Targeting HCV Entry For Development of Therapeutics

{kind=link}
Abstract
:1. Introduction
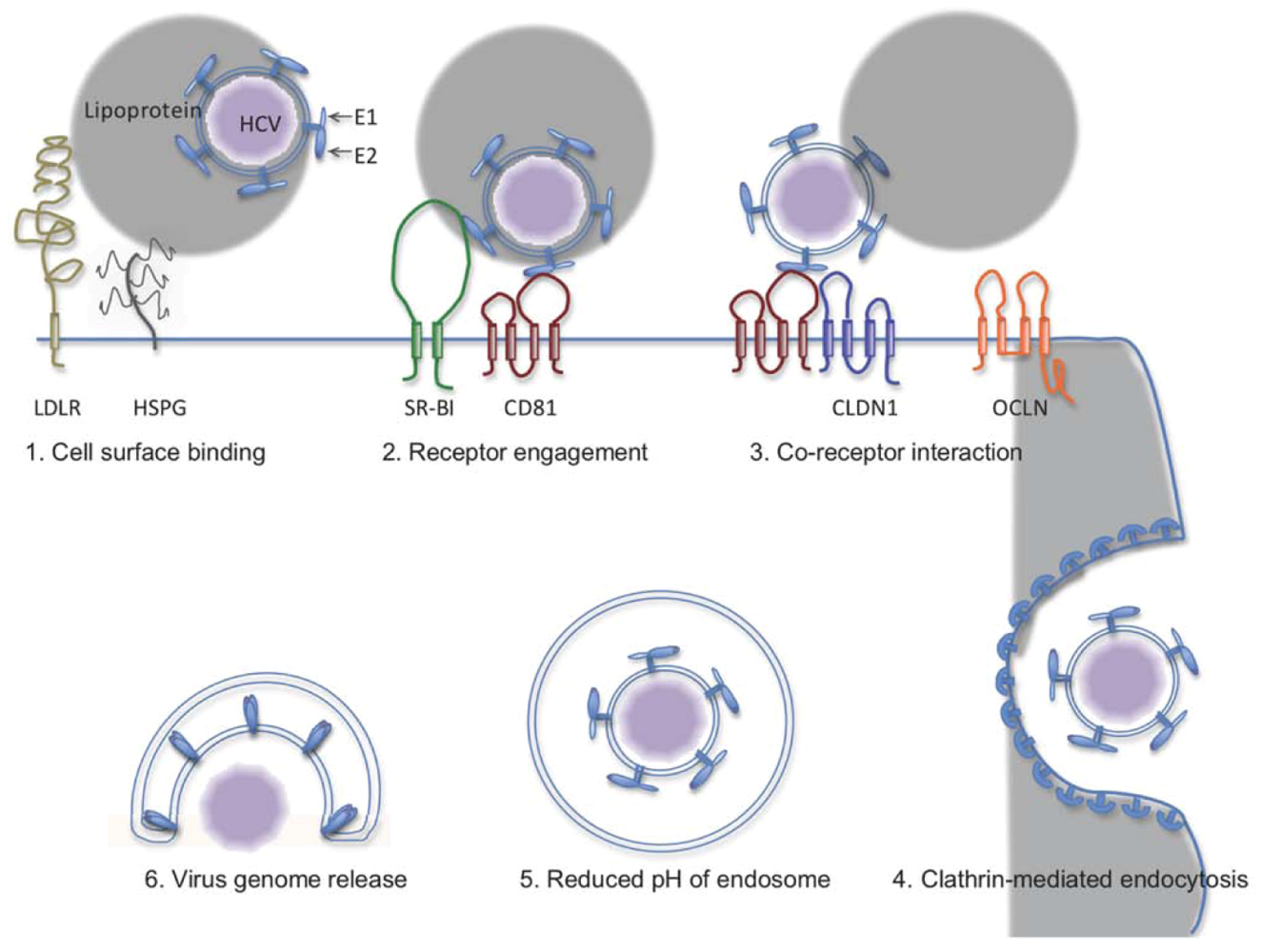
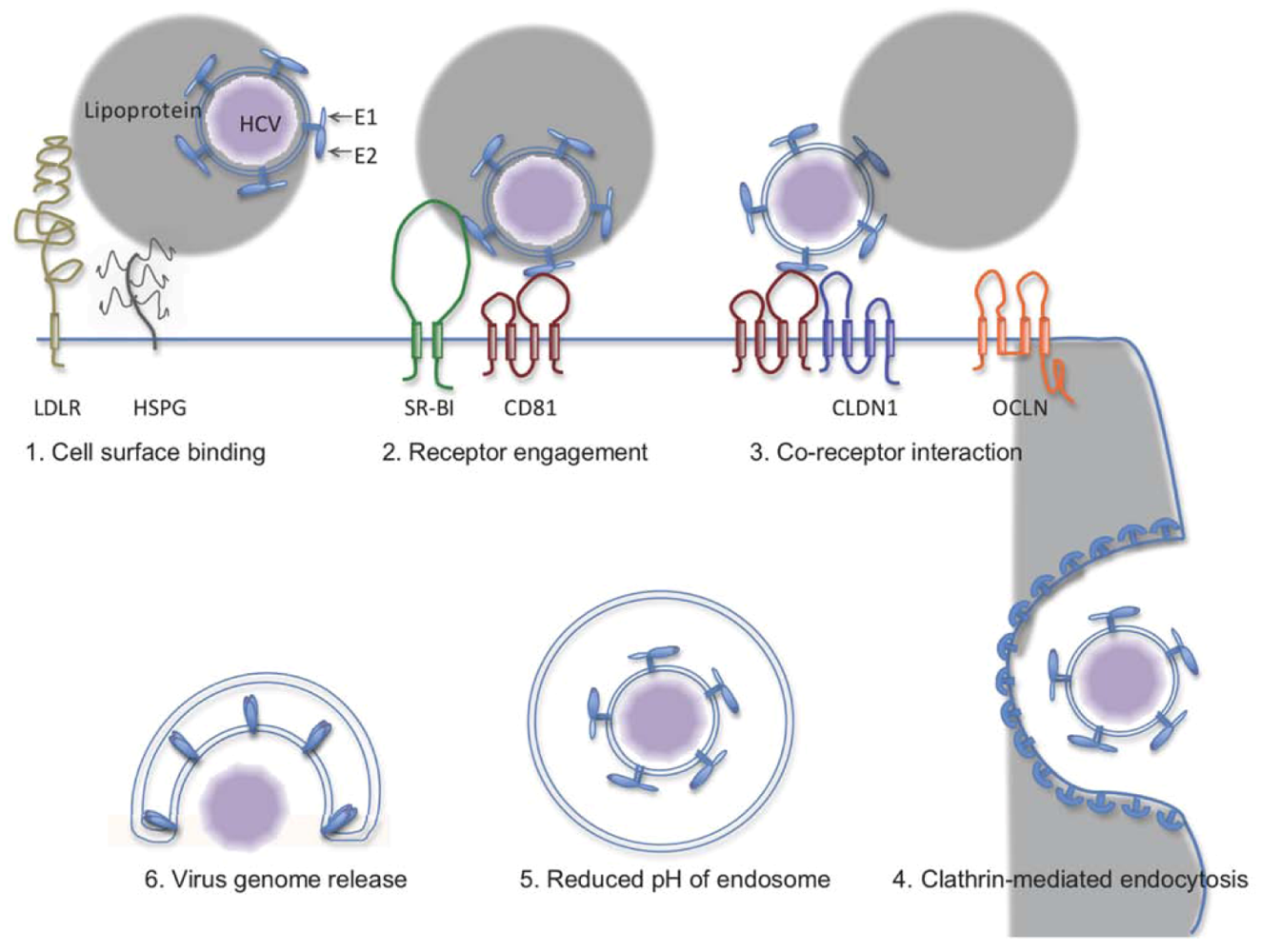
2. The HCV Entry Process
3. Interventions at Virus Entry
3.1. Targeting the Viral Envelope Proteins
3.2. Targeting Host Proteins Involved in Entry.
4. Random Screening for HCV Entry Inhibitors
5. Perspective
References
- Falck-Ytter, Y.; Kale, H.; Mullen, K.D.; Sarbah, S.A.; Sorescu, L.; McCullough, A.J. Surprisingly small effect of antiviral treatment in patients with hepatitis C. Ann. Intern. Med. 2002, 136, 288–292. [Google Scholar] [PubMed]
- Lohmann, V.; Korner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Dubuisson, J.; Cosset, F.L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 2003, 197, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.; Zhang, J.; Flint, M.; Logvinoff, C.; Cheng-Mayer, C.; Rice, C.M.; McKeating, J.A. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 7271–7276. [Google Scholar] [CrossRef] [PubMed]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; Bartenschlager, R.; Liang, T.J. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T. L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; Rice, C.M. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 9294–9299. [Google Scholar] [CrossRef] [PubMed]
- Gastaminza, P.; Cheng, G.; Wieland, S.; Zhong, J.; Liao, W.; Chisari, F.V. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 2008, 82, 2120–2129. [Google Scholar] [CrossRef] [PubMed]
- Modi, A.A.; Hoofnagle, J.H. New therapies for hepatitis C. Hepatology 2007, 46, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; McHutchison, J.G. Review article: investigational agents for chronic hepatitis C. Aliment. Pharmacol. Ther. 2009, 29, 689–705. [Google Scholar] [CrossRef] [PubMed]
- Behrens, S.E.; Tomei, L.; De Francesco, R. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 1996, 15, 12–22. [Google Scholar] [PubMed]
- Shudo, E.; Ribeiro, R.M.; Perelson, A.S. Modeling HCV kinetics under therapy using PK and PD information. Expert. Opin. Drug Metab. Toxicol. 2009, 5, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Foster, G.R.; Rockstroh, J.K.; Zeuzem, S.; Zoulim, F.; Houghton, M. The way forward in HCV treatment--finding the right path. Nat. Rev. Drug Discov. 2007, 6, 991–1000. [Google Scholar] [CrossRef]
- van der Valk, M.; Reiss, P.; van Leth, F.C.; Ackermans, M.T.; Endert, E.; Romijn, J.A.; Heijligenberg, R.; Sauerwein, H. Highly active antiretroviral therapy-induced lipodystrophy has minor effects on human immunodeficiency virus-induced changes in lipolysis, but normalizes resting energy expenditure. J. Clin. Endocrinol. Metab. 2002, 87, 5066–5071. [Google Scholar] [CrossRef] [PubMed]
