Gene Therapy with Helper-Dependent Adenoviral Vectors: Current Advances and Future Perspectives

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
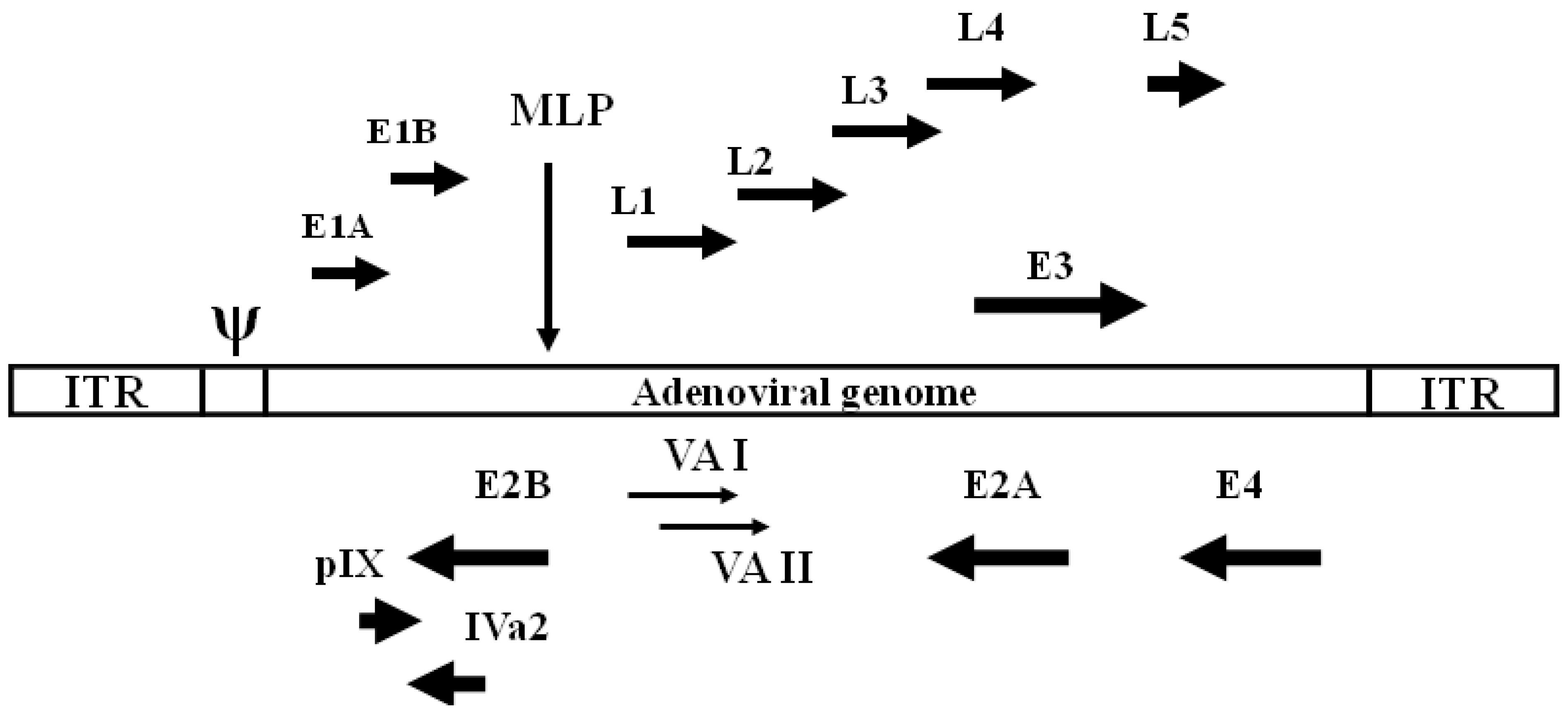
2. Adenoviruses
3. Early-generation Adenoviral vectors
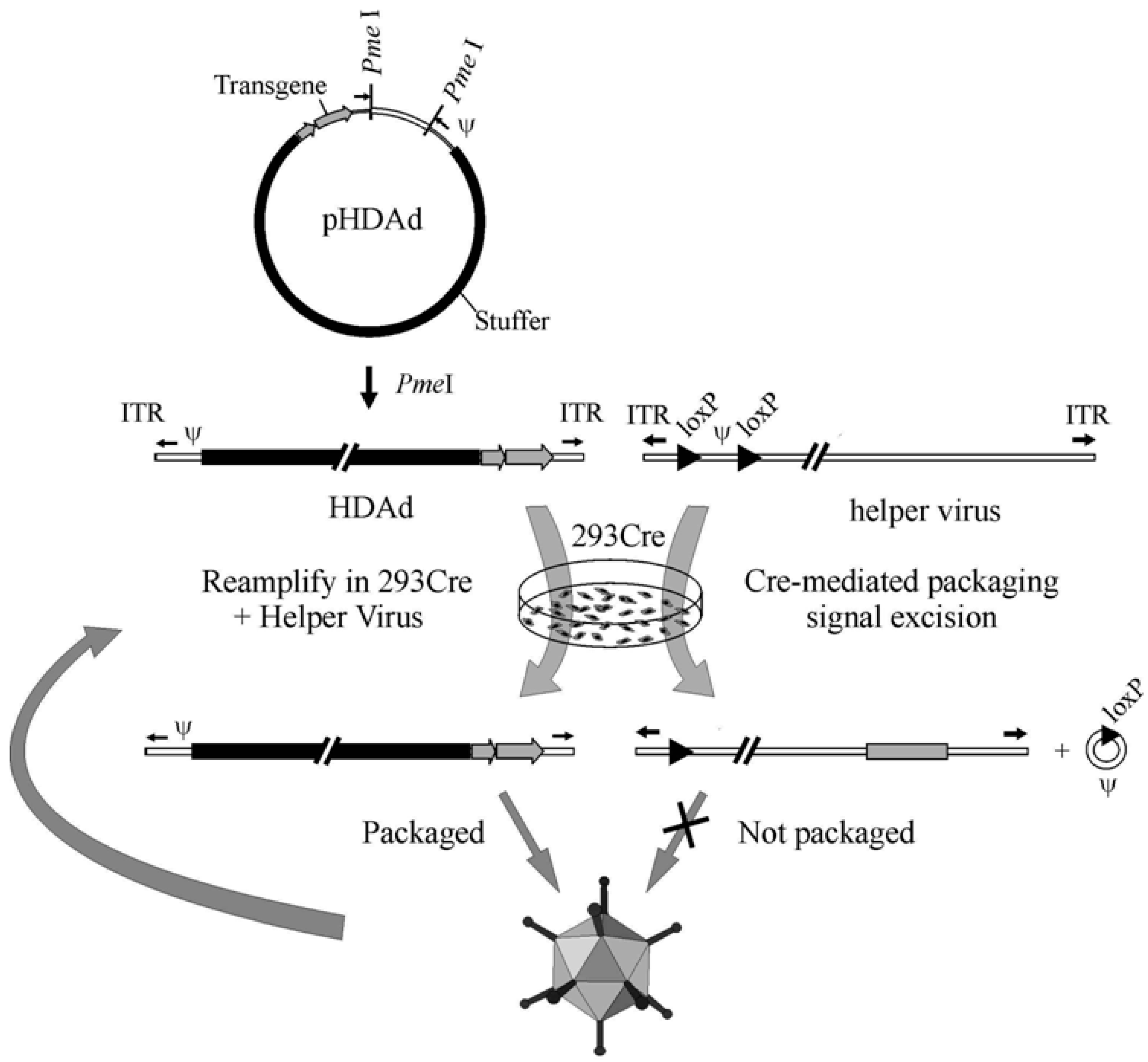
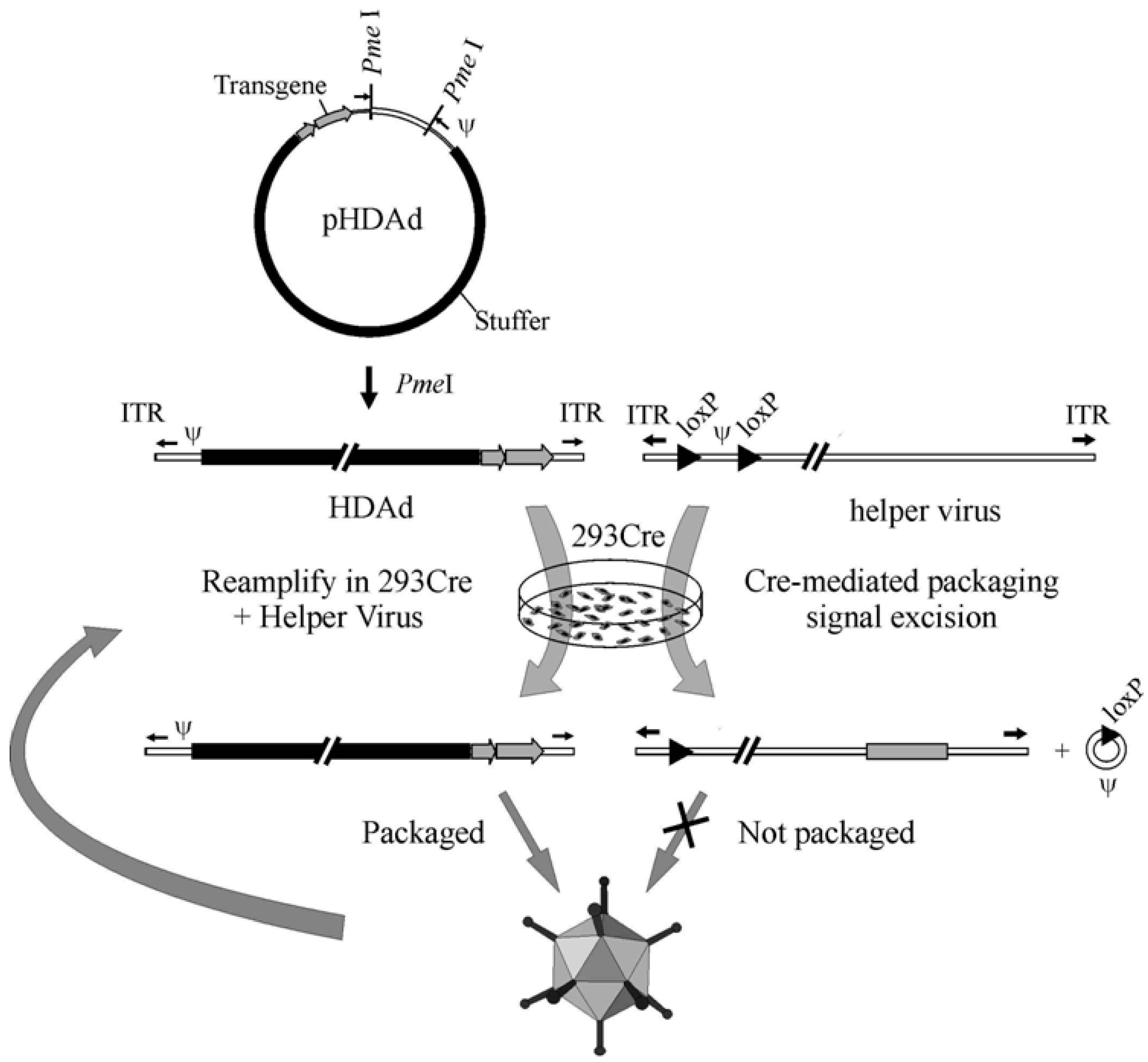
4. HDAd
5. HDAd in vivo studies
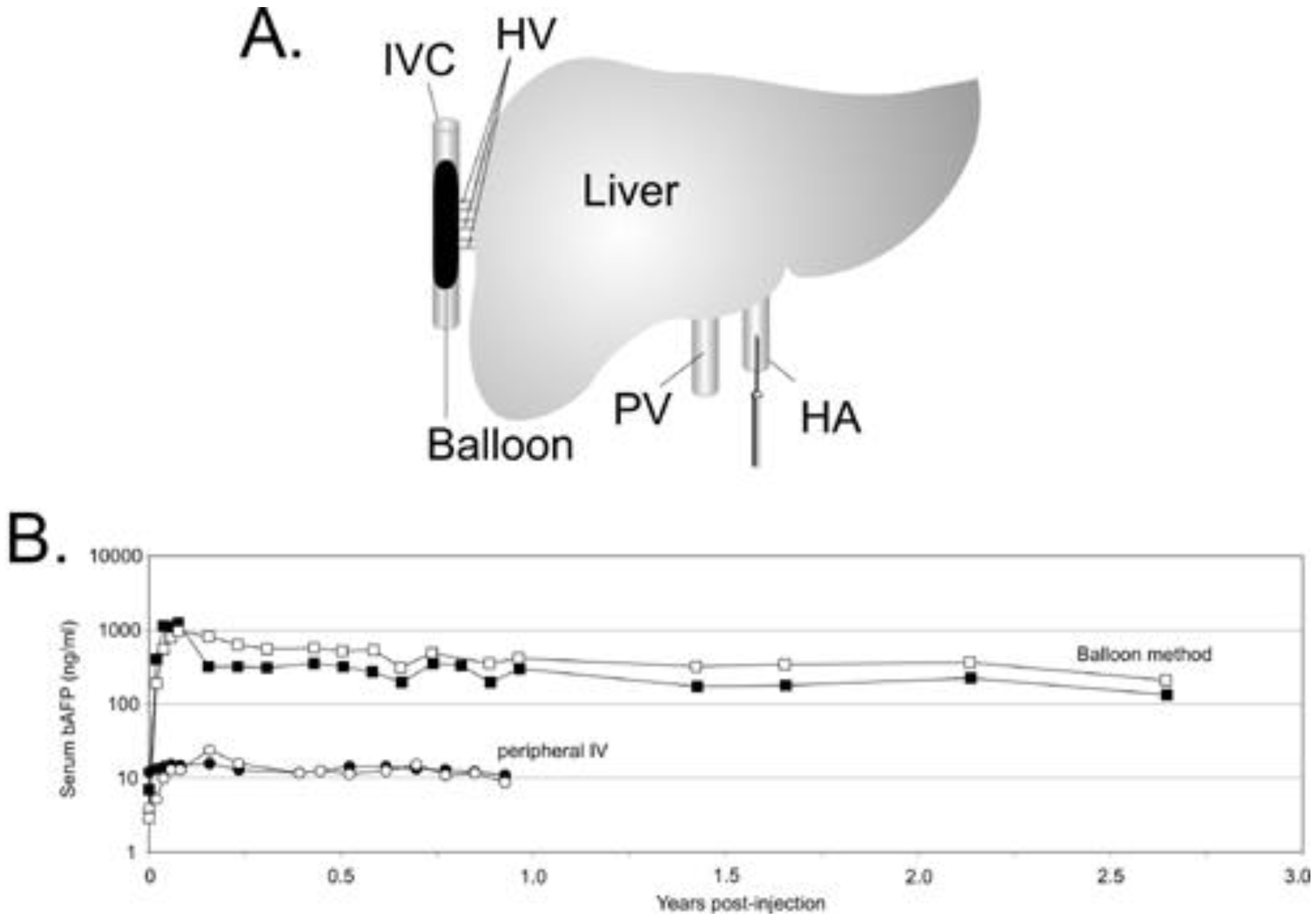
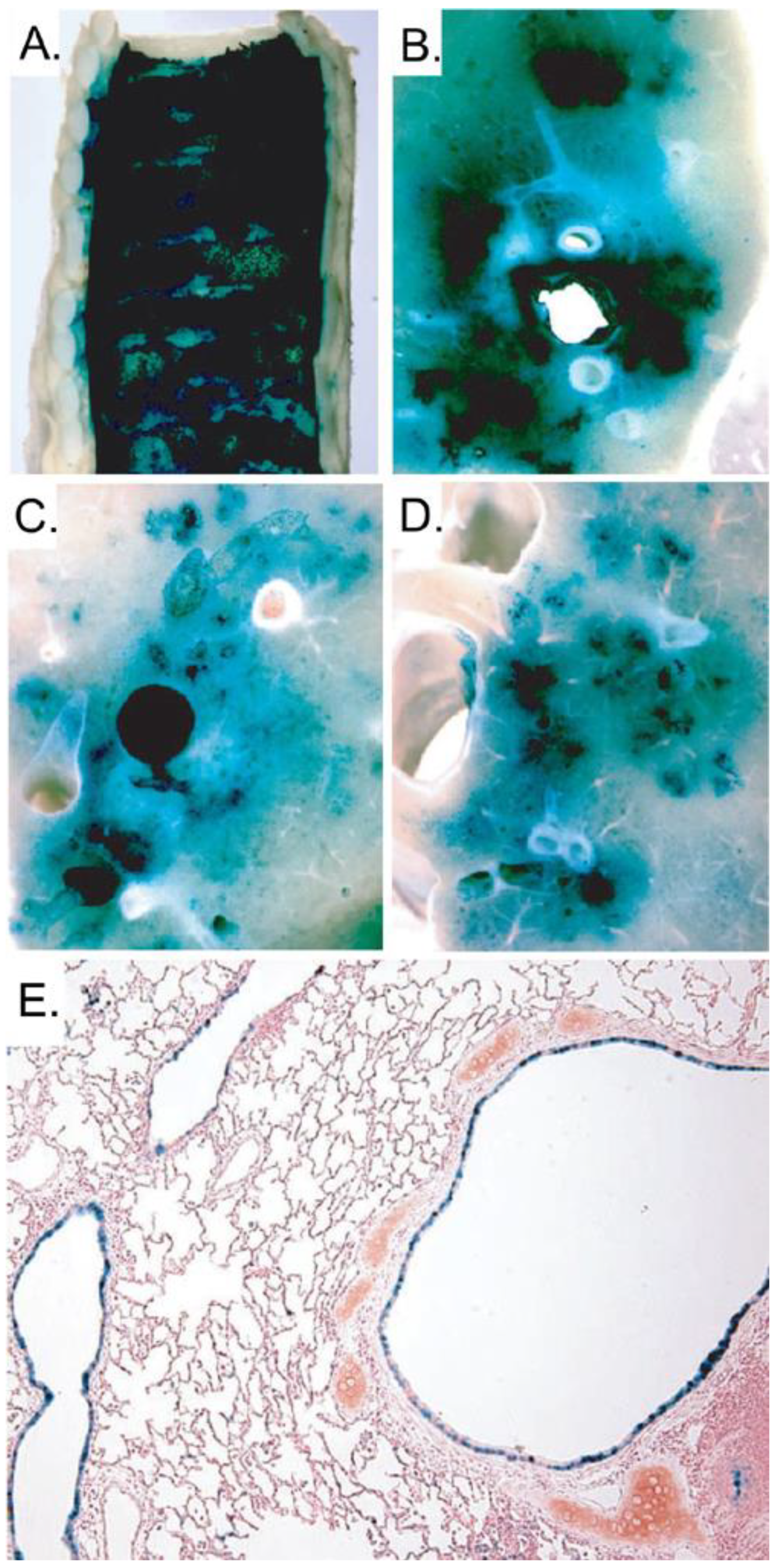
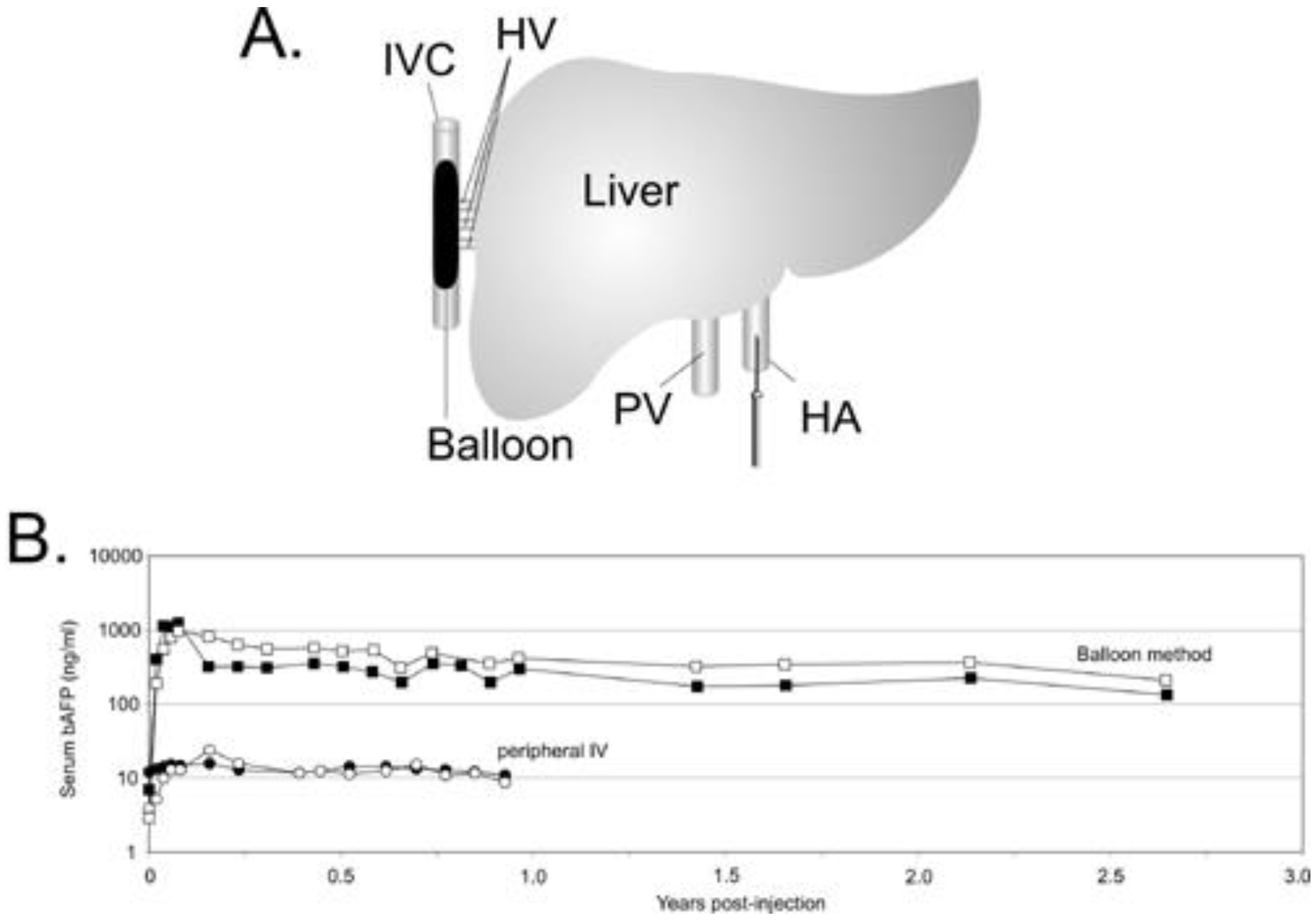
5.1. Liver directed gene therapy
5.1.1. Nonlinear dose-response to hepatocyte transduction
5.1.2. Immunobiology of HDAd and pathogenesis of the acute toxicity
5.1.3. Overcoming the threshold effect and the acute toxicity
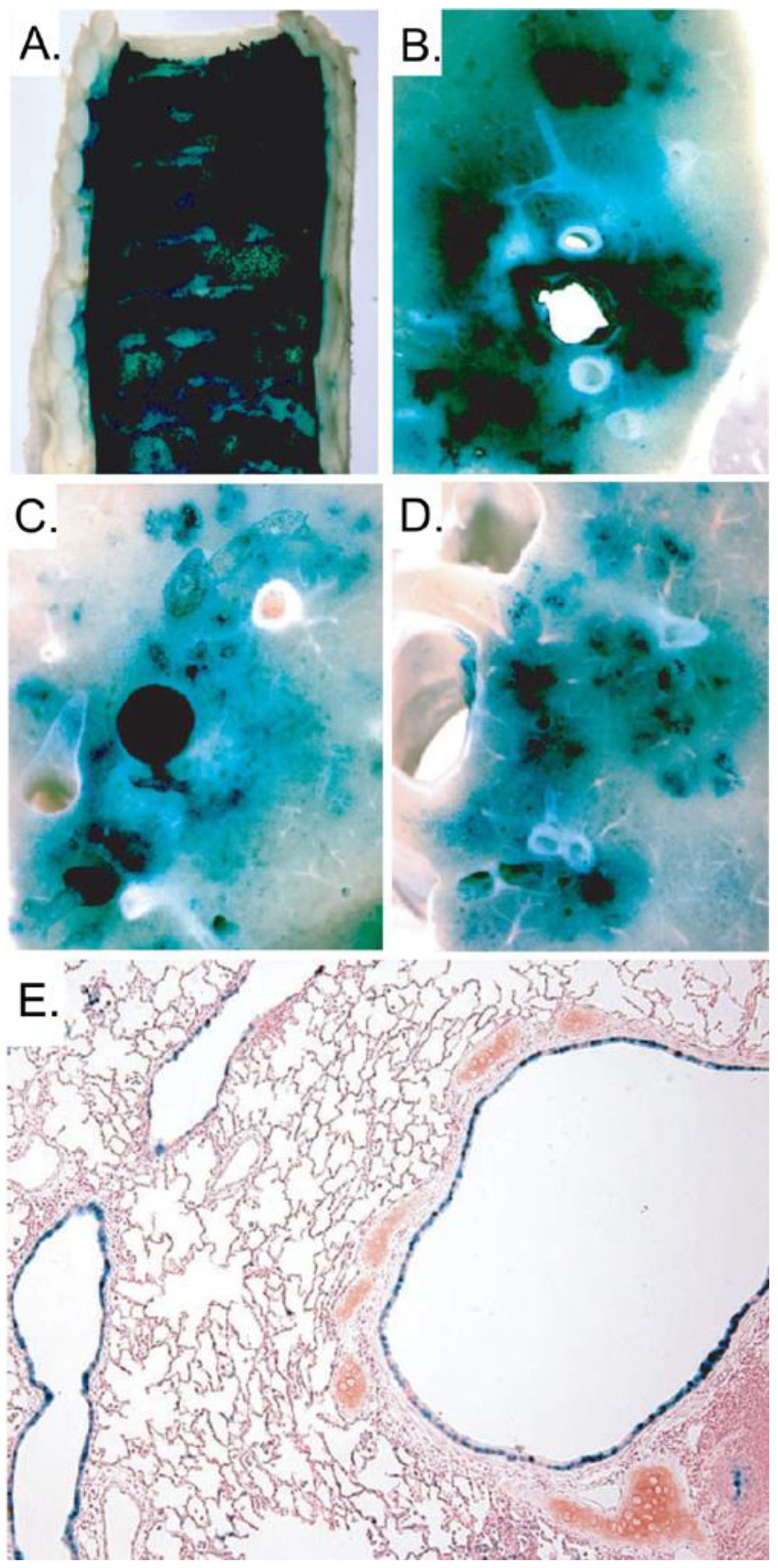
5.2. Gene therapy of Cystic Fibrosis lung disease
5.3. HDAd for muscle-directed gene therapy
5.4. Gene therapy for brain and eye
5.5. Ex vivo gene therapy in human patients
5.6. HDAd as genetic vaccines
6. Concluding remarks and future perspectives
References and Notes
- Paielli, D.L.; Wing, M.S.; Rogulski, K.R.; Gilbert, J.D.; Kolozsvary, A.; Kim, J.H.; Hughes, J.; Schnell, M.; Thompson, T.; Freytag, S.O. Evaluation of the biodistribution, persistence, toxicity, and potential of germ-line transmission of a replication-competent human adenovirus following intraprostatic administration in the mouse. Mol. Ther. 2000, 1, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Muruve, D.A.; Barnes, M.J.; Stillman, I.E.; Libermann, T.A. Adenoviral gene therapy leads to rapid induction of multiple chemokines and acute neutrophil-dependent hepatic injury in vivo. Hum. Gene Ther. 1999, 10, 965–976. [Google Scholar] [CrossRef]
- Muruve, D.A.; Cotter, M.J.; Zaiss, A.K.; White, L.R.; Liu, Q.; Chan, T.; Clark, S.A.; Ross, P.J.; Meulenbroek, R.A.; Maelandsmo, G.M.; Parks, R.J. Helper-dependent adenovirus vectors elicit intact innate but attenuated adaptive host immune responses in vivo. J. Virol. 2004, 78, 5966–5972. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Ng, P. Progress and prospects: gene therapy for genetic diseases with helper-dependent adenoviral vectors. Gene Ther. 2008, 15, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Toietta, G.; Koehler, D.R.; Finegold, M.J.; Lee, B.; Hu, J.; Beaudet, A.L. Reduced inflammation and improved airway expression using helper-dependent adenoviral vectors with a K18 promoter. Mol. Ther. 2003, 7, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Simonet, W.S.; Bucay, N.; Lauer, S.J.; Taylor, J.M. A far-downstream hepatocyte-specific control region directs expression of the linked human apolipoprotein E and C-I genes in transgenic mice. J. Biol. Chem. 1993, 268, 8221–8229. [Google Scholar] [CrossRef]
- Benihoud, K.; Yeh, P.; Perricaudet, M. Adenovirus vectors for gene delivery. Curr. Opin. Biotechnol. 1999, 10, 440–447. [Google Scholar] [CrossRef]
- Jozkowicz, A.; Dulak, J. Helper-dependent adenoviral vectors in experimental gene therapy. Acta Biochim. Pol. 2005, 52, 589–599. [Google Scholar] [CrossRef]
- Hong, S.S.; Karayan, L.; Tournier, J.; Curiel, D.T.; Boulanger, P.A. Adenovirus type 5 fiber knob binds to MHC class I alpha2 domain at the surface of human epithelial and B lymphoblastoid cells. EMBO J. 1997, 16, 2294–2306. [Google Scholar] [CrossRef]
- Tomko, R.P.; Xu, R.; Philipson, L. HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 3352–3356. [Google Scholar] [CrossRef]
- Tomko, R.P.; Johansson, C.B.; Totrov, M.; Abagyan, R.; Frisen, J.; Philipson, L. Expression of the adenovirus receptor and its interaction with the fiber knob. Exp. Cell Res. 2000, 255, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M. Receptors mediating adenovirus attachment and internalization. Biochem. Pharmacol. 1999, 57, 975–979. [Google Scholar] [CrossRef]
- Wickham, T.J.; Mathias, P.; Cheresh, D.A.; Nemerow, G.R. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 1993, 73, 309–319. [Google Scholar] [CrossRef]
- Wickham, T.J.; Carrion, M.E.; Kovesdi, I. Targeting of adenovirus penton base to new receptors through replacement of its RGD motif with other receptor-specific peptide motifs. Gene Ther. 1995, 2, 750–756. [Google Scholar]
- Wickham, T.J. Targeting adenovirus. Gene Ther. 2000, 7, 110–114. [Google Scholar] [CrossRef]
- Leopold, P.L.; Ferris, B.; Grinberg, I.; Worgall, S.; Hackett, N.R.; Crystal, R.G. Fluorescent virions: dynamic tracking of the pathway of adenoviral gene transfer vectors in living cells. Hum. Gene Ther. 1998, 9, 367–378. [Google Scholar] [CrossRef]
- Leopold, P.L.; Kreitzer, G.; Miyazawa, N.; Rempel, S.; Pfister, K.K.; Rodriguez-Boulan, E.; Crystal, R.G. Dynein- and microtubule-mediated translocation of adenovirus serotype 5 occurs after endosomal lysis. Hum. Gene Ther. 2000, 11, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F.; Willetts, M.; Webster, P.; Helenius, A. Stepwise dismantling of adenovirus 2 during entry into cells. Cell 1993, 75, 477–486. [Google Scholar] [CrossRef]
- Ruben, M.; Bacchetti, S.; Graham, F. Covalently closed circles of adenovirus 5 DNA. Nature 1983, 301, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Amalfitano, A.; Hauser, M.A.; Hu, H.; Serra, D.; Begy, C.R.; Chamberlain, J.S. Production and characterization of improved adenovirus vectors with the E1, E2b, and E3 genes deleted. J. Virol. 1998, 72, 926–933. [Google Scholar] [CrossRef]
- Imperiale, M.J.; Kao, H.T.; Feldman, L.T.; Nevins, J.R.; Strickland, S. Common control of the heat shock gene and early adenovirus genes: evidence for a cellular E1A-like activity. Mol. Cell Biol. 1984, 4, 867–874. [Google Scholar]
