, 1675–1684. [Google Scholar] [CrossRef]
- Kuzmin, A.I.; Finegold, M.J.; Eisensmith, R.C. Macrophage depletion increases the safety, efficacy and persistence of adenovirus-mediated gene transfer in vivo. Gene Ther. 1997, 4, 309–316. [Google Scholar] [CrossRef]
- Appledorn, D.M.; Patial, S.; McBride, A.; Godbehere, S.; Van Rooijen, N.; Parameswaran, N.; Amalfitano, A. Adenovirus vector-induced innate inflammatory mediators, MAPK signaling, as well as adaptive immune responses are dependent upon both TLR2 and TLR9 in vivo. J. Immunol. 2008, 181, 2134–2144. [Google Scholar] [CrossRef]
- van Rooijen, N.; van Kesteren-Hendrikx, E. Clodronate liposomes: perspectives in research and therapeutics. J. Liposome Res. 2002, 12, 81–94. [Google Scholar] [CrossRef]
- Cerullo, V.; Seiler, M.P.; Mane, V.; Brunetti-Pierri, N.; Clarke, C.; Bertin, T.K.; Rodgers, J.R.; Lee, B. Toll-like receptor 9 triggers an innate immune response to helper-dependent adenoviral vectors. Mol. Ther. 2007, 15, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Wilderman, M.J.; Kim, S.; Gillespie, C.T.; Sun, J.; Kapoor, V.; Vachani, A.; Sterman, D.H.; Kaiser, L.R.; Albelda, S.M. Blockade of TNF-alpha decreases both inflammation and efficacy of intrapulmonary Ad.IFNbeta immunotherapy in an orthotopic model of bronchogenic lung cancer. Mol. Ther. 2006, 13, 910–917. [Google Scholar] [CrossRef]
- Benihoud, K.; Esselin, S.; Descamps, D.; Jullienne, B.; Salone, B.; Bobe, P.; Bonardelle, D.; Connault, E.; Opolon, P.; Saggio, I.; Perricaudet, M. Respective roles of TNF-alpha and IL-6 in the immune response-elicited by adenovirus-mediated gene transfer in mice. Gene Ther. 2007, 14, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Tibbles, L.A.; Spurrell, J.C.; Bowen, G.P.; Liu, Q.; Lam, M.; Zaiss, A.K.; Robbins, S.M.; Hollenberg, M.D.; Wickham, T.J.; Muruve, D.A. Activation of p38 and ERK signaling during adenovirus vector cell entry lead to expression of the C-X-C chemokine IP-10. J. Virol. 2002, 76, 1559–1568. [Google Scholar] [CrossRef]
- Schumann, M.; Dobbelstein, M. Adenovirus-induced extracellular signal-regulated kinase phosphorylation during the late phase of infection enhances viral protein levels and virus progeny. Cancer Res. 2006, 66, 1282–1288. [Google Scholar] [CrossRef]
- Kay, M.A.; Meuse, L.; Gown, A.M.; Linsley, P.; Hollenbaugh, D.; Aruffo, A.; Ochs, H.D.; Wilson, C.B. Transient immunomodulation with anti-CD40 ligand antibody and CTLA4Ig enhances persistence and secondary adenovirus-mediated gene transfer into mouse liver. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 4686–4691. [Google Scholar] [CrossRef]
- Dai, Y.; Schwarz, E.M.; Gu, D.; Zhang, W.W.; Sarvetnick, N.; Verma, I.M. Cellular and humoral immune responses to adenoviral vectors containing factor IX gene: tolerization of factor IX and vector antigens allows for long-term expression. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 1401–1405. [Google Scholar] [CrossRef]
- Engelhardt, J.F.; Ye, X.; Doranz, B.; Wilson, J.M. Ablation of E2A in recombinant adenoviruses improves transgene persistence and decreases inflammatory response in mouse liver. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 6196–6200. [Google Scholar] [CrossRef]
