HIV-1 Subtype C Phylodynamics in the Global Epidemic

Abstract
:1. Introduction
2. Results and Discussion
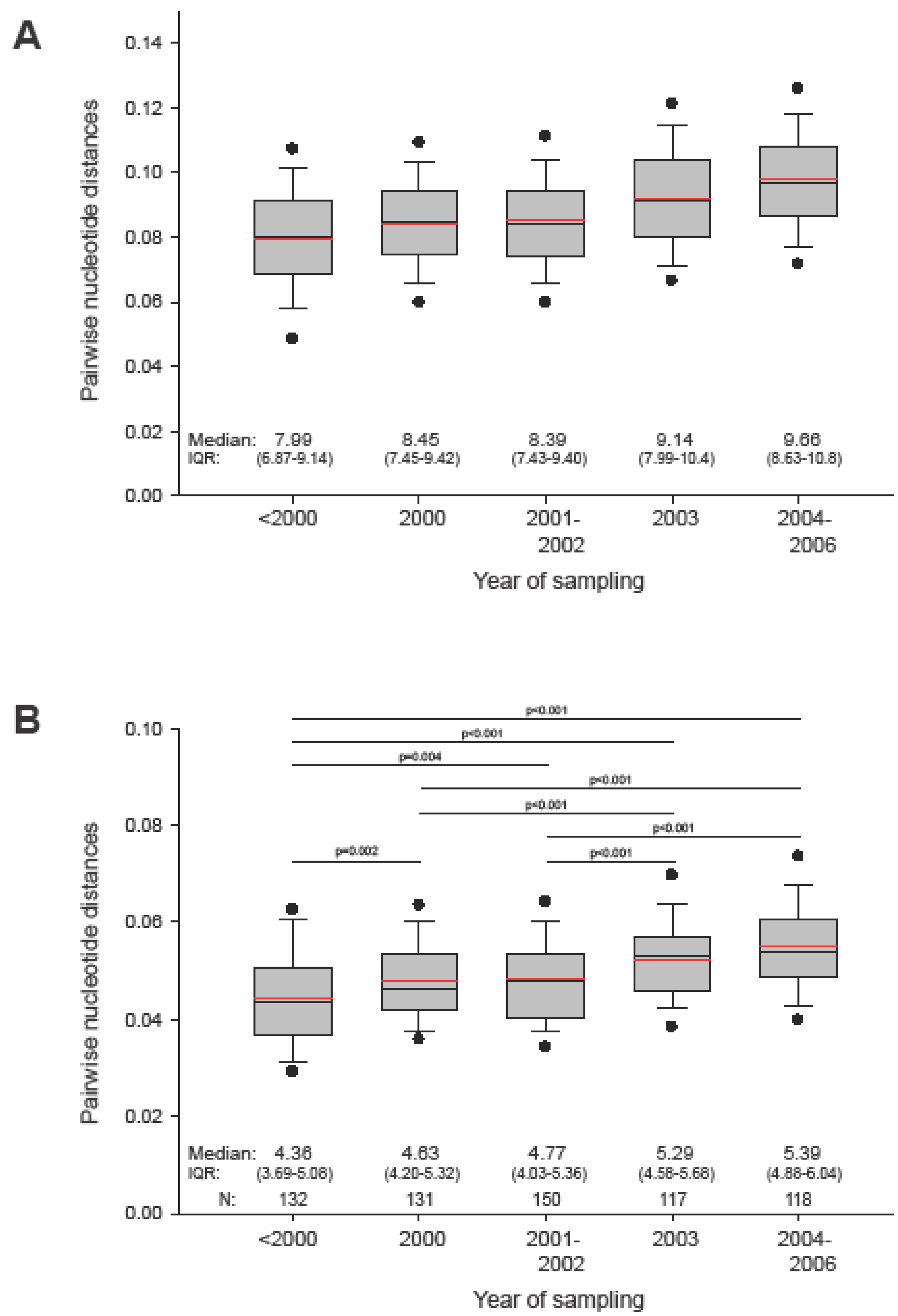
2.1. The in vivo dynamics of gag diversity
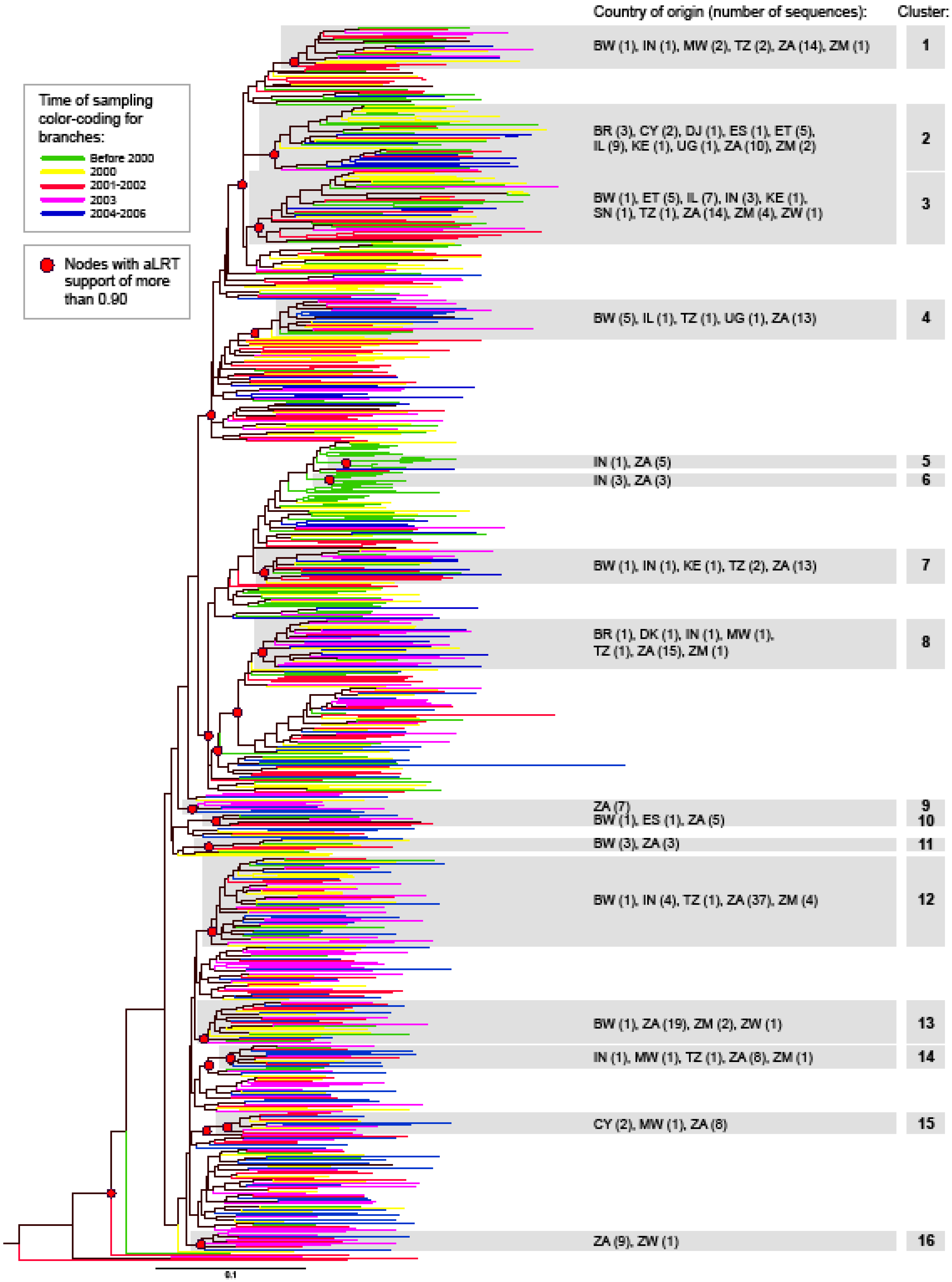

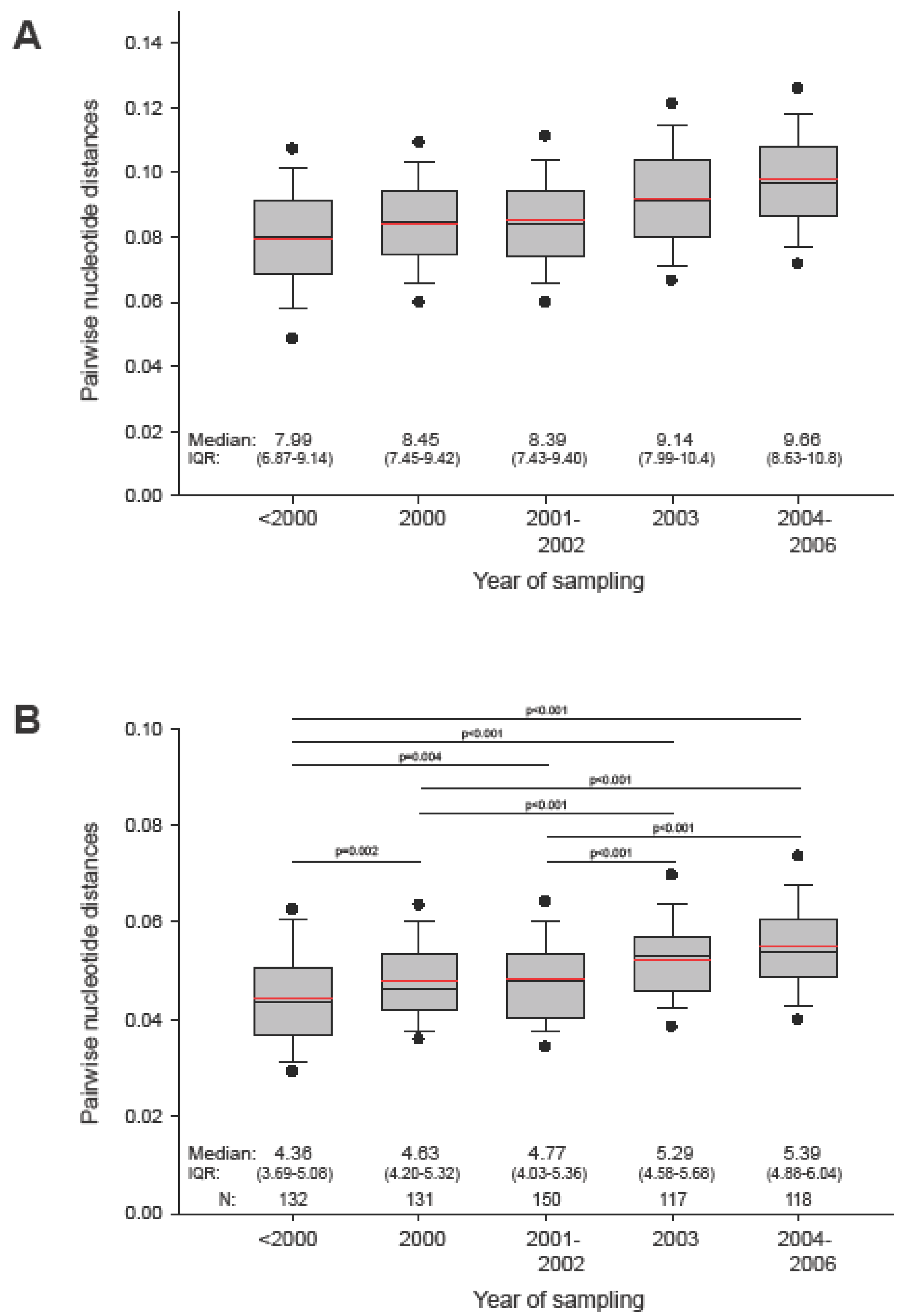
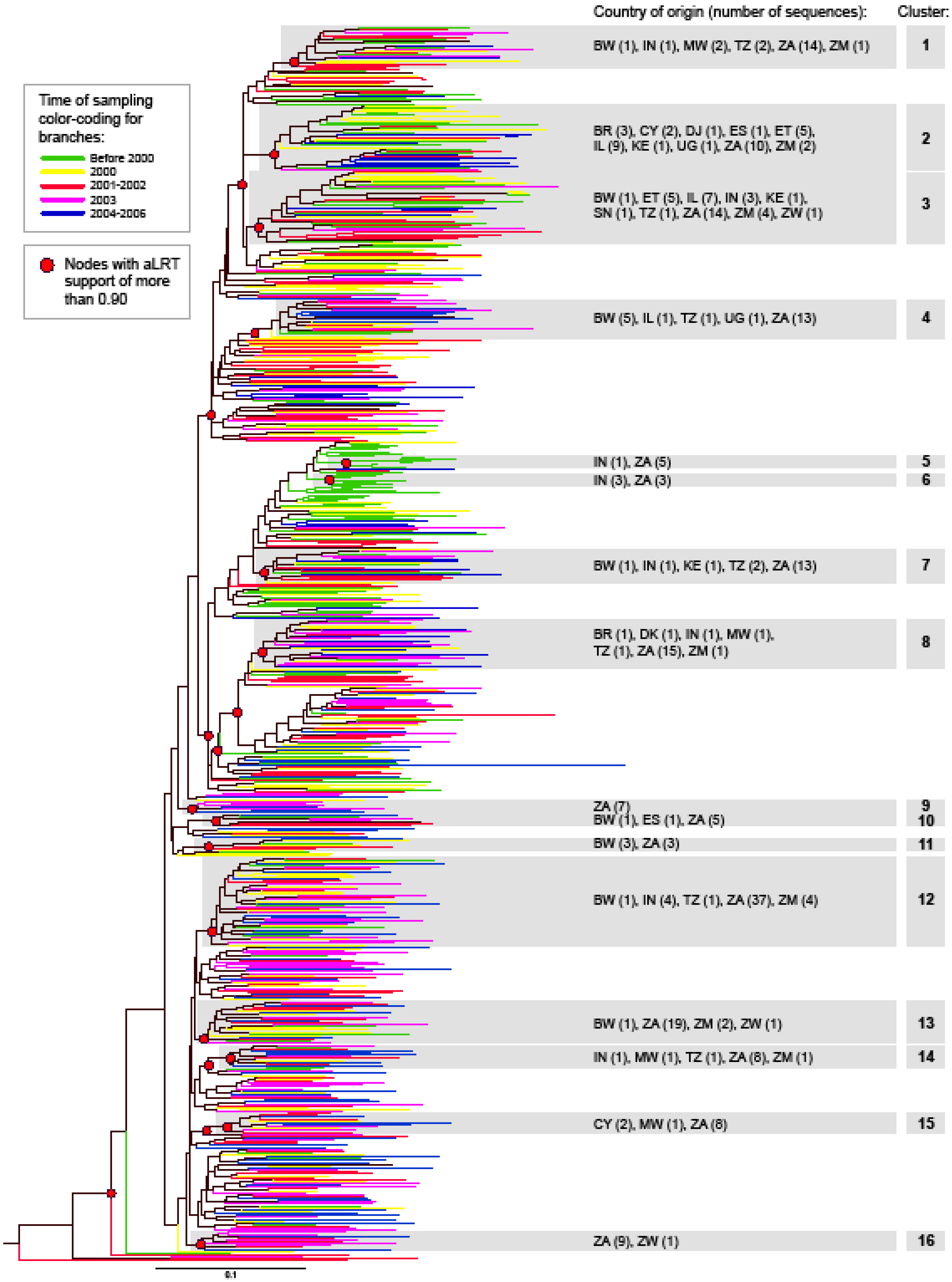
2.2. Amino acid frequency in HIV-1 subtype C Gag

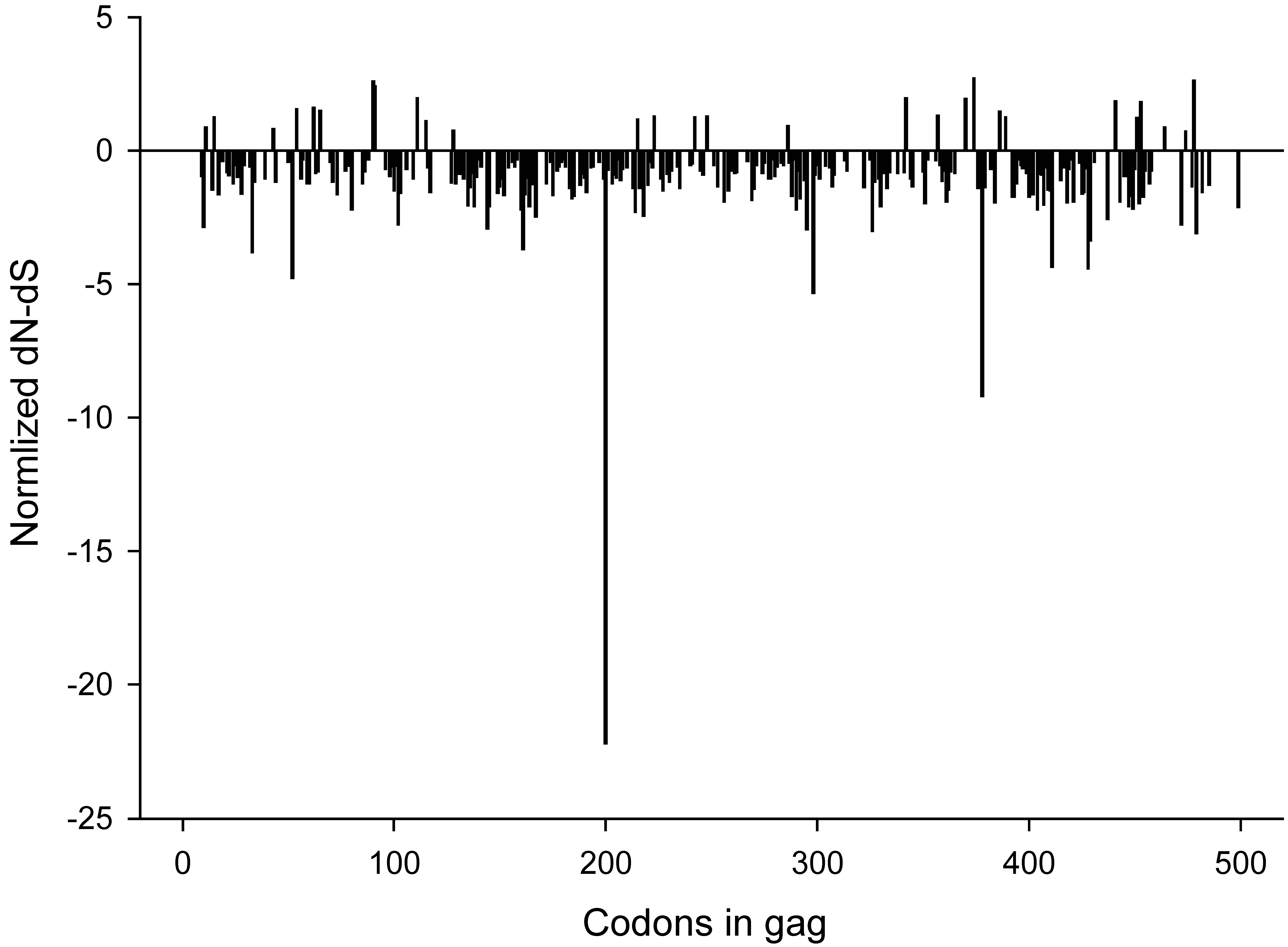
2.3. Synonymous and non-synonymous changes
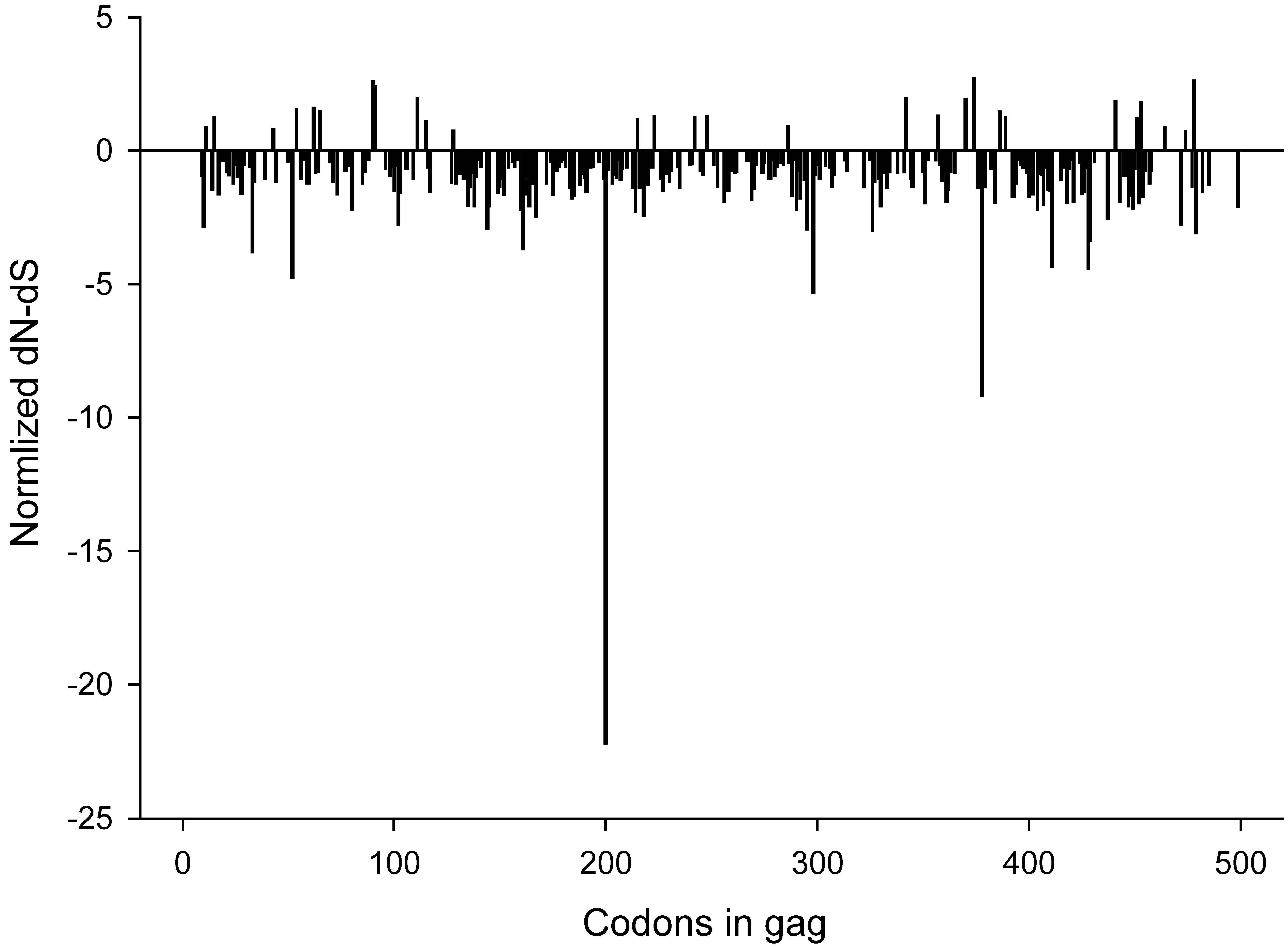
2.4. Changing amino acids and CTL epitopes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gag cleavage product | Amino acid position | Number of associated CTL epitopes | Epitope | HXB2 | HIV-1 Subtype | HLA restriction | |
|---|---|---|---|---|---|---|---|
| start | end | ||||||
| p17 | 11 | 1 | ASILRGGKLDK | 5 | 15 | C | |
| 28 | 10 | RLRPGGKKHY | 20 | 29 | C | A*3002 | |