- Reddy, P.S.; Sakhuja, K.; Ganesh, S.; Yang, L.; Kayda, D.; Brann, T.; Pattison, S.; Golightly, D.; Idamakanti, N.; Pinkstaff, A.; Kaloss, M.; Barjot, C.; Chamberlain, J.S.; Kaleko, M.; Connelly, S. Sustained human factor VIII expression in hemophilia A mice following systemic delivery of a gutless adenoviral vector. Mol. Ther. 2002, 5, 63–73. [Google Scholar] [CrossRef]
- Yang, Y.; Nunes, F.A.; Berencsi, K.; Gonczol, E.; Engelhardt, J.F.; Wilson, J.M. Inactivation of E2a in recombinant adenoviruses improves the prospect for gene therapy in cystic fibrosis. Nat. Genet. 1994, 7, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Nunes, F.A.; Berencsi, K.; Furth, E.E.; Gonczol, E.; Wilson, J.M. Cellular immunity to viral antigens limits E1-deleted adenoviruses for gene therapy. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 4407–4411. [Google Scholar] [CrossRef]
- Engelhardt, J.F.; Ye, X.; Doranz, B.; Wilson, J.M. Ablation of E2A in recombinant adenoviruses improves transgene persistence and decreases inflammatory response in mouse liver. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 6196–6200. [Google Scholar] [CrossRef]
- Engelhardt, J.F.; Litzky, L.; Wilson, J.M. Prolonged transgene expression in cotton rat lung with recombinant adenoviruses defective in E2a. Hum. Gene Ther. 1994, 5, 1217–1229. [Google Scholar] [CrossRef]
- Fang, B.; Wang, H.; Gordon, G.; Bellinger, D.A.; Read, M.S.; Brinkhous, K.M.; Woo, S.L.; Eisensmith, R.C. Lack of persistence of E1- recombinant adenoviral vectors containing a temperature-sensitive E2A mutation in immunocompetent mice and hemophilia B dogs. Gene Ther. 1996, 3, 217–222. [Google Scholar]
- Lusky, M.; Christ, M.; Rittner, K.; Dieterle, A.; Dreyer, D.; Mourot, B.; Schultz, H.; Stoeckel, F.; Pavirani, A.; Mehtali, M. In vitro and in vivo biology of recombinant adenovirus vectors with E1, E1/E2A, or E1/E4 deleted. J. Virol. 1998, 72, 2022–2032. [Google Scholar] [CrossRef] [PubMed]
- O'Neal, W.K.; Zhou, H.; Morral, N.; guilar-Cordova, E.; Pestaner, J.; Langston, C.; Mull, B.; Wang, Y.; Beaudet, A.L.; Lee, B. Toxicological comparison of E2a-deleted and first-generation adenoviral vectors expressing alpha1-antitrypsin after systemic delivery. Hum. Gene Ther. 1998, 9, 1587–1598. [Google Scholar] [CrossRef] [PubMed]
- Parks, R.J.; Chen, L.; Anton, M.; Sankar, U.; Rudnicki, M.A.; Graham, F.L. A helper-dependent adenovirus vector system: removal of helper virus by Cre-mediated excision of the viral packaging signal. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 13565–13570. [Google Scholar] [CrossRef]
- Parks, R.J.; Graham, F.L. A helper-dependent system for adenovirus vector production helps define a lower limit for efficient DNA packaging. J. Virol. 1997, 71, 3293–3298. [Google Scholar] [CrossRef] [PubMed]
- Parks, R.J.; Bramson, J.L.; Wan, Y.; Addison, C.L.; Graham, F.L. Effects of stuffer DNA on transgene expression from helper-dependent adenovirus vectors. J. Virol. 1999, 73, 8027–8034. [Google Scholar] [CrossRef]
- Palmer, D.; Ng, P. Improved system for helper-dependent adenoviral vector production. Mol. Ther. 2003, 8, 846–852. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Palmer, D.J.; Beaudet, A.L.; Carey, K.D.; Finegold, M.; Ng, P. Acute toxicity after high-dose systemic injection of helper-dependent adenoviral vectors into nonhuman primates. Hum. Gene Ther. 2004, 15, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Nichols, T.C.; McCorquodale, S.; Merricks, E.; Palmer, D.J.; Beaudet, A.L.; Ng, P. Sustained phenotypic correction of canine hemophilia B after systemic administration of helper-dependent adenoviral vector. Hum. Gene Ther. 2005, 16, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Ng, T.; Iannitti, D.A.; Palmer, D.J.; Beaudet, A.L.; Finegold, M.J.; Carey, K.D.; Cioffi, W.G.; Ng, P. Improved hepatic transduction, reduced systemic vector dissemination, and long-term transgene expression by delivering helper-dependent adenoviral vectors into the surgically isolated liver of nonhuman primates. Hum. Gene Ther. 2006, 17, 391–404. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Stapleton, G.E.; Palmer, D.J.; Zuo, Y.; Mane, V.P.; Finegold, M.J.; Beaudet, A.L.; Leland, M.M.; Mullins, C.E.; Ng, P. Pseudo-hydrodynamic delivery of helper-dependent adenoviral vectors into non-human primates for liver-directed gene therapy. Mol. Ther. 2007, 15, 732–740. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Stapleton, G.E.; Law, M.; Breinholt, J.; Palmer, D.J.; Zuo, Y.; Grove, N.C.; Finegold, M.J.; Rice, K.; Beaudet, A.L.; Mullins, C.E.; Ng, P. Efficient, long-term hepatic gene transfer using clinically relevant HDAd doses by balloon occlusion catheter delivery in nonhuman primates. Mol. Ther. 2009, 17, 327–333. [Google Scholar] [CrossRef]
- McCormack, W.M., Jr.; Seiler, M.P.; Bertin, T.K.; Ubhayakar, K.; Palmer, D.J.; Ng, P.; Nichols, T.C.; Lee, B. Helper-dependent adenoviral gene therapy mediates long-term correction of the clotting defect in the canine hemophilia A model. J. Thromb. Haemost. 2006, 4, 1218–1225. [Google Scholar] [CrossRef]
- Ng, P.; Beauchamp, C.; Evelegh, C.; Parks, R.; Graham, F.L. Development of a FLP/frt system for generating helper-dependent adenoviral vectors. Mol. Ther. 2001, 3, 809–815. [Google Scholar] [CrossRef]