- Kuriyama, S.; Tominaga, K.; Mitoro, A.; Tsujinoue, H.; Nakatani, T.; Yamazaki, M.; Nagao, S.; Toyokawa, Y.; Okamoto, S.; Fukui, H. Immunomodulation with FK506 around the time of intravenous re-administration of an adenoviral vector facilitates gene transfer into primed rat liver. Int. J. Cancer 2000, 85, 839–844. [Google Scholar] [CrossRef]
- Smith, T.A.; White, B.D.; Gardner, J.M.; Kaleko, M.; McClelland, A. Transient immunosuppression permits successful repetitive intravenous administration of an adenovirus vector. Gene Ther. 1996, 3, 496–502. [Google Scholar] [PubMed]
- Maguire, A.M.; Simonelli, F.; Pierce, E.A.; Pugh, E.N., Jr.; Mingozzi, F.; Bennicelli, J.; Banfi, S.; Marshall, K.A.; Testa, F.; Surace, E.M.; Rossi, S.; Lyubarsky, A.; Arruda, V.R.; Konkle, B.; Stone, E.; Sun, J.; Jacobs, J.; Dell’Osso, L.; Hertle, R.; Ma, J.X.; Redmond, T.M.; Zhu, X.; Hauck, B.; Zelenaia, O.; Shindler, K.S.; Maguire, M.G.; Wright, J.F.; Volpe, N.J.; McDonnell, J.W.; Auricchio, A.; High, K.A.; Bennett, J. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N. Engl. J. Med. 2008, 358, 2240–2248. [Google Scholar] [CrossRef] [PubMed]
- Hauswirth, W.W.; Aleman, T.S.; Kaushal, S.; Cideciyan, A.V.; Schwartz, S.B.; Wang, L.; Conlon, T.J.; Boye, S.L.; Flotte, T.R.; Byrne, B.J.; Jacobson, S.G. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum. Gene Ther. 2008, 19, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Hauswirth, W.W.; Aleman, T.S.; Kaushal, S.; Schwartz, S.B.; Boye, S.L.; Windsor, E.A.; Conlon, T.J.; Sumaroka, A.; Pang, J.J.; Roman, A.J.; Byrne, B.J.; Jacobson, S.G. Human RPE65 gene therapy for Leber congenital amaurosis: persistence of early visual improvements and safety at 1 year. Hum. Gene Ther. 2009, 20, 999–1004. [Google Scholar] [CrossRef]
- Weaver, T.A.; Charafeddine, A.H.; Kirk, A.D. Costimulation blockade: towards clinical application. Front. Biosci. 2008, 13, 2120–2139. [Google Scholar] [CrossRef]
- Ozkaynak, E.; Gao, W.; Shemmeri, N.; Wang, C.; Gutierrez-Ramos, J.C.; Amaral, J.; Qin, S.; Rottman, J.B.; Coyle, A.J.; Hancock, W.W. Importance of ICOS-B7RP-1 costimulation in acute and chronic allograft rejection. Nat. Immunol. 2001, 2, 591–596. [Google Scholar] [CrossRef]
- Wilson, C.B.; Embree, L.J.; Schowalter, D.; Albert, R.; Aruffo, A.; Hollenbaugh, D.; Linsley, P.; Kay, M.A. Transient inhibition of CD28 and CD40 ligand interactions prolongs adenovirus-mediated transgene expression in the lung and facilitates expression after secondary vector administration. J. Virol. 1998, 72, 7542–7550. [Google Scholar] [CrossRef]
- Scaria, A.; St George, J.A.; Gregory, R.J.; Noelle, R.J.; Wadsworth, S.C.; Smith, A.E.; Kaplan, J.M. Antibody to CD40 ligand inhibits both humoral and cellular immune responses to adenoviral vectors and facilitates repeated administration to mouse airway. Gene Ther. 1997, 4, 611–617. [Google Scholar] [CrossRef]
- Chirmule, N.; Raper, S.E.; Burkly, L.; Thomas, D.; Tazelaar, J.; Hughes, J.V.; Wilson, J.M. Readministration of adenovirus vector in nonhuman primate lungs by blockade of CD40-CD40 ligand interactions. J. Virol. 2000, 74, 3345–3352. [Google Scholar] [CrossRef]
- Sawchuk, S.J.; Boivin, G.P.; Duwel, L.E.; Ball, W.; Bove, K.; Trapnell, B.; Hirsch, R. Anti-T cell receptor monoclonal antibody prolongs transgene expression following adenovirus-mediated in vivo gene transfer to mouse synovium. Hum. Gene Ther. 1996, 7, 499–506. [Google Scholar] [CrossRef]