| RLRPGGKKHYM | 20 | 30 | C | ||||
| RPGGKKHY | 22 | 29 | A, C, D | B42, B7 | |||
| RPGGKKRYM | 22 | 30 | C | B35, Cw*0602 | |||
| RPGGKKKYML | 22 | 31 | A, C, D | B*0702, B*5801, B*8101 | |||
| KRYMIKHLV | 27 | 35 | C | Cw*0602 | |||
| HYMLKHIVW | 28 | 36 | A, C | A*2301 | |||
| 34 | 7 | HYMLKHLVW | 28 | 36 | A, B, C | A*2301, A*2402, A24 | |
| HYMLNHIVW | 28 | 36 | A, C, D | B*0702, B*5801, B*8101 | |||
| HYMLKHLVWAS | 28 | 38 | C | ||||
| HLVWASREL | 33 | 41 | C | Cw*0602, Cw*0804 | |||
| LVWASRELERF | 34 | 44 | C | A*3002, A30, B*5703, B57 | |||
| 76 | 2 | EELRSLYNTV | 73 | 82 | C | B*4006 | |
| RSLYNTVATLY | 76 | 86 | B, C | A*30, A*3002, A30, B57, B58, B63 | |||
| 111 | 0 | ||||||
| 119 | 0 | ||||||
| p24 | 342 | 4 | RALGPGATL | 335 | 343 | A, B, C, D | B7 |
| ALGPGASLEEM | 336 | 346 | C | ||||
| GPGATLEEM | 338 | 346 | A, C, D | B*0702, B*5801, B*8101 | |||