- Umana, P.; Gerdes, C.A.; Stone, D.; Davis, J.R.; Ward, D.; Castro, M.G.; Lowenstein, P.R. Efficient FLPe recombinase enables scalable production of helper-dependent adenoviral vectors with negligible helper-virus contamination. Nat. Biotechnol. 2001, 19, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, A.; Xu, H.; Kay, M.A. Episomal persistence of recombinant adenoviral vector genomes during the cell cycle in vivo. J. Virol. 2003, 77, 7689–7695. [Google Scholar] [CrossRef] [PubMed]
- Jager, L.; Ehrhardt, A. Persistence of high-capacity adenoviral vectors as replication-defective monomeric genomes in vitro and in murine liver. Hum. Gene Ther. 2009, 20, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.A.; Parks, R.J. Adenovirus virion stability and the viral genome: size matters. Mol. Ther. 2009, 17, 1664–1666. [Google Scholar] [CrossRef] [PubMed]
- Schiedner, G.; Hertel, S.; Johnston, M.; Biermann, V.; Dries, V.; Kochanek, S. Variables affecting in vivo performance of high-capacity adenovirus vectors. J. Virol. 2002, 76, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Soifer, H.; Higo, C.; Kazazian, H.H., Jr.; Moran, J.V.; Mitani, K.; Kasahara, N. Stable integration of transgenes delivered by a retrotransposon-adenovirus hybrid vector. Hum. Gene Ther. 2001, 12, 1417–1428. [Google Scholar] [CrossRef]
- Kim, I.H.; Jozkowicz, A.; Piedra, P.A.; Oka, K.; Chan, L. Lifetime correction of genetic deficiency in mice with a single injection of helper-dependent adenoviral vector. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 13282–13287. [Google Scholar] [CrossRef]
- Palinski, W.; Ord, V.A.; Plump, A.S.; Breslow, J.L.; Steinberg, D.; Witztum, J.L. ApoE-deficient mice are a model of lipoprotein oxidation in atherogenesis. Demonstration of oxidation-specific epitopes in lesions and high titers of autoantibodies to malondialdehyde-lysine in serum. Arterioscler. Thromb. 1994, 14, 605–616. [Google Scholar] [CrossRef]
- Strauss, K.A.; Robinson, D.L.; Vreman, H.J.; Puffenberger, E.G.; Hart, G.; Morton, D.H. Management of hyperbilirubinemia and prevention of kernicterus in 20 patients with Crigler-Najjar disease. Eur. J. Pediatr. 2006, 165, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Toietta, G.; Mane, V.P.; Norona, W.S.; Finegold, M.J.; Ng, P.; McDonagh, A.F.; Beaudet, A.L.; Lee, B. Lifelong elimination of hyperbilirubinemia in the Gunn rat with a single injection of helper-dependent adenoviral vector. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 3930–3935. [Google Scholar] [CrossRef]
- Kojima, H.; Fujimiya, M.; Matsumura, K.; Younan, P.; Imaeda, H.; Maeda, M.; Chan, L. NeuroD-betacellulin gene therapy induces islet neogenesis in the liver and reverses diabetes in mice. Nat. Med. 2003, 9, 596–603. [Google Scholar] [CrossRef]
- Witting, S.R.; Brown, M.; Saxena, R.; Nabinger, S.; Morral, N. Helper-dependent adenovirus-mediated short hairpin RNA expression in the liver activates the interferon response. J. Biol. Chem. 2008, 283, 2120–2128. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, R.; Witting, S.R.; Saxena, R.; Morral, N. Robust hepatic gene silencing for functional studies using helper-dependent adenovirus vectors. Hum. Gene Ther. 2008. [Google Scholar] [CrossRef]
- Grimm, D.; Streetz, K.L.; Jopling, C.L.; Storm, T.A.; Pandey, K.; Davis, C.R.; Marion, P.; Salazar, F.; Kay, M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006, 441, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.D.; Shi, C.X.; Powell, S.; Hurlbut, D.; Graham, F.L.; Lillicrap, D. Helper-dependent adenoviral vectors mediate therapeutic factor VIII expression for several months with minimal accompanying toxicity in a canine model of severe hemophilia A. Blood 2004, 103, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Morral, N.; O'Neal, W.; Rice, K.; Leland, M.; Kaplan, J.; Piedra, P.A.; Zhou, H.; Parks, R.J.; Velji, R.; guilar-Cordova, E.; Wadsworth, S.; Graham, F.L.; Kochanek, S.; Carey, K.D.; Beaudet, A.L. Administration of helper-dependent adenoviral vectors and sequential delivery of different vector serotype for long-term liver-directed gene transfer in baboons. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 12816–12821. [Google Scholar] [CrossRef]
- Di Paolo, N.C.; Van, R.N.; Shayakhmetov, D.M. Redundant and synergistic mechanisms control the sequestration of blood-born adenovirus in the liver. Mol. Ther. 2009, 17, 675–684. [Google Scholar] [CrossRef]
- Xu, Z.; Tian, J.; Smith, J.S.; Byrnes, A.P. Clearance of adenovirus by Kupffer cells is mediated by scavenger receptors, natural antibodies, and complement. J. Virol. 2008, 82, 11705–11713. [Google Scholar] [CrossRef]
- Parker, A.L.; Waddington, S.N.; Nicol, C.G.; Shayakhmetov, D.M.; Buckley, S.M.; Denby, L.; Kemball-Cook, G.; Ni, S.; Lieber, A.; McVey, J.H.; Nicklin, S.A.; Baker, A.H. Multiple vitamin K-dependent coagulation zymogens promote adenovirus-mediated gene delivery to hepatocytes. Blood 2006, 108, 2554–2561. [Google Scholar] [CrossRef]
- Parker, A.L.; McVey, J.H.; Doctor, J.H.; Lopez-Franco, O.; Waddington, S.N.; Havenga, M.J.; Nicklin, S.A.; Baker, A.H. Influence of coagulation factor zymogens on the infectivity of adenoviruses pseudotyped with fibers from subgroup D. J. Virol. 2007, 81, 3627–3631. [Google Scholar] [CrossRef]
- Shayakhmetov, D.M.; Gaggar, A.; Ni, S.; Li, Z.Y.; Lieber, A. Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J. Virol. 2005, 79, 7478–7491. [Google Scholar] [CrossRef] [PubMed]
- Waddington, S.N.; Parker, A.L.; Havenga, M.; Nicklin, S.A.; Buckley, S.M.; McVey, J.H.; Baker, A.H. Targeting of adenovirus serotype 5 (Ad5) and 5/47 pseudotyped vectors in vivo: fundamental involvement of coagulation factors and redundancy of CAR binding by Ad5. J. Virol. 2007, 81, 9568–9571. [Google Scholar] [CrossRef] [PubMed]