- McHutchison, J.G.; Everson, G.T.; Gordon, S.C.; Jacobson, I.M.; Sulkowski, M.; Kauffman, R.; McNair, L.; Alam, J.; Muir, A.J. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N. Engl. J. Med. 2009, 360, 1827–1838. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. Molecular biology of flaviviruses. Adv. Virus Res. 2003, 59, 23–61. [Google Scholar] [PubMed]
- Fletcher, N.F.; Yang, J.P.; Farquhar, M.J.; Hu, K.; Davis, C.; He, Q.; Dowd, K.; Ray, S.C.; Krieger, S.E.; Neyts, J.; Baumert, T.F.; Balfe, P.; McKeating, J.A.; Wong-Staal, F. Hepatitis C virus infection of neuroepithelioma cell lines . Gastroenterology 2010. [Google Scholar]
- Deleersnyder, V.; Pillez, A.; Wychowski, C.; Blight, K.; Xu, J.; Hahn, Y.S.; Rice, C.M.; Dubuisson, J. Formation of native hepatitis C virus glycoprotein complexes. J. Virol. 1997, 71, 697–704. [Google Scholar] [PubMed]
- Patel, A.H.; Wood, J.; Penin, F.; Dubuisson, J.; McKeating, J.A. Construction and characterization of chimeric hepatitis C virus E2 glycoproteins: analysis of regions critical for glycoprotein aggregation and CD81 binding. J. Gen. Virol. 2000, 81, 2873–2883. [Google Scholar] [PubMed]
- Cocquerel, L.; Wychowski, C.; Minner, F.; Penin, F.; Dubuisson, J. Charged residues in the transmembrane domains of hepatitis C virus glycoproteins play a major role in the processing, subcellular localization, and assembly of these envelope proteins. J. Virol. 2000, 74, 3623–3633. [Google Scholar] [CrossRef] [PubMed]
- Brazzoli, M.; Helenius, A.; Foung, S.K.; Houghton, M.; Abrignani, S.; Merola, M. Folding and dimerization of hepatitis C virus E1 and E2 glycoproteins in stably transfected CHO cells. Virology 2005, 332, 438–453. [Google Scholar] [CrossRef] [PubMed]
- Krey, T.; d'Alayer, J.; Kikuti, C.M.; Saulnier, A.; Damier-Piolle, L.; Petitpas, I.; Johansson, D.X.; Tawar, R. G.; Baron, B.; Robert, B.; England, P.; Persson, M.A.; Martin, A.; Rey, F.A. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog. 2010, 6, e1000762. [Google Scholar] [CrossRef] [PubMed]
- Farci, P.; Shimoda, A.; Wong, D.; Cabezon, T.; De Gioannis, D.; Strazzera, A.; Shimizu, Y.; Shapiro, M.; Alter, H.J.; Purcell, R.H. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 15394–15399. [Google Scholar] [CrossRef] [PubMed]
- Forns, X.; Thimme, R.; Govindarajan, S.; Emerson, S.U.; Purcell, R.H.; Chisari, F.V.; Bukh, J. Hepatitis C virus lacking the hypervariable region 1 of the second envelope protein is infectious and causes acute resolving or persistent infection in chimpanzees. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 13318–13323. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J. L.; Sodoyer, M.; Pol, S.; Brechot, C.; Paranhos-Baccala, G.; Lotteau, V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 2002, 76, 6919–6928. [Google Scholar] [CrossRef] [PubMed]