| ATLEEMMTA | 341 | 349 | B, C, CRF01_AE | A*0201, A*0206, A*0220, A*0234, A*0236, A2 | |||
| p2 | 375 | 0 | |||||
| Gag cleavage product | Amino acid position | Number of associated CTL epitopes | Epitope | HXB2 | HIV-1 Subtype | HLA restriction | |
|---|---|---|---|---|---|---|---|
| start | end | ||||||
| p17 | 7 | 1 | ASILRGGKLDK | 5 | 15 | C | |
| 15 | 2 | ELDRWEKIRL | 12 | 21 | B, C | B63 | |
| 18 | 2 | KIRLRPGGKK | 18 | 27 | B, C, multiple | A*0301, A11, A3, B27 | |
| 20 | 4 | RLRPGGKKHY | 20 | 29 | C | A*3002 | |
| 30 | 7 | RLRPGGKKHYM | 20 | 30 | C | ||
| RPGGKKRYM | 22 | 30 | C | B35, Cw*0602 | |||
| 31 | 6 | RPGGKKKYML | 22 | 31 | A, C, D | B*0702, B*5801, B*8101 | |
| KRYMIKHLV | 27 | 35 | C | Cw*0602 | |||
| HYMLKHIVW | 28 | 36 | A, C | A*2301 | |||
| HYMLKHLVW | 28 | 36 | A, B, C | A*2301, A*2402, A24 | |||