- Waddington, S.N.; McVey, J.H.; Bhella, D.; Parker, A.L.; Barker, K.; Atoda, H.; Pink, R.; Buckley, S.M.; Greig, J.A.; Denby, L.; Custers, J.; Morita, T.; Francischetti, I.M.; Monteiro, R.Q.; Barouch, D.H.; Van, R.N.; Napoli, C.; Havenga, M.J.; Nicklin, S.A.; Baker, A.H. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 2008, 132, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Alba, R.; Bradshaw, A.C.; Parker, A.L.; Bhella, D.; Waddington, S.N.; Nicklin, S.A.; Van, R.N.; Custers, J.; Goudsmit, J.; Barouch, D.H.; McVey, J.H.; Baker, A.H. Identification of coagulation factor (F)X binding sites on the adenovirus serotype 5 hexon: effect of mutagenesis on FX interactions and gene transfer. Blood 2009, 114, 965–971. [Google Scholar] [CrossRef]
- Sakurai, F.; Mizuguchi, H.; Yamaguchi, T.; Hayakawa, T. Characterization of in vitro and in vivo gene transfer properties of adenovirus serotype 35 vector. Mol. Ther. 2003, 8, 813–821. [Google Scholar] [CrossRef]
- Tao, N.; Gao, G.P.; Parr, M.; Johnston, J.; Baradet, T.; Wilson, J.M.; Barsoum, J.; Fawell, S.E. Sequestration of adenoviral vector by Kupffer cells leads to a nonlinear dose response of transduction in liver. Mol. Ther. 2001, 3, 28–35. [Google Scholar] [CrossRef]
- Yu, Q.; Que, L.G.; Rockey, D.C. Adenovirus-mediated gene transfer to nonparenchymal cells in normal and injured liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G565–G572. [Google Scholar] [CrossRef]
- Tian, J.; Xu, Z.; Smith, J.S.; Hofherr, S.E.; Barry, M.A.; Byrnes, A.P. Adenovirus activates complement by distinctly different mechanisms in vitro and in vivo: indirect complement activation by virions in vivo. J. Virol. 2009, 83, 5648–5658. [Google Scholar] [CrossRef]
- Stone, D.; Liu, Y.; Shayakhmetov, D.; Li, Z.Y.; Ni, S.; Lieber, A. Adenovirus-platelet interaction in blood causes virus sequestration to the reticuloendothelial system of the liver. J. Virol. 2007, 81, 4866–4871. [Google Scholar] [CrossRef]
- Schiedner, G.; Hertel, S.; Johnston, M.; Dries, V.; Van, R.N.; Kochanek, S. Selective depletion or blockade of Kupffer cells leads to enhanced and prolonged hepatic transgene expression using high-capacity adenoviral vectors. Mol. Ther. 2003, 7, 35–43. [Google Scholar] [CrossRef]
- Morral, N.; O'Neal, W.K.; Rice, K.; Leland, M.M.; Piedra, P.A.; guilar-Cordova, E.; Carey, K.D.; Beaudet, A.L.; Langston, C. Lethal toxicity, severe endothelial injury, and a threshold effect with high doses of an adenoviral vector in baboons. Hum. Gene Ther. 2002, 13, 143–154. [Google Scholar] [CrossRef]
- Bowen, G.P.; Borgland, S.L.; Lam, M.; Libermann, T.A.; Wong, N.C.; Muruve, D.A. Adenovirus vector-induced inflammation: capsid-dependent induction of the C-C chemokine RANTES requires NF-kappa B. Hum. Gene Ther. 2002, 13, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Huang, X.; Yang, Y. Innate immune response to adenoviral vectors is mediated by both Toll-like receptor-dependent and -independent pathways. J. Virol. 2007, 81, 3170–3180. [Google Scholar] [CrossRef] [PubMed]
- Appledorn, D.M.; Patial, S.; McBride, A.; Godbehere, S.; Van, R.N.; Parameswaran, N.; Amalfitano, A. Adenovirus vector-induced innate inflammatory mediators, MAPK signaling, as well as adaptive immune responses are dependent upon both TLR2 and TLR9 in vivo. J. Immunol. 2008, 181, 2134–2144. [Google Scholar] [CrossRef]
- Appledorn, D.M.; Patial, S.; Godbehere, S.; Parameswaran, N.; Amalfitano, A. TRIF, and TRIF-interacting TLRs differentially modulate several adenovirus vector-induced immune responses. J. Innate. Immun. 2009, 1, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Cerullo, V.; Seiler, M.P.; Mane, V.; Brunetti-Pierri, N.; Clarke, C.; Bertin, T.K.; Rodgers, J.R.; Lee, B. Toll-like receptor 9 triggers an innate immune response to helper-dependent adenoviral vectors. Mol. Ther. 2007, 15, 378–385. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Black, E.P.; Amalfitano, A. Adenoviral infection induces a multi-faceted innate cellular immune response that is mediated by the toll-like receptor pathway in A549 cells. Virology 2007, 358, 357–372. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Kiang, A.; Everett, R.S.; Serra, D.; Yang, X.Y.; Clay, T.M.; Amalfitano, A. Adenovirus infection triggers a rapid, MyD88-regulated transcriptome response critical to acute-phase and adaptive immune responses in vivo. J. Virol. 2007, 81, 1796–1812. [Google Scholar] [CrossRef]
- Zhu, J.; Huang, X.; Yang, Y. Type I IFN signaling on both B and CD4 T cells is required for protective antibody response to adenovirus. J. Immunol. 2007, 178, 3505–3510. [Google Scholar] [CrossRef]
- Zhu, J.; Huang, X.; Yang, Y. A critical role for type I IFN-dependent NK cell activation in innate immune elimination of adenoviral vectors in vivo. Mol. Ther. 2008, 16, 1300–1307. [Google Scholar] [CrossRef]
- Nayak, S.; Herzog, R.W. Progress and prospects: immune responses to viral vectors. Gene Ther. 2010, 17, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Nociari, M.; Ocheretina, O.; Schoggins, J.W.; Falck-Pedersen, E. Sensing infection by adenovirus: Toll-like receptor-independent viral DNA recognition signals activation of the interferon regulatory factor 3 master regulator. J. Virol. 2007, 81, 4145–4157. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, N.C.; Miao, E.A.; Iwakura, Y.; Murali-Krishna, K.; Aderem, A.; Flavell, R.A.; Papayannopoulou, T.; Shayakhmetov, D.M. Virus binding to a plasma membrane receptor triggers interleukin-1 alpha-mediated proinflammatory macrophage response in vivo. Immunity 2009, 31, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Cichon, G.; Boeckh-Herwig, S.; Schmidt, H.H.; Wehnes, E.; Muller, T.; Pring-Akerblom, P.; Burger, R. Complement activation by recombinant adenoviruses. Gene Ther. 2001, 8, 1794–1800. [Google Scholar] [CrossRef]