- Guerette, B.; Vilquin, J.T.; Gingras, M.; Gravel, C.; Wood, K.J.; Tremblay, J.P. Prevention of immune reactions triggered by first-generation adenoviral vectors by monoclonal antibodies and CTLA4Ig. Hum. Gene Ther. 1996, 7, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Shean, M.K.; Baskin, G.; Sullivan, D.; Schurr, J.; Cavender, D.E.; Shellito, J.E.; Schwarzenberger, P.O.; Kolls, J.K. Immunomodulation and adenoviral gene transfer to the lungs of nonhuman primates. Hum. Gene Ther. 2000, 11, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Appledorn, D.M.; Aldhamen, Y.A.; Depas, W.; Seregin, S.S.; Liu, C.J.; Schuldt, N.; Quach, D.; Quiroga, D.; Godbehere, S.; Zlatkin, I.; Kim, S.; McCormick, J.J.; Amalfitano, A. A new adenovirus based vaccine vector expressing an Eimeria tenella derived TLR agonist improves cellular immune responses to an antigenic target. PLoS One 2010, 5, e9579. [Google Scholar] [CrossRef]
- Stampfli, M.R.; Wiley, R.E.; Neigh, G.S.; Gajewska, B.U.; Lei, X.F.; Snider, D.P.; Xing, Z.; Jordana, M. GM-CSF transgene expression in the airway allows aerosolized ovalbumin to induce allergic sensitization in mice. J. Clin. Invest. 1998, 102, 1704–1714. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Ju, D.W.; Sun, Y.; Tao, Q.; Qian, S.; Mi, J.; Hamada, H.; Cao, X. The potent antitumor effects of combined p16 gene and GM-CSF gene therapy through efficient induction of antitumor immunity. J. Cancer Res. Clin. Oncol. 2001, 127, 101–108. [Google Scholar] [CrossRef]
- Hull, G.W.; McCurdy, M.A.; Nasu, Y.; Bangma, C.H.; Yang, G.; Shimura, S.; Lee, H.M.; Wang, J.; Albani, J.; Ebara, S.; Sato, T.; Timme, T.L.; Thompson, T.C. Prostate cancer gene therapy: comparison of adenovirus-mediated expression of interleukin 12 with interleukin 12 plus B7-1 for in situ gene therapy and gene-modified, cell-based vaccines. Clin. Cancer Res. 2000, 6, 4101–4109. [Google Scholar]
- Hirschowitz, E.A.; Crystal, R.G. Adenovirus-mediated expression of interleukin-12 induces natural killer cell activity and complements adenovirus-directed gp75 treatment of melanoma lung metastases. Am. J. Respir. Cell Mol. Biol. 1999, 20, 935–941. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, J.H.; Choi, K.J.; Choi, I.K.; Kim, H.; Cho, S.; Cho, B.C.; Yun, C.O. Enhanced antitumor effect of oncolytic adenovirus expressing interleukin-12 and B7-1 in an immunocompetent murine model. Clin. Cancer Res. 2006, 12, 5859–5868. [Google Scholar] [CrossRef]
- Loskog, A.; Dzojic, H.; Vikman, S.; Ninalga, C.; Essand, M.; Korsgren, O.; Totterman, T.H. Adenovirus CD40 ligand gene therapy counteracts immune escape mechanisms in the tumor Microenvironment. J. Immunol. 2004, 172, 7200–7205. [Google Scholar] [CrossRef]
- Verwaerde, C.; Naud, M.C.; Delanoye, A.; Wood, M.; Thillaye-Goldenberg, B.; Auriault, C.; de Kozak, Y. Ocular transfer of retinal glial cells transduced ex vivo with adenovirus expressing viral IL-10 or CTLA4-Ig inhibits experimental autoimmune uveoretinitis. Gene Ther. 2003, 10, 1970–1981. [Google Scholar] [CrossRef]
- Schowalter, D.B.; Meuse, L.; Wilson, C.B.; Linsley, P.S.; Kay, M.A. Constitutive expression of murine CTLA4Ig from a recombinant adenovirus vector results in prolonged transgene expression. Gene Ther. 1997, 4, 853–860. [Google Scholar] [CrossRef]
- Liu, W.; Gao, C.; Zhou, B.G.; Li, W.M. Effects of adenovirus-mediated gene transfer of ICOSIg and CTLA4Ig fusion protein on experimental autoimmune myocarditis. Autoimmunity 2006, 39, 83–92. [Google Scholar] [CrossRef]