| HYMLNHIVW | 28 | 36 | A, C, D | B*0702, B*5801, B*8101 | |||
| HYMLKHLVWAS | 28 | 38 | C | ||||
| 61 | 0 | ||||||
| 62 | 0 | ||||||
| 65 | 0 | ||||||
| 69 | 0 | ||||||
| 79 | 6 | EELRSLYNTV | 73 | 82 | C | B*4006 | |
| RSLYNTVATLY | 76 | 86 | B, C | A*30, A*3002, A30, B57, B58, B63 | |||
| SLYNTVATL | 77 | 85 | A, B, C, CRF02_AG, D, F, G, K | A*02.01, A*0201, A*0202, A*0205, A*0214, A*0220, A*0234, A*0236, A*68, A02, A2, B*1503 | |||
| SLFNTVATLY | 77 | 86 | C | ||||
| LYNTVATLY | 78 | 86 | C | A*2902, A29, B*4403 | |||
| LFNTVATLY | 78 | 86 | C | A*2902 | |||
| p17 | 90 | 1 | YCVHAGIEVRD | 86 | 96 | C | |
| 91 | 1 | ||||||
| 93 | 1 | ||||||
| 95 | 1 | ||||||
| 120 | 0 | ||||||
| p24 | 163 | 8 | VKVIEEKAF | 156 | 164 | C | B*1503 |
| VKVVEEKAF | 156 | 164 | B, C | B*1503 | |||
| IEEKAFSPEV | 159 | 168 | C | B*4006 | |||