- Søren, U.; Nielsen, M.F.B.; Alastair, D.; Burt, C.M.; Pumeechockchai, W.; Toms, G.L. Association between Hepatitis C Virus and Very-Low-Density Lipoprotein (VLDL)/LDL Analyzed in Iodixanol Density Gradients J. Virol. 2006, 80, 2418–2428. [Google Scholar] [CrossRef]
- Bartosch, B.; Verney, G.; Dreux, M.; Donot, P.; Morice, Y.; Penin, F.; Pawlotsky, J.M.; Lavillette, D.; Cosset, F.L. An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J. Virol. 2005, 79, 8217–8229. [Google Scholar] [CrossRef] [PubMed]
- Voisset, C.; Op de Beeck, A.; Horellou, P.; Dreux, M.; Gustot, T.; Duverlie, G.; Cosset, F.L.; Vu-Dac, N.; Dubuisson, J. High-density lipoproteins reduce the neutralizing effect of hepatitis C virus (HCV)-infected patient antibodies by promoting HCV entry. J. Gen. Virol. 2006, 87, 2577–2581. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. Unravelling hepatitis C virus replication from genome to function. Nature 2005, 436, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Schnober, E.K.; Zhang, F.; Linhardt, R.J.; Depla, E.; Boson, B.; Cosset, F.L.; Patel, A.H.; Blum, H.E.; Baumert, T.F. Viral and cellular determinants of the hepatitis C virus envelope-heparan sulfate interaction. J. Virol. 2006, 80, 10579–10590. [Google Scholar] [CrossRef] [PubMed]
- Koutsoudakis, G.; Kaul, A.; Steinmann, E.; Kallis, S.; Lohmann, V.; Pietschmann, T.; Bartenschlager, R. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 2006, 80, 5308–5320. [Google Scholar] [CrossRef] [PubMed]
- Molina, S.; Castet, V.; Fournier-Wirth, C.; Pichard-Garcia, L.; Avner, R.; Harats, D.; Roitelman, J.; Barbaras, R.; Graber, P.; Ghersa, P.; Smolarsky, M.; Funaro, A.; Malavasi, F.; Larrey, D.; Coste, J.; Fabre, J.M.; Sa-Cunha, A.; Maurel, P. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J. Hepatol. 2007, 46, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.M.; Huang, H.; Ye, J.; Gale Jr., M. Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology 2009, 394, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Rigotti, A.; Miettinen, H.E.; Krieger, M. The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr. Rev. 2003, 24, 357–387. [Google Scholar] [CrossRef] [PubMed]
- Catanese, M.T.; Ansuini, H.; Graziani, R.; Huby, T.; Moreau, M.; Ball, J.K.; Paonessa, G.; Rice, C.M.; Cortese, R.; Vitelli, A.; Nicosia, A. Role of scavenger receptor class B type I in hepatitis C virus entry: kinetics and molecular determinants. J. Virol. 2010, 84, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Dao Thi, V.L.; Fresquet, J.; Guerin, M.; Julia, Z.; Verney, G.; Durantel, D.; Zoulim, F.; Lavillette, D.; Cosset, F.L.; Bartosch, B. Receptor complementation and mutagenesis reveal SR-BI as an essential HCV entry factor and functionally imply its intra- and extra-cellular domains. PLoS Pathog. 2009, 5, e1000310. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Vitelli, A.; Granier, C.; Goujon, C.; Dubuisson, J.; Pascale, S.; Scarselli, E.; Cortese, R.; Nicosia, A.; Cosset, F.L. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 2003, 278, 41624–41630. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.; Shoham, T. The tetraspanin web modulates immune-signalling complexes. Nat. Rev. Immunol. 2005, 5, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; Abrignani, S. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Randall, G.; Higginbottom, A.; Monk, P.; Rice, C.M.; McKeating, J.A. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J. Virol. 2004, 78, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Cormier, E.G.; Durso, R.J.; Tsamis, F.; Boussemart, L.; Manix, C.; Olson, W.C.; Gardner, J.P.; Dragic, T. L-SIGN (CD209L) and DC-SIGN (CD209) mediate transinfection of liver cells by hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 14067–14072. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, D.; Date, T.; Morikawa, K.; Murayama, A.; Miyamoto, M.; Kaga, M.; Barth, H.; Baumert, T.F.; Dubuisson, J.; Wakita, T. CD81 expression is important for the permissiveness of Huh7 cell clones for heterogeneous hepatitis C virus infection. J. Virol. 2007, 81, 5036–5045. [Google Scholar] [CrossRef] [PubMed]
- Cormier, E.G.; Tsamis, F.; Kajumo, F.; Durso, R.J.; Gardner, J.P.; Dragic, T. CD81 is an entry coreceptor for hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 7270–7274. [Google Scholar] [CrossRef] [PubMed]
- McKeating, J.A.; Zhang, L.Q.; Logvinoff, C.; Flint, M.; Zhang, J.; Yu, J.; Butera, D.; Ho, D.D.; Dustin, L.B.; Rice, C.M.; Balfe, P. Diverse hepatitis C virus glycoproteins mediate viral infection in a CD81-dependent manner. J. Virol. 2004, 78, 8496–8505. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.; Brimacombe, C.; Syder, A.J.; Flores, N.V.; Krieger, S.E.; Timpe, J.M.; Baumert, T.F.; Tellinghuisen, T.L.; Wong-Staal, F.; Balfe, P.; McKeating, J.A. Neutralizing antibody resistant hepatitis C virus transmission . Society of General Microbiology: Edinburgh, UK, 2010. [Google Scholar]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wolk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.J.; Farquhar, M.J.; Mee, C.J.; Davis, C.; Reynolds, G.M.; Jennings, A.; Hu, K.; Yuan, F.; Deng, H.; Hubscher, S.G.; Han, J.H.; Balfe, P.; McKeating, J.A. CD81 and claudin 1 coreceptor association: role in hepatitis C virus entry. J. Virol. 2008, 82, 5007–5020. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, G.M.; Harris, H.J.; Jennings, A.; Hu, K.; Grove, J.; Lalor, P.F.; Adams, D.H.; Balfe, P.; Hubscher, S.G.; McKeating, J.A. Hepatitis C virus receptor expression in normal and diseased liver tissue. Hepatology 2008, 47, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.J.; Davis, C.; Mullins, J.G.; Hu, K.; Goodall, M.; Farquhar, M.J.; Mee, C. J.; McCaffrey, K.; Young, S.; Drummer, H.; Balfe, P.; McKeating, J.A. Claudin association with CD81 defines hepatitis C virus entry . J. Biol. Chem. 2010, M110.104836. [Google Scholar]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; de Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yang, W.; Shen, L.; Turner, J.R.; Coyne, C.B.; Wang, T. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J. Virol. 2009, 83, 2011–2014. [Google Scholar] [CrossRef] [PubMed]
- Benedicto, I.; Molina-Jimenez, F.; Bartosch, B.; Cosset, F.L.; Lavillette, D.; Prieto, J.; Moreno-Otero, R.; Valenzuela-Fernandez, A.; Aldabe, R.; Lopez-Cabrera, M.; Majano, P.L. The tight junction-associated protein occludin is required for a postbinding step in hepatitis C virus entry and infection. J. Virol. 2009, 83, 8012–8020. [Google Scholar] [CrossRef] [PubMed]
- Kohaar, I.; Ploss, A.; Korol, E.; Mu, K.; Schoggins, J.W.; O'Brien, T.R.; Rice, C.M.; Prokunina-Olsson, L. Splicing diversity of human OCLN gene and its biological significance for hepatitis C virus (HCV) entry . J. Virol. 2010, 84, 6987–6994. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, E.; Belouzard, S.; Goueslain, L.; Wakita, T.; Dubuisson, J.; Wychowski, C.; Rouille, Y. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 2006, 80, 6964–6972. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.; Bertaux, C.; Dragic, T. Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J. Virol. 2006, 80, 11571–11578. [Google Scholar] [CrossRef] [PubMed]
- Coller, K.E.; Berger, K.L.; Heaton, N.S.; Cooper, J.D.; Yoon, R.; Randall, G. RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLoS Pathog. 2009, 5, e1000702. [Google Scholar] [CrossRef] [PubMed]
- Tscherne, D.M.; Jones, C.T.; Evans, M.J.; Lindenbach, B.D.; McKeating, J.A.; Rice, C.M. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 2006, 80, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Koutsoudakis, G.; Herrmann, E.; Kallis, S.; Bartenschlager, R.; Pietschmann, T. The level of CD81 cell surface expression is a key determinant for productive entry of hepatitis C virus into host cells. J. Virol. 2007, 81, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.G.; Walker, C.M. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature 2005, 436, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Law, M.; Maruyama, T.; Lewis, J.; Giang, E.; Tarr, A.W.; Stamataki, Z.; Gastaminza, P.; Chisari, F.V.; Jones, I.M.; Fox, R.I.; Ball, J.K.; McKeating, J.A.; Kneteman, N.M.; Burton, D.R. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 2008, 14, 25–27. [Google Scholar] [CrossRef] [PubMed]