- Kiang, A.; Hartman, Z.C.; Everett, R.S.; Serra, D.; Jiang, H.; Frank, M.M.; Amalfitano, A. Multiple innate inflammatory responses induced after systemic adenovirus vector delivery depend on a functional complement system. Mol. Ther. 2006, 14, 588–598. [Google Scholar] [CrossRef]
- Othman, M.; Labelle, A.; Mazzetti, I.; Elbatarny, H.S.; Lillicrap, D. Adenovirus-induced thrombocytopenia: the role of von Willebrand factor and P-selectin in mediating accelerated platelet clearance. Blood 2007, 109, 2832–2839. [Google Scholar] [CrossRef]
- Harding, T.C.; Koprivnikar, K.E.; Tu, G.H.; Zayek, N.; Lew, S.; Subramanian, A.; Sivakumaran, A.; Frey, D.; Ho, K.; VanRoey, M.J.; Nichols, T.C.; Bellinger, D.A.; Yendluri, S.; Waugh, J.; McArthur, J.; Veres, G.; Donahue, B.A. Intravenous administration of an AAV-2 vector for the expression of factor IX in mice and a dog model of hemophilia B. Gene Ther. 2004, 11, 204–213. [Google Scholar] [CrossRef]
- Chao, H.; Liu, Y.; Rabinowitz, J.; Li, C.; Samulski, R.J.; Walsh, C.E. Several log increase in therapeutic transgene delivery by distinct adeno-associated viral serotype vectors. Mol. Ther. 2000, 2, 619–623. [Google Scholar] [CrossRef]
- Cordier, L.; Hack, A.A.; Scott, M.O.; Barton-Davis, E.R.; Gao, G.; Wilson, J.M.; McNally, E.M.; Sweeney, H.L. Rescue of skeletal muscles of gamma-sarcoglycan-deficient mice with adeno-associated virus-mediated gene transfer. Mol. Ther. 2000, 1, 119–129. [Google Scholar] [CrossRef]
- Dinculescu, A.; Glushakova, L.; Min, S.H.; Hauswirth, W.W. Adeno-associated virus-vectored gene therapy for retinal disease. Hum. Gene Ther. 2005, 16, 649–663. [Google Scholar] [CrossRef]
- Zaiss, A.K.; Muruve, D.A. Immunity to adeno-associated virus vectors in animals and humans: a continued challenge. Gene Ther. 2008, 15, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; Maus, M.V.; Hui, D.J.; Sabatino, D.E.; Murphy, S.L.; Rasko, J.E.; Ragni, M.V.; Manno, C.S.; Sommer, J.; Jiang, H.; Pierce, G.F.; Ertl, H.C.; High, K.A. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 2007, 13, 419–422. [Google Scholar] [CrossRef]
- Pien, G.C.; Basner-Tschakarjan, E.; Hui, D.J.; Mentlik, A.N.; Finn, J.D.; Hasbrouck, N.C.; Zhou, S.; Murphy, S.L.; Maus, M.V.; Mingozzi, F.; Orange, J.S.; High, K.A. Capsid antigen presentation flags human hepatocytes for destruction after transduction by adeno-associated viral vectors. J. Clin. Invest 2009, 119, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.D.; Cheng, Q.; Harui, A.; Basak, S.K.; Mitani, K.; Low, T.A.; Kiertscher, S.M. Helper-dependent adenoviral vectors efficiently express transgenes in human dendritic cells but still stimulate antiviral immune responses. J. Immunol. 2002, 169, 4651–4656. [Google Scholar] [CrossRef] [PubMed]
- Kafri, T.; Morgan, D.; Krahl, T.; Sarvetnick, N.; Sherman, L.; Verma, I. Cellular immune response to adenoviral vector infected cells does not require de novo viral gene expression: implications for gene therapy. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 11377–11382. [Google Scholar] [CrossRef]
- Kushwah, R.; Cao, H.; Hu, J. Potential of helper-dependent adenoviral vectors in modulating airway innate immunity. Cell Mol. Immunol. 2007, 4, 81–89. [Google Scholar]
- Kushwah, R.; Cao, H.; Hu, J. Characterization of pulmonary T cell response to helper-dependent adenoviral vectors following intranasal delivery. J. Immunol. 2008, 180, 4098–4108. [Google Scholar] [CrossRef]
- Jacobs, F.; Feng, Y.; Van, C.E.; Lievens, J.; Snoeys, J.; De, G.B. Species differences in hepatocyte-directed gene transfer: implications for clinical translation. Curr. Gene Ther. 2009, 9, 83–90. [Google Scholar] [CrossRef]
- Jacobs, F.; Wisse, E.; De, G.B. The role of liver sinusoidal cells in hepatocyte-directed gene transfer. Am. J. Pathol. 2010, 176, 14–21. [Google Scholar] [CrossRef]
- Vetrini, F.; Brunetti-Pierri, N.; Palmer, D.J.; Bertin, T.; Grove, N.C.; Finegold, M.J.; Ng, P. Vasoactive Intestinal Peptide Increases Hepatic Transduction and Reduces Innate Immune Response Following Administration of Helper-dependent Ad. Mol. Ther. 2010. [Google Scholar] [CrossRef]
- Ajuf'ev, B.N.; Dizhe, E.B.; Efremov, A.M.; Mogilenko, D.A.; Oleinikova, G.N.; Lapikov, I.A.; Zhdanova, O.I.; Kidgotko, O.V.; Orlov, S.V.; Perevozchikov, A.P. [Hydrodynamics-based transfer of human apolipoprotein A-I gene into mice: study of factors involving an efficacy and duration of the transferred gene expression in animals' liver]. Mol. Biol. (Mosk) 2004, 38, 1076–1084. [Google Scholar] [PubMed]
- Brunetti-Pierri, N.; Palmer, D.J.; Mane, V.; Finegold, M.; Beaudet, A.L.; Ng, P. Increased hepatic transduction with reduced systemic dissemination and proinflammatory cytokines following hydrodynamic injection of helper-dependent adenoviral vectors. Mol. Ther. 2005, 12, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Wolff, L.J.; Wolff, J.A.; Sebestyen, M.G. Effect of tissue-specific promoters and microRNA recognition elements on stability of transgene expression after hydrodynamic naked plasmid DNA delivery. Hum. Gene Ther. 2009, 20, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Yotnda, P.; Chen, D.H.; Chiu, W.; Piedra, P.A.; Davis, A.; Templeton, N.S.; Brenner, M.K. Bilamellar cationic liposomes protect adenovectors from preexisting humoral immune responses. Mol. Ther. 2002, 5, 233–241. [Google Scholar] [CrossRef]