| IEEKAFSPEVI | 159 | 169 | C | B*4501 | |||
| EEKAFSPEV | 160 | 168 | A, C | B*4415, B*4501 | |||
| EKAFSPEV | 161 | 168 | C | Cw*0602 | |||
| KAFSPEVIPMF | 162 | 172 | A, B, C, CRF02_AG, G | A*310102, A*6603, B*440302, B*5701, B*5703, B*5801, B57, B58, B63, B8, Cw*040101, Cw*07 | |||
| AFSPEVIPMFT | 163 | 173 | C | ||||
| 215 | 3 | AAEWDRLHPVH | 209 | 219 | C | ||
| AEWDRLHPV | 210 | 218 | B, C | A2, B*04, B*4006, Cw*0602 | |||
| 223 | 4 | RLHPVHAGPIA | 214 | 224 | C | ||
| HPVHAGPIA | 216 | 224 | B, C | B*3910, B07, B35, B7 | |||
| HPVHAGPVA | 216 | 224 | A, B, C, D | B7 | |||
| GPIAPGQM | 221 | 228 | C, D | B35 | |||
| 248 | 3 | TSTLQEQIGW | 240 | 249 | B, C, HIV-2 | A*310102, A*6603, B*440302, B*5701, B*5703, B*58, B*5801, B27, B35, B57, B58, B63, B7, Cw*040101, Cw*07 | |
| TSTLQEQIAW | 240 | 249 | B, C | B*5701, B*5703, B*5801, B57 | |||