- Fernandes, J.R.; Duvivier-Kali, V.F.; Keegan, M.; Hollister-Lock, J.; Omer, A.; Su, S.; Bonner-Weir, S.; Feng, S.; Lee, J.S.; Mulligan, R.C.; Weir, G.C. Transplantation of islets transduced with CTLA4-Ig and TGFbeta using adenovirus and lentivirus vectors. Transpl. Immunol. 2004, 13, 191–200. [Google Scholar] [CrossRef]
- Londrigan, S.L.; Sutherland, R.M.; Brady, J.L.; Zhan, Y.; Li, R.; Estella, E.; Kay, T.W.; Lew, A.M. Prolonged local expression of anti-CD4 antibody by adenovirally transduced allografts can promote long-term graft survival. J. Gene Med. 2006, 8, 42–52. [Google Scholar] [CrossRef]
- Jiang, Z.; Feingold, E.; Kochanek, S.; Clemens, P.R. Systemic delivery of a high-capacity adenoviral vector expressing mouse CTLA4Ig improves skeletal muscle gene therapy. Mol. Ther. 2002, 6, 369–376. [Google Scholar] [CrossRef]
- Wold, W.S.; Doronin, K.; Toth, K.; Kuppuswamy, M.; Lichtenstein, D.L.; Tollefson, A.E. Immune responses to adenoviruses: viral evasion mechanisms and their implications for the clinic. Curr. Opin. Immunol. 1999, 11, 380–386. [Google Scholar] [CrossRef]
- Hayder, H.; Mullbacher, A. Molecular basis of immune evasion strategies by adenoviruses. Immunol. Cell Biol. 1996, 74, 504–512. [Google Scholar] [CrossRef]
- Bruder, J.T.; Jie, T.; McVey, D.L.; Kovesdi, I. Expression of gp19K increases the persistence of transgene expression from an adenovirus vector in the mouse lung and liver. J. Virol. 1997, 71, 7623–7628. [Google Scholar] [CrossRef]
- Ilan, Y.; Droguett, G.; Chowdhury, N.R.; Li, Y.; Sengupta, K.; Thummala, N.R.; Davidson, A.; Chowdhury, J.R.; Horwitz, M.S. Insertion of the adenoviral E3 region into a recombinant viral vector prevents antiviral humoral and cellular immune responses and permits long-term gene expression. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 2587–2592. [Google Scholar] [CrossRef]
- Ribacka, C.; Pesonen, S.; Hemminki, A. Cancer, stem cells, and oncolytic viruses. Ann. Med. 2008, 40, 496–505. [Google Scholar] [CrossRef]
- Joshi, A.; Tang, J.; Kuzma, M.; Wagner, J.; Mookerjee, B.; Filicko, J.; Carabasi, M.; Flomenberg, N.; Flomenberg, P. Adenovirus DNA polymerase is recognized by human CD8+ T cells. J. Gen. Virol. 2009, 90, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Gabitzsch, E.S.; Xu, Y.; Yoshida, L.H.; Balint, J.; Gayle, R.B.; Amalfitano, A.; Jones, F.R. A preliminary and comparative evaluation of a novel Ad5 [E1-, E2b-] recombinant-based vaccine used to induce cell mediated immune responses. Immunol. Lett. 2009, 122, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Gall, J.; Kass-Eisler, A.; Leinwand, L.; Falck-Pedersen, E. Adenovirus type 5 and 7 capsid chimera: fiber replacement alters receptor tropism without affecting primary immune neutralization epitopes. J. Virol. 1996, 70, 2116–2123. [Google Scholar] [CrossRef] [PubMed]
- Gall, J.G.; Crystal, R.G.; Falck-Pedersen, E. Construction and characterization of hexon-chimeric adenoviruses: specification of adenovirus serotype. J. Virol. 1998, 72, 10260–10264. [Google Scholar] [CrossRef]
- Molinier-Frenkel, V.; Lengagne, R.; Gaden, F.; Hong, S.S.; Choppin, J.; Gahery-Segard, H.; Boulanger, P.; Guillet, J.G. Adenovirus hexon protein is a potent adjuvant for activation of a cellular immune response. J. Virol. 2002, 76, 127–135. [Google Scholar] [CrossRef]