- Croyle, M.A.; Chirmule, N.; Zhang, Y.; Wilson, J.M. PEGylation of E1-deleted adenovirus vectors allows significant gene expression on readministration to liver. Hum. Gene Ther. 2002, 13, 1887–1900. [Google Scholar] [CrossRef]
- Hofherr, S.E.; Mok, H.; Gushiken, F.C.; Lopez, J.A.; Barry, M.A. Polyethylene glycol modification of adenovirus reduces platelet activation, endothelial cell activation, and thrombocytopenia. Hum. Gene Ther. 2007, 18, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Mok, H.; Palmer, D.J.; Ng, P.; Barry, M.A. Evaluation of polyethylene glycol modification of first-generation and helper-dependent adenoviral vectors to reduce innate immune responses. Mol. Ther. 2005, 11, 66–79. [Google Scholar] [CrossRef]
- De, G.B.; Snoeys, J.; Van, L.S.; Lievens, J.; Collen, D. Elimination of innate immune responses and liver inflammation by PEGylation of adenoviral vectors and methylprednisolone. Hum. Gene Ther. 2005, 16, 1439–1451. [Google Scholar]
- Seregin, S.S.; Appledorn, D.M.; McBride, A.J.; Schuldt, N.J.; Aldhamen, Y.A.; Voss, T.; Wei, J.; Bujold, M.; Nance, W.; Godbehere, S.; Amalfitano, A. Transient pretreatment with glucocorticoid ablates innate toxicity of systemically delivered adenoviral vectors without reducing efficacy. Mol. Ther. 2009, 17, 685–696. [Google Scholar] [CrossRef]
- Culling, B.; Ogle, R. Genetic counselling issues in cystic fibrosis. Paediatr. Respir. Rev. 2010, 11, 75–79. [Google Scholar] [CrossRef]
- O'Riordan, S.M.; Dattani, M.T.; Hindmarsh, P.C. Cystic fibrosis-related diabetes in childhood. Horm. Res. Paediatr. 2010, 73, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Pickles, R.J.; McCarty, D.; Matsui, H.; Hart, P.J.; Randell, S.H.; Boucher, R.C. Limited entry of adenovirus vectors into well-differentiated airway epithelium is responsible for inefficient gene transfer. J. Virol. 1998, 72, 6014–6023. [Google Scholar] [CrossRef] [PubMed]
- Pickles, R.J.; Fahrner, J.A.; Petrella, J.M.; Boucher, R.C.; Bergelson, J.M. Retargeting the coxsackievirus and adenovirus receptor to the apical surface of polarized epithelial cells reveals the glycocalyx as a barrier to adenovirus-mediated gene transfer. J. Virol. 2000, 74, 6050–6057. [Google Scholar] [CrossRef]
- Engelhardt, J.F.; Simon, R.H.; Yang, Y.; Zepeda, M.; Weber-Pendleton, S.; Doranz, B.; Grossman, M.; Wilson, J.M. Adenovirus-mediated transfer of the CFTR gene to lung of nonhuman primates: biological efficacy study. Hum. Gene Ther. 1993, 4, 759–769. [Google Scholar] [CrossRef]
- Engelhardt, J.F.; Yang, Y.; Stratford-Perricaudet, L.D.; Allen, E.D.; Kozarsky, K.; Perricaudet, M.; Yankaskas, J.R.; Wilson, J.M. Direct gene transfer of human CFTR into human bronchial epithelia of xenografts with E1-deleted adenoviruses. Nat. Genet. 1993, 4, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.H.; Engelhardt, J.F.; Yang, Y.; Zepeda, M.; Weber-Pendleton, S.; Grossman, M.; Wilson, J.M. Adenovirus-mediated transfer of the CFTR gene to lung of nonhuman primates: toxicity study. Hum. Gene Ther. 1993, 4, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, J.B.; Robinson, C.B.; McCoy, K.S.; Shell, R.; Sferra, T.J.; Chirmule, N.; Magosin, S.A.; Propert, K.J.; Brown-Parr, E.C.; Hughes, J.V.; Tazelaar, J.; Baker, C.; Goldman, M.J.; Wilson, J.M. A phase I study of adenovirus-mediated transfer of the human cystic fibrosis transmembrane conductance regulator gene to a lung segment of individuals with cystic fibrosis. Hum. Gene Ther. 1999, 10, 2973–2985. [Google Scholar] [CrossRef]
- Chu, Q.; St, G.J.; Lukason, M.; Cheng, S.H.; Scheule, R.K.; Eastman, S.J. EGTA enhancement of adenovirus-mediated gene transfer to mouse tracheal epithelium in vivo. Hum. Gene Ther. 2001, 12, 455–467. [Google Scholar] [CrossRef]
- Kaplan, J.M.; Pennington, S.E.; St George, J.A.; Woodworth, L.A.; Fasbender, A.; Marshall, J.; Cheng, S.H.; Wadsworth, S.C.; Gregory, R.J.; Smith, A.E. Potentiation of gene transfer to the mouse lung by complexes of adenovirus vector and polycations improves therapeutic potential. Hum. Gene Ther. 1998, 9, 1469–1479. [Google Scholar] [CrossRef]
- Wang, G.; Zabner, J.; Deering, C.; Launspach, J.; Shao, J.; Bodner, M.; Jolly, D.J.; Davidson, B.L.; McCray, P.B., Jr. Increasing epithelial junction permeability enhances gene transfer to airway epithelia In vivo. Am. J. Respir. Cell Mol. Biol. 2000, 22, 129–138. [Google Scholar] [CrossRef]
- Chirmule, N.; Hughes, J.V.; Gao, G.P.; Raper, S.E.; Wilson, J.M. Role of E4 in eliciting CD4 T-cell and B-cell responses to adenovirus vectors delivered to murine and nonhuman primate lungs. J. Virol. 1998, 72, 6138–6145. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Litzky, L.A.; Engelhardt, J.F.; Wilson, J.M. Transfer of the CFTR gene to the lung of nonhuman primates with E1-deleted, E2a-defective recombinant adenoviruses: a preclinical toxicology study. Hum. Gene Ther. 1995, 6, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Koehler, D.R.; Sajjan, U.; Chow, Y.H.; Martin, B.; Kent, G.; Tanswell, A.K.; McKerlie, C.; Forstner, J.F.; Hu, J. Protection of Cftr knockout mice from acute lung infection by a helper-dependent adenoviral vector expressing Cftr in airway epithelia. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 15364–15369. [Google Scholar] [CrossRef]