| TLQEQIGWM | 242 | 250 | B, C | A*0201, A*0220, A*0234, A*0236, A2 | |||
| 260 | 6 | PPIPVGDIY | 254 | 262 | B, C | B*3501, B*3502, B35 | |
| PPVPVGDIY | 254 | 262 | C | B35 | |||
| PPIPVGEIY | 254 | 262 | A, B, C, D | B35, B53, B7 supertype | |||
| PVGDIYKRWII | 257 | 267 | C | ||||
| GEIYKRWII | 259 | 267 | A, B, C, CRF02_AG, D | A*01, A*6801, B*0801, B*51, B8, Cw*07, Cw15, DQ2, DQ3, DR3, DR4 | |||
| DIYKRWII | 260 | 267 | B, C | B*0801, B8 | |||
| 286 | 0 | ||||||
| 312 | 3 | TLRAEQATQD | 303 | 312 | C | Cw*0304 | |
| RAEQATQDVKN | 305 | 315 | C | ||||
| QATQDVKNW | 308 | 316 | C | B*5301, B*5801, B57 | |||
| 319 | 0 | ||||||
| 335 | 2 | NPDCKTILRAL | 327 | 337 | C | B*3910 | |
| 336 | 1 | RALGPGATL | 335 | 343 | A, B, C, D | B7 | |
| 339 | 3 | ALGPGASLEEM | 336 | 346 | C | ||
| GPGATLEEM | 338 | 346 | A, C, D | B*0702, B*5801, B*8101 | |||