- Roy, S.; Shirley, P.S.; McClelland, A.; Kaleko, M. Circumvention of immunity to the adenovirus major coat protein hexon. J. Virol. 1998, 72, 6875–6879. [Google Scholar] [CrossRef]
- Wu, H.; Dmitriev, I.; Kashentseva, E.; Seki, T.; Wang, M.; Curiel, D.T. Construction and characterization of adenovirus serotype 5 packaged by serotype 3 hexon. J. Virol. 2002, 76, 12775–12782. [Google Scholar] [CrossRef]
- Everett, R.S.; Hodges, B.L.; Ding, E.Y.; Xu, F.; Serra, D.; Amalfitano, A. Liver toxicities typically induced by first-generation adenoviral vectors can be reduced by use of E1, E2b-deleted adenoviral vectors. Hum. Gene Ther. 2003, 14, 1715–1726. [Google Scholar] [CrossRef]
- Alba, R.; Bosch, A.; Chillon, M. Gutless adenovirus: last-generation adenovirus for gene therapy. Gene Ther. 2005, 12 (Suppl. 1), S18–27. [Google Scholar] [CrossRef]
- Appledorn, D.M.; Seregin, S.; Amalfitano, A. Adenovirus vectors for renal-targeted gene delivery. Contrib. Nephrol. 2008, 159, 47–62. [Google Scholar]
- O’Riordan, C.R.; Lachapelle, A.; Delgado, C.; Parkes, V.; Wadsworth, S.C.; Smith, A.E.; Francis, G.E. PEGylation of adenovirus with retention of infectivity and protection from neutralizing antibody in vitro and in vivo. Hum. Gene Ther. 1999, 10, 1349–1358. [Google Scholar] [CrossRef]
- Matthews, C.; Jenkins, G.; Hilfinger, J.; Davidson, B. Poly-L-lysine improves gene transfer with adenovirus formulated in PLGA microspheres. Gene Ther. 1999, 6, 1558–1564. [Google Scholar] [CrossRef]
- Lee, S.G.; Yoon, S.J.; Kim, C.D.; Kim, K.; Lim, D.S.; Yeom, Y.I.; Sung, M.W.; Heo, D.S.; Kim, N.K. Enhancement of adenoviral transduction with polycationic liposomes in vivo. Cancer Gene Ther. 2000, 7, 1329–1335. [Google Scholar] [CrossRef]
- Wonganan, P.; Croyle, M. PEGylated Adenoviruses: From mice to Monkeys. Viruses 2010, 2, 468–502. [Google Scholar] [CrossRef]
- Croyle, M.A.; Chirmule, N.; Zhang, Y.; Wilson, J.M. PEGylation of E1-deleted adenovirus vectors allows significant gene expression on readministration to liver. Hum. Gene Ther. 2002, 13, 1887–1900. [Google Scholar] [CrossRef]
- Croyle, M.A.; Le, H.T.; Linse, K.D.; Cerullo, V.; Toietta, G.; Beaudet, A.; Pastore, L. PEGylated helper-dependent adenoviral vectors: highly efficient vectors with an enhanced safety profile. Gene Ther. 2005, 12, 579–587. [Google Scholar] [CrossRef]
- De Geest, B.; Snoeys, J.; Van Linthout, S.; Lievens, J.; Collen, D. Elimination of innate immune responses and liver inflammation by PEGylation of adenoviral vectors and methylprednisolone. Hum. Gene Ther. 2005, 16, 1439–1451. [Google Scholar] [CrossRef]
- Mok, H.; Palmer, D.J.; Ng, P.; Barry, M.A. Evaluation of polyethylene glycol modification of first-generation and helper-dependent adenoviral vectors to reduce innate immune responses. Mol. Ther. 2005, 11, 66–79. [Google Scholar] [CrossRef]
- Ogawara, K.; Rots, M.G.; Kok, R.J.; Moorlag, H.E.; Van Loenen, A.M.; Meijer, D.K.; Haisma, H.J.; Molema, G. A novel strategy to modify adenovirus tropism and enhance transgene delivery to activated vascular endothelial cells in vitro and in vivo. Hum. Gene Ther. 2004, 15, 433–443. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Stapleton, G.E.; Palmer, D.J.; Zuo, Y.; Mane, V.P.; Finegold, M.J.; Beaudet, A.L.; Leland, M.M.; Mullins, C.E.; Ng, P. Pseudo-hydrodynamic delivery of helper-dependent adenoviral vectors into non-human primates for liver-directed gene therapy. Mol. Ther. 2007, 15, 732–740. [Google Scholar] [CrossRef]