- Koehler, D.R.; Frndova, H.; Leung, K.; Louca, E.; Palmer, D.; Ng, P.; McKerlie, C.; Cox, P.; Coates, A.L.; Hu, J. Aerosol delivery of an enhanced helper-dependent adenovirus formulation to rabbit lung using an intratracheal catheter. J. Gene Med. 2005, 7, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Ng, P. Progress towards liver and lung-directed gene therapy with helper-dependent adenoviral vectors. Curr. Gene Ther. 2009, 9, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Mellis, C.M.; Wood, R.E. Edetate sodium aerosol in Pseudomonas lung infection in cystic fibrosis. Am. J. Dis. Child 1985, 139, 836–839. [Google Scholar] [CrossRef]
- Rogers, C.S.; Stoltz, D.A.; Meyerholz, D.K.; Ostedgaard, L.S.; Rokhlina, T.; Taft, P.J.; Rogan, M.P.; Pezzulo, A.A.; Karp, P.H.; Itani, O.A.; Kabel, A.C.; Wohlford-Lenane, C.L.; Davis, G.J.; Hanfland, R.A.; Smith, T.L.; Samuel, M.; Wax, D.; Murphy, C.N.; Rieke, A.; Whitworth, K.; Uc, A.; Starner, T.D.; Brogden, K.A.; Shilyansky, J.; McCray, P.B., Jr.; Zabner, J.; Prather, R.S.; Welsh, M.J. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 2008, 321, 1837–1841. [Google Scholar] [CrossRef]
- Moss, R.B.; Rodman, D.; Spencer, L.T.; Aitken, M.L.; Zeitlin, P.L.; Waltz, D.; Milla, C.; Brody, A.S.; Clancy, J.P.; Ramsey, B.; Hamblett, N.; Heald, A.E. Repeated adeno-associated virus serotype 2 aerosol-mediated cystic fibrosis transmembrane regulator gene transfer to the lungs of patients with cystic fibrosis: a multicenter, double-blind, placebo-controlled trial. Chest 2004, 125, 509–521. [Google Scholar] [CrossRef]
- Johnson, L.G.; Olsen, J.C.; Sarkadi, B.; Moore, K.L.; Swanstrom, R.; Boucher, R.C. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat. Genet. 1992, 2, 21–25. [Google Scholar] [CrossRef]
- Segura, M.M.; Alba, R.; Bosch, A.; Chillon, M. Advances in helper-dependent adenoviral vector research. Curr. Gene Ther. 2008, 8, 222–235. [Google Scholar] [CrossRef]
- Dudley, R.W.; Lu, Y.; Gilbert, R.; Matecki, S.; Nalbantoglu, J.; Petrof, B.J.; Karpati, G. Sustained improvement of muscle function one year after full-length dystrophin gene transfer into mdx mice by a gutted helper-dependent adenoviral vector. Hum. Gene Ther. 2004, 15, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.; Nalbantoglu, J.; Howell, J.M.; Davies, L.; Fletcher, S.; Amalfitano, A.; Petrof, B.J.; Kamen, A.; Massie, B.; Karpati, G. Dystrophin expression in muscle following gene transfer with a fully deleted ("gutted") adenovirus is markedly improved by trans-acting adenoviral gene products. Hum. Gene Ther. 2001, 12, 1741–1755. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Feingold, E.; Kochanek, S.; Clemens, P.R. Systemic delivery of a high-capacity adenoviral vector expressing mouse CTLA4Ig improves skeletal muscle gene therapy. Mol. Ther. 2002, 6, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Schiedner, G.; Van, R.N.; Liu, C.C.; Kochanek, S.; Clemens, P.R. Sustained muscle expression of dystrophin from a high-capacity adenoviral vector with systemic gene transfer of T cell costimulatory blockade. Mol. Ther. 2004, 10, 688–696. [Google Scholar] [CrossRef]
- Bilbao, R.; Reay, D.P.; Hughes, T.; Biermann, V.; Volpers, C.; Goldberg, L.; Bergelson, J.; Kochanek, S.; Clemens, P.R. Fetal muscle gene transfer is not enhanced by an RGD capsid modification to high-capacity adenoviral vectors. Gene Ther. 2003, 10, 1821–1829. [Google Scholar] [CrossRef]
- Smith, P.E.; Calverley, P.M.; Edwards, R.H.; Evans, G.A.; Campbell, E.J. Practical problems in the respiratory care of patients with muscular dystrophy. N. Engl. J. Med. 1987, 316, 1197–1205. [Google Scholar] [CrossRef]
- Smith, P.E.; Calverley, P.M.; Edwards, R.H. Hypoxemia during sleep in Duchenne muscular dystrophy. Am. Rev. Respir. Dis. 1988, 137, 884–888. [Google Scholar] [CrossRef]
- Matecki, S.; Dudley, R.W.; Divangahi, M.; Gilbert, R.; Nalbantoglu, J.; Karpati, G.; Petrof, B.J. Therapeutic gene transfer to dystrophic diaphragm by an adenoviral vector deleted of all viral genes. Am. J. Physiol Lung Cell Mol. Physiol 2004, 287, L569–L576. [Google Scholar] [CrossRef]
- Maione, D.; Della, R.C.; Giannetti, P.; D'Arrigo, R.; Liberatoscioli, L.; Franlin, L.L.; Sandig, V.; Ciliberto, G.; La, M.N.; Savino, R. An improved helper-dependent adenoviral vector allows persistent gene expression after intramuscular delivery and overcomes preexisting immunity to adenovirus. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 5986–5991. [Google Scholar] [CrossRef]
- Bramson, J.L.; Grinshtein, N.; Meulenbroek, R.A.; Lunde, J.; Kottachchi, D.; Lorimer, I.A.; Jasmin, B.J.; Parks, R.J. Helper-dependent adenoviral vectors containing modified fiber for improved transduction of developing and mature muscle cells. Hum. Gene Ther. 2004, 15, 179–188. [Google Scholar] [CrossRef]
- Persson, A.; Fan, X.; Widegren, B.; Englund, E. Cell type- and region-dependent coxsackie adenovirus receptor expression in the central nervous system. J. Neurooncol. 2006, 78, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.L.; Allen, E.D.; Kozarsky, K.F.; Wilson, J.M.; Roessler, B.J. A model system for in vivo gene transfer into the central nervous system using an adenoviral vector. Nat. Genet. 1993, 3, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Doran, S.E.; Roessler, B.J.; Hartman, J.W.; Hoff, J.T.; Shewach, D.S.; Davidson, B.L. Adenovirus-mediated in vivo gene transfer into the central nervous system of a nonhuman primate (resident award paper). Clin. Neurosurg. 1994, 41, 242–257. [Google Scholar] [PubMed]