| p2 | 370 | 1 | CLAEAMSQV | 362 | 370 | B, C | A*0201, A*0220, A*0234, A*0236 |
| 371 | 0 | ||||||
| 374 | 0 | ||||||
| p7 | 377 | 0 | |||||
| 380 | 0 | ||||||
| 381 | 0 | ||||||
| 389 | 0 | ||||||
| 401 | 1 | IAKNCRAPRKK | 401 | 411 | C | ||
| p1 | 441 | 1 | FLGKIWPSHK | 433 | 442 | A, B, C, CRF01_AE | A*0201, A*0205, A2 |
| p6 | 451 | 0 | |||||
| 467 | 0 | ||||||
| 472 | 0 | ||||||
| 490 | 1 | PLTSLKSLFGS | 485 | 495 | C | ||
| 498 | 0 | ||||||
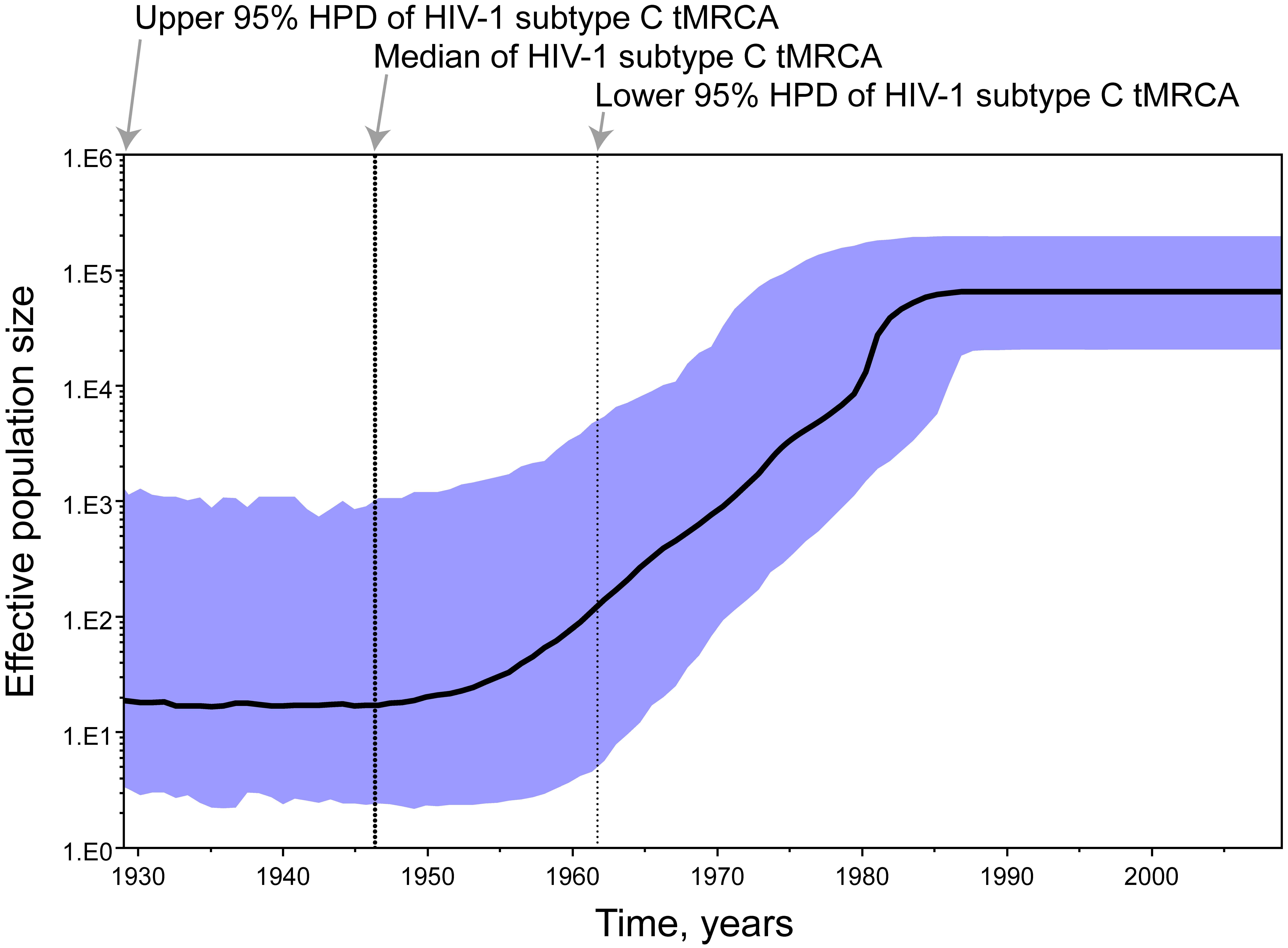
2.5. Dating of HIV-1 subtype C divergence
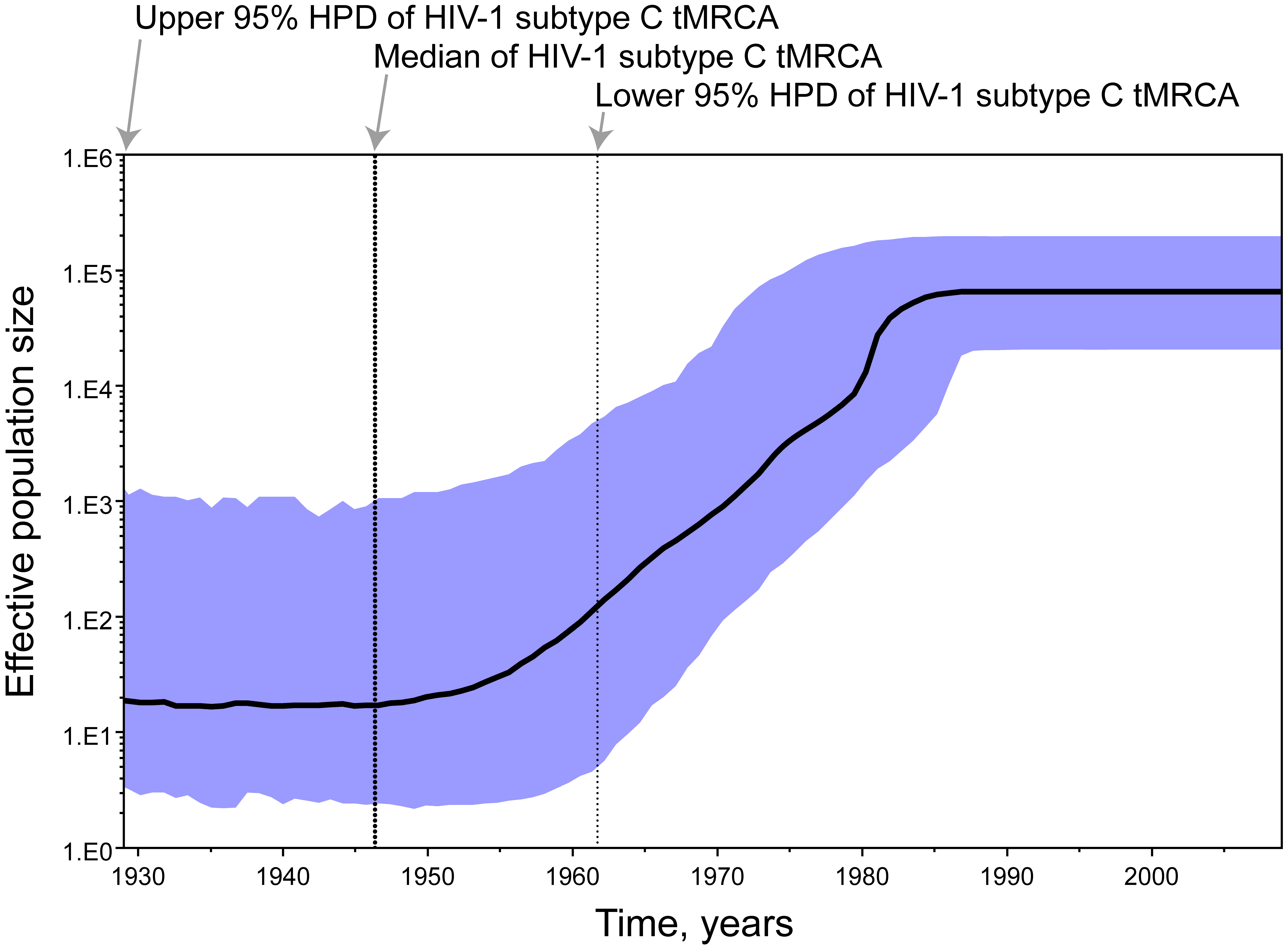
3. Experimental Section
| Country | n | 1986 | 1987 | 1988 | 1989 | 1990 | 1991 | 1992 | 1993 | 1994 | 1995 | 1996 | 1997 | 1998 | 1999 | 2000 | 2001 | 2002 | 2003 | 2004 | 2005 | 2006 | 2007 | 2008 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Botswana | 49 | 10 | 7 | 4 | 25 | 3 | ||||||||||||||||||