- von Hahn, T.; Yoon, J.C.; Alter, H.; Rice, C.M.; Rehermann, B.; Balfe, P.; McKeating, J.A. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology 2007, 132, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Shilagard, T.; Xiao, S.Y.; Snyder, N.; Lau, D.; Cicalese, L.; Weiss, H.; Vargas, G.; Lemon, S.M. Visualizing hepatitis C virus infections in human liver by two-photon microscopy. Gastroenterology 2009, 137, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Durantel, D. Celgosivir, an alpha-glucosidase I inhibitor for the potential treatment of HCV infection. Curr. Opin. Investig. Drugs 2009, 10, 860–870. [Google Scholar] [PubMed]
- Boriskin, Y.S.; Pecheur, E.I.; Polyak, S.J. Arbidol: a broad-spectrum antiviral that inhibits acute and chronic HCV infection. Virol. J. 2006, 3, 56. [Google Scholar] [CrossRef] [PubMed]
- Helle, F.; Wychowski, C.; Vu-Dac, N.; Gustafson, K.R.; Voisset, C.; Dubuisson, J. Cyanovirin-N inhibits hepatitis C virus entry by binding to envelope protein glycans. J. Biol. Chem. 2006, 281, 25177–25183. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Hu, Z.; Kato, T.; Dreux, M.; Zhang, Y.Y.; Imamura, M.; Hiraga, N.; Juteau, J.M.; Cosset, F.L.; Chayama, K.; Vaillant, A.; Liang, T.J. Amphipathic DNA polymers inhibit hepatitis C virus infection by blocking viral entry. Gastroenterology 2009, 137, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Barth, H.; Schuster, C.; Baumert, T.F. Hepatitis C virus entry: molecular mechanisms and targets for antiviral therapy. Front. Biosci. 2009, 14, 3274–3285. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, P.; Hesselgesser, J.; Paulson, M.; Vanwolleghem, T.; Desombere, I.; Reiser, H.; Leroux-Roels, G. Anti-CD81 antibodies can prevent a hepatitis C virus infection in vivo. Hepatology 2008, 48, 1761–1768. [Google Scholar] [CrossRef] [PubMed]
- Syder, A.J.; Lee, H.; Zeisel, M.B.; Grove, J.; Soulier, E.; Macdonald, J.; Chow, S.; Chang, J.; Baumert, T.F.; McKeating, J.A.; McKelvy, J.; Wong-Staal, F. Small Molecule Scavenger Receptor BI Antagonists are Potent HCV Entry Inhibitors . J. Hepatol. 2010, in press. [Google Scholar]
- Grove, J.; Nielsen, S.; Zhong, J.; Bassendine, M.F.; Drummer, H.E.; Balfe, P.; McKeating, J.A. Identification of a residue in hepatitis C virus E2 glycoprotein that determines scavenger receptor BI and CD81 receptor dependency and sensitivity to neutralizing antibodies. J. Virol. 2008, 82, 12020–12029. [Google Scholar] [CrossRef] [PubMed]
- Masson, D.; Koseki, M.; Ishibashi, M.; Larson, C.J.; Miller, S.G.; King, B.D.; Tall, A.R. Increased HDL cholesterol and apoA-I in humans and mice treated with a novel SR-BI inhibitor. Arterioscler. Thromb Vasc. Biol. 2009, 29, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- McKelvy, J.; Lee, H.; Syder, A.J.; Chang, J.; MacDonald, J.; Wong-Staal, F. ITX5061, an orally bioavailable, clinical stage compound, targets scavenger receptor BI to inhibit HCV entry. 16th Int. Symp. on Hepatitis C Virus & Related Viruses, Nice, France; 2009. [Google Scholar]