- Rizzo, M.; Berneis, K.; Corrado, E.; Novo, S. The significance of low-density-lipoproteins size in vascular diseases. Int. Angiol. 2006, 25, 4–9. [Google Scholar] [PubMed]
- Bays, H.E. "Sick fat," metabolic disease, and atherosclerosis. Am. J. Med. 2009, 122, S26–37. [Google Scholar] [CrossRef] [PubMed]
- De Geest, B.R.; Van Linthout, S.A.; Collen, D. Humoral immune response in mice against a circulating antigen induced by adenoviral transfer is strictly dependent on expression in antigen-presenting cells. Blood 2003, 101, 2551–2556. [Google Scholar] [CrossRef] [PubMed]
- Schiedner, G.; Morral, N.; Parks, R.J.; Wu, Y.; Koopmans, S.C.; Langston, C.; Graham, F.L.; Beaudet, A.L.; Kochanek, S. Genomic DNA transfer with a high-capacity adenovirus vector results in improved in vivo gene expression and decreased toxicity. Nat. Genet. 1998, 18, 180–183. [Google Scholar] [CrossRef]
- Balague, C.; Zhou, J.; Dai, Y.; Alemany, R.; Josephs, S.F.; Andreason, G.; Hariharan, M.; Sethi, E.; Prokopenko, E.; Jan, H.Y.; Lou, Y.C.; Hubert-Leslie, D.; Ruiz, L.; Zhang, W.W. Sustained high-level expression of full-length human factor VIII and restoration of clotting activity in hemophilic mice using a minimal adenovirus vector. Blood 2000, 95, 820–828. [Google Scholar] [CrossRef]
- Waddington, S.N.; McVey, J.H.; Bhella, D.; Parker, A.L.; Barker, K.; Atoda, H.; Pink, R.; Buckley, S.M.; Greig, J.A.; Denby, L.; Custers, J.; Morita, T.; Francischetti, I.M.; Monteiro, R.Q.; Barouch, D.H.; van Rooijen, N.; Napoli, C.; Havenga, M.J.; Nicklin, S.A.; Baker, A.H. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 2008, 132, 397–409. [Google Scholar] [CrossRef]
- Coico, R.; Sunshine, G. Immunology: a short course, 6th ed.; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2009. [Google Scholar]
- Coughlan, L.; Vallath, S.; Saha, A.; Flak, M.; McNeish, I.A.; Vassaux, G.; Marshall, J.F.; Hart, I.R.; Thomas, G.J. In vivo retargeting of adenovirus type 5 to alphavbeta6 integrin results in reduced hepatotoxicity and improved tumor uptake following systemic delivery. J. Virol. 2009, 83, 6416–6428. [Google Scholar] [CrossRef]
- Rein, D.T.; Breidenbach, M.; Curiel, D.T. Current developments in adenovirus-based cancer gene therapy. Future Oncol. 2006, 2, 137–143. [Google Scholar] [CrossRef]
- Hawlisch, H.; Kohl, J. Complement and Toll-like receptors: key regulators of adaptive immune responses. Mol. Immunol. 2006, 43, 13–21. [Google Scholar] [CrossRef]
- Kemper, C.; Atkinson, J.P. T-cell regulation: with complements from innate immunity. Nat. Rev. Immunol. 2007, 7, 9–18. [Google Scholar] [CrossRef]
- Morgan, B.P.; Marchbank, K.J.; Longhi, M.P.; Harris, C.L.; Gallimore, A.M. Complement: central to innate immunity and bridging to adaptive responses. Immunol. Lett. 2005, 97, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Xu, Z.; Smith, J.S.; Hofherr, S.E.; Barry, M.A.; Byrnes, A.P. Adenovirus activates complement by distinctly different mechanisms in vitro and in vivo: indirect complement activation by virions in vivo. J. Virol. 2009, 83, 5648–5658. [Google Scholar] [CrossRef] [PubMed]