- Doran, S.E.; Ren, X.D.; Betz, A.L.; Pagel, M.A.; Neuwelt, E.A.; Roessler, B.J.; Davidson, B.L. Gene expression from recombinant viral vectors in the central nervous system after blood-brain barrier disruption. Neurosurgery 1995, 36, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, A.P.; Wood, M.J.; Charlton, H.M. Role of T cells in inflammation caused by adenovirus vectors in the brain. Gene Ther. 1996, 3, 644–651. [Google Scholar]
- Byrnes, A.P.; MacLaren, R.E.; Charlton, H.M. Immunological instability of persistent adenovirus vectors in the brain: peripheral exposure to vector leads to renewed inflammation, reduced gene expression, and demyelination. J. Neurosci. 1996, 16, 3045–3055. [Google Scholar] [CrossRef]
- Thomas, C.E.; Schiedner, G.; Kochanek, S.; Castro, M.G.; Lowenstein, P.R. Peripheral infection with adenovirus causes unexpected long-term brain inflammation in animals injected intracranially with first-generation, but not with high-capacity, adenovirus vectors: toward realistic long-term neurological gene therapy for chronic diseases. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 7482–7487. [Google Scholar]
- Zou, L.; Yuan, X.; Zhou, H.; Lu, H.; Yang, K. Helper-dependent adenoviral vector-mediated gene transfer in aged rat brain. Hum. Gene Ther. 2001, 12, 181–191. [Google Scholar] [CrossRef]
- Butti, E.; Bergami, A.; Recchia, A.; Brambilla, E.; Franciotta, D.; Cattalini, A.; Stornaiuolo, A.; Lachapelle, F.; Comi, G.; Mavilio, F.; Martino, G.; Furlan, R. Absence of an intrathecal immune reaction to a helper-dependent adenoviral vector delivered into the cerebrospinal fluid of non-human primates. Gene Ther. 2008, 15, 233–238. [Google Scholar] [CrossRef]
- Butti, E.; Bergami, A.; Recchia, A.; Brambilla, E.; Del, C.U.; Amadio, S.; Cattalini, A.; Esposito, M.; Stornaiuolo, A.; Comi, G.; Pluchino, S.; Mavilio, F.; Martino, G.; Furlan, R. IL4 gene delivery to the CNS recruits regulatory T cells and induces clinical recovery in mouse models of multiple sclerosis. Gene Ther. 2008, 15, 504–515. [Google Scholar] [CrossRef]
- Huang, B.; Schiefer, J.; Sass, C.; Landwehrmeyer, G.B.; Kosinski, C.M.; Kochanek, S. High-capacity adenoviral vector-mediated reduction of huntingtin aggregate load in vitro and in vivo. Hum. Gene Ther. 2007, 18, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Schiefer, J.; Sass, C.; Kosinski, C.M.; Kochanek, S. Inducing huntingtin inclusion formation in primary neuronal cell culture and in vivo by high-capacity adenoviral vectors expressing truncated and full-length huntingtin with polyglutamine expansion. J. Gene Med. 2008, 10, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, A.K.; Puntel, M.; Candolfi, M.; Salem, A.; Yagiz, K.; Farrokhi, C.; Kroeger, K.M.; Xiong, W.; Curtin, J.F.; Liu, C.; Lawrence, K.; Bondale, N.S.; Lerner, J.; Baker, G.J.; Foulad, D.; Pechnick, R.N.; Palmer, D.; Ng, P.; Lowenstein, P.R.; Castro, M.G. Study of the Efficacy, Biodistribution, and Safety Profile of Therapeutic Gutless Adenovirus Vectors as a Prelude to a Phase I Clinical Trial for Glioblastoma. Clin. Pharmacol. Ther. 2010, 88, 204-13. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Singh, R. Barriers for retinal gene therapy: separating fact from fiction. Vision Res. 2008, 48, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Kreppel, F.; Luther, T.T.; Semkova, I.; Schraermeyer, U.; Kochanek, S. Long-term transgene expression in the RPE after gene transfer with a high-capacity adenoviral vector. Invest Ophthalmol. Vis. Sci. 2002, 43, 1965–1970. [Google Scholar] [PubMed]
- Harui, A.; Roth, M.D.; Kiertscher, S.M.; Mitani, K.; Basak, S.K. Vaccination with helper-dependent adenovirus enhances the generation of transgene-specific CTL. Gene Ther. 2004, 11, 1617–1626. [Google Scholar] [CrossRef]
- Weaver, E.A.; Nehete, P.N.; Buchl, S.S.; Senac, J.S.; Palmer, D.; Ng, P.; Sastry, K.J.; Barry, M.A. Comparison of replication-competent, first generation, and helper-dependent adenoviral vaccines. PLoS. One 2009, 4, 5059. [Google Scholar] [CrossRef]
- Weaver, E.A.; Nehete, P.N.; Nehete, B.P.; Buchl, S.J.; Palmer, D.; Montefiori, D.C.; Ng, P.; Sastry, K.J.; Barry, M.A. Protection against Mucosal SHIV Challenge by Peptide and Helper-Dependent Adenovirus Vaccines. Viruses. 2009, 1, 920–938. [Google Scholar] [CrossRef]
- Stern, BS.; Shoshani, W.; Pearlman, AL.; Ng, P.; Nissenson, AR.; Galun, E.; Besarab, A.; Rimler, A.; Goltzmann, H.; Miari, R.O; kun, A.; Bookay, H.; Jibly, T.; Abrameto, JA; Elhalel, M. Erythropoeisis Sustained 1 Year by the EPODURE BioPump in Patients with Chronic Kideney Disease: Further Results of PhaseI/II Proof of Concept Trial. Mol. Ther. 2010, 18, S239. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2010 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vetrini, F.; Ng, P. Gene Therapy with Helper-Dependent Adenoviral Vectors: Current Advances and Future Perspectives. Viruses 2010, 2, 1886-1917. https://doi.org/10.3390/v2091886
Vetrini F, Ng P. Gene Therapy with Helper-Dependent Adenoviral Vectors: Current Advances and Future Perspectives. Viruses. 2010; 2(9):1886-1917. https://doi.org/10.3390/v2091886
Chicago/Turabian StyleVetrini, Francesco, and Philip Ng. 2010. "Gene Therapy with Helper-Dependent Adenoviral Vectors: Current Advances and Future Perspectives" Viruses 2, no. 9: 1886-1917. https://doi.org/10.3390/v2091886
APA StyleVetrini, F., & Ng, P. (2010). Gene Therapy with Helper-Dependent Adenoviral Vectors: Current Advances and Future Perspectives. Viruses, 2(9), 1886-1917. https://doi.org/10.3390/v2091886