- Luchoomun, J.V.; Villegas, D; Killion, S; Hirst, T; Adelman, S; Collins, H; Hellerstein, M; Turner, S. Differential effects of SR-B1 inhibition on reverse cholesterol transport pathways in CETP and non-CETP species. In American Heart Association Meetings Abstracts; 2010. [Google Scholar]
- Yang, J.P.; Zhou, D.; Wong-Staal, F. Screening of small-molecule compounds as inhibitors of HCV entry. Methods Mol. Biol. 2009, 510, 295–304. [Google Scholar] [PubMed]
- Wong-Staal, F.; Liu, G.; McKelvy, J. HCV Viral Entry Inhibitors. In Antiviral Drugs: Biology, Chemistry, Clinic; Kazmierski, W.M.; John Wiley and Sons: Hoboken, New Jersey, USA; in press.
- Qian, D.C.; Han, A.Q.; de Muys, J.M.; Gauss, C; Provoncha, K; Canfield, M; Paul, D; Mohamed, S; Moorji, S; Fisch, D; Murga, J; Rotshteyn, Y; Maddon, P.J.; Olson, W.C. Preclinical characterization of PRO 206, an orally active HCV entry inhibitor. EASL 44th Annual Meeting, Copenhagen, Denmark; 2009. [Google Scholar]
- de Muys, J. M. Coburn.; Han, A.; Provoncha, K.; Paul, D.; Moorji, S.; Fisch, D.; Murga, J.; Qian, D.C.; Maddon, P.J.; Olson, W.C. Discovery of potent and selective inhibitors of Hepatitis C virus (HCV) entry. 16th Int. Symp. on Hepatitis C Virus & Related Virus, Nice, France; 2009. [Google Scholar]
- Ge, D.; Fellay, J.; Thompson, A.J.; Simon, J.S.; Shianna, K.V.; Urban, T.J.; Heinzen, E.L.; Qiu, P.; Bertelsen, A.H.; Muir, A.J.; Sulkowski, M.; McHutchison, J.G.; Goldstein, D.B. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009, 461, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.J.; Portmann, B.C.; Naoumov, N.V.; Smith, H.M.; Underhill, J.A.; Donaldson, P.T.; Maertens, G.; Williams, R. Long-term outcome of hepatitis C infection after liver transplantation. N. Engl. J. Med. 1996, 334, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Rowe, I.A.; Webb, K.; Gunson, B.K.; Mehta, N.; Haque, S.; Neuberger, J. The impact of disease recurrence on graft survival following liver transplantation: a single centre experience. Transpl. Int. 2008, 21, 459–465. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Wong-Staal, F.; Syder, A.J.; McKelvy, J.F. Targeting HCV Entry For Development of Therapeutics. Viruses 2010, 2, 1718-1733. https://doi.org/10.3390/v2081718
Wong-Staal F, Syder AJ, McKelvy JF. Targeting HCV Entry For Development of Therapeutics. Viruses. 2010; 2(8):1718-1733. https://doi.org/10.3390/v2081718
Chicago/Turabian StyleWong-Staal, Flossie, Andrew J. Syder, and Jeffrey F. McKelvy. 2010. "Targeting HCV Entry For Development of Therapeutics" Viruses 2, no. 8: 1718-1733. https://doi.org/10.3390/v2081718
APA StyleWong-Staal, F., Syder, A. J., & McKelvy, J. F. (2010). Targeting HCV Entry For Development of Therapeutics. Viruses, 2(8), 1718-1733. https://doi.org/10.3390/v2081718