- Seregin, S.S.; Hartman, Z.C.; Appledorn, D.M.; Godbehere, S.; Jiang, H.; Frank, M.M.; Amalfitano, A. Novel Adenovirus vectors “capsid-displaying” a human complement inhibitor. J. Inn. Imm. 2010, 2, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Seregin, S.S.; Aldhamen, Y.A.; Appledorn, D.M.; Hartman, Z.C.; Schuldt, N.J.; Scott, J.; Godbehere, S.; Jiang, H.; Frank, M.M.; Amalfitano, A. Adenovirus capsid-display of the retro-oriented human complement inhibitor DAF reduces Ad-vector triggered immune responses in vitro and in vivo. Blood 2010, 116, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Curiel, D.T. Fiber-modified adenoviruses for targeted gene therapy. Methods Mol. Biol. 2008, 434, 113–132. [Google Scholar]
- Noureddini, S.C.; Curiel, D.T. Genetic targeting strategies for adenovirus. Mol. Pharm. 2005, 2, 341–347. [Google Scholar] [CrossRef]
- Youil, R.; Toner, T.J.; Su, Q.; Chen, M.; Tang, A.; Bett, A.J.; Casimiro, D. Hexon gene switch strategy for the generation of chimeric recombinant adenovirus. Hum. Gene Ther. 2002, 13, 311–320. [Google Scholar] [CrossRef]
- Roberts, D.M.; Nanda, A.; Havenga, M.J.; Abbink, P.; Lynch, D.M.; Ewald, B.A.; Liu, J.; Thorner, A.R.; Swanson, P.E.; Gorgone, D.A.; Lifton, M.A.; Lemckert, A.A.; Holterman, L.; Chen, B.; Dilraj, A.; Carville, A.; Mansfield, K.G.; Goudsmit, J.; Barouch, D.H. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature 2006, 441, 239–243. [Google Scholar] [CrossRef]
- Liu, J.; O’Brien, K.L.; Lynch, D.M.; Simmons, N.L.; La Porte, A.; Riggs, A.M.; Abbink, P.; Coffey, R.T.; Grandpre, L.E.; Seaman, M.S.; Landucci, G.; Forthal, D.N.; Montefiori, D.C.; Carville, A.; Mansfield, K.G.; Havenga, M.J.; Pau, M.G.; Goudsmit, J.; Barouch, D.H. Immune control of an SIV challenge by a T-cell-based vaccine in rhesus monkeys. Nature 2009, 457, 87–91. [Google Scholar] [CrossRef]
- McCoy, K.; Tatsis, N.; Korioth-Schmitz, B.; Lasaro, M.O.; Hensley, S.E.; Lin, S.W.; Li, Y.; Giles-Davis, W.; Cun, A.; Zhou, D.; Xiang, Z.; Letvin, N.L.; Ertl, H.C. Effect of preexisting immunity to adenovirus human serotype 5 antigens on the immune responses of nonhuman primates to vaccine regimens based on human- or chimpanzee-derived adenovirus vectors. J. Virol. 2007, 81, 6594–6604. [Google Scholar] [CrossRef]
- Barouch, D.H.; Pau, M.G.; Custers, J.H.; Koudstaal, W.; Kostense, S.; Havenga, M.J.; Truitt, D.M.; Sumida, S.M.; Kishko, M.G.; Arthur, J.C.; Korioth-Schmitz, B.; Newberg, M.H.; Gorgone, D.A.; Lifton, M.A.; Panicali, D.L.; Nabel, G.J.; Letvin, N.L.; Goudsmit, J. Immunogenicity of recombinant adenovirus serotype 35 vaccine in the presence of pre-existing anti-Ad5 immunity. J. Immunol. 2004, 172, 6290–6297. [Google Scholar] [CrossRef]
- Liu, J.; Ewald, B.A.; Lynch, D.M.; Denholtz, M.; Abbink, P.; Lemckert, A.A.; Carville, A.; Mansfield, K.G.; Havenga, M.J.; Goudsmit, J.; Barouch, D.H. Magnitude and phenotype of cellular immune responses elicited by recombinant adenovirus vectors and heterologous prime-boost regimens in rhesus monkeys. J. Virol. 2008, 82, 4844–4852. [Google Scholar] [CrossRef]
- Pinto, A.R.; Fitzgerald, J.C.; Giles-Davis, W.; Gao, G.P.; Wilson, J.M.; Ertl, H.C. Induction of CD8+ T cells to an HIV-1 antigen through a prime boost regimen with heterologous E1-deleted adenoviral vaccine carriers. J. Immunol. 2003, 171, 6774–6779. [Google Scholar] [CrossRef]
- Clinical Trials.gov: registry of federally and privately supported clinical trials conducted in the United States and around the world. Safety of and Immune Response to an Adenoviral (Ad26.ENVA.01) HIV-1 Vaccine in Healthy Adults. http://clinicaltrials.gov/ct2/show/NCT00618605 (accessed on 2 September 2010).
- Clinical Trials.gov: registry of federally and privately supported clinical trials conducted in the United States and around the world. Evaluating the Safety and Immune Response of an Adenovirus-Based (Ad26.ENVA.01) HIV Vaccine in HIV-Uninfected Adults. http://clinicaltrials.gov/ct2/show/NCT00618605 (accessed on 2 September 2010).
- Hartman, Z.C.; Appledorn, D.M.; Serra, D.; Glass, O.; Mendelson, T.B.; Clay, T.M.; Amalfitano, A. Replication-attenuated Human Adenoviral Type 4 vectors elicit capsid dependent enhanced innate immune responses that are partially dependent upon interactions with the complement system. Virology 2008, 374, 453–467. [Google Scholar] [CrossRef]
- Hensley, S.E.; Cun, A.S.; Giles-Davis, W.; Li, Y.; Xiang, Z.; Lasaro, M.O.; Williams, B.R.; Silverman, R.H.; Ertl, H.C. Type I interferon inhibits antibody responses induced by a chimpanzee adenovirus vector. Mol. Ther. 2007, 15, 393–403. [Google Scholar] [CrossRef]
- Appledorn, D.M.; Patial, S.; Godbehere, S.; Parameswaran, N.; Amalfitano, A. TRIF, and TRIF-Interacting TLRs Differentially Modulate Several Adenovirus Vector-Induced Immune Responses. J. Inn. Imm. 2009, 1, 376–388. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Black, E.P.; Amalfitano, A. Adenoviral infection induces a multi-faceted innate cellular immune response that is mediated by the toll-like receptor pathway in A549 cells. Virology 2007, 358, 357–372. [Google Scholar] [CrossRef]
- Schagen, F.H.; Ossevoort, M.; Toes, R.E.; Hoeben, R.C. Immune responses against adenoviral vectors and their transgene products: a review of strategies for evasion. Crit. Rev. Oncol. Hematol. 2004, 50, 51–70. [Google Scholar] [CrossRef]
© 2010 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Seregin, S.S.; Amalfitano, A. Improving Adenovirus Based Gene Transfer: Strategies to Accomplish Immune Evasion. Viruses 2010, 2, 2013-2036. https://doi.org/10.3390/v2092013
Seregin SS, Amalfitano A. Improving Adenovirus Based Gene Transfer: Strategies to Accomplish Immune Evasion. Viruses. 2010; 2(9):2013-2036. https://doi.org/10.3390/v2092013
Chicago/Turabian StyleSeregin, Sergey S., and Andrea Amalfitano. 2010. "Improving Adenovirus Based Gene Transfer: Strategies to Accomplish Immune Evasion" Viruses 2, no. 9: 2013-2036. https://doi.org/10.3390/v2092013
APA StyleSeregin, S. S., & Amalfitano, A. (2010). Improving Adenovirus Based Gene Transfer: Strategies to Accomplish Immune Evasion. Viruses, 2(9), 2013-2036. https://doi.org/10.3390/v2092013