| Ethiopia | 12 | 1 | 4 | 3 | 1 | 1 | 2 | |||||||||||||||||
| India | 41 | 4 | 5 | 2 | 23 | 4 | 2 | 1 | ||||||||||||||||
| Israel | 22 | 1 | 4 | 17 | ||||||||||||||||||||
| Malawi | 11 | 1 | 3 | 7 | ||||||||||||||||||||
| South Africa | 431 | 3 | 17 | 27 | 72 | 73 | 22 | 111 | 79 | 24 | 3 | |||||||||||||
| Tanzania | 18 | 2 | 11 | 5 | ||||||||||||||||||||
| Zambia | 30 | 1 | 1 | 6 | 11 | 8 | 3 | |||||||||||||||||
| Others: | 39 | 1 | 2 | 2 | 1 | 5 | 0 | 4 | 6 | 2 | 2 | 5 | 2 | 5 | 1 | 1 | ||||||||
| Total: | 653 | 1 | 0 | 5 | 1 | 2 | 2 | 1 | 5 | 5 | 0 | 14 | 4 | 34 | 58 | 131 | 111 | 39 | 117 | 84 | 26 | 8 | 4 | 1 |
 |
4. Conclusions
Acknowledgments
References
- Edwards, B.H.; Bansal, A.; Sabbaj, S.; Bakari, J.; Mulligan, M.J.; Goepfert, P.A. Magnitude of Functional CD8+ T-Cell Responses to the Gag Protein of Human Immunodeficiency Virus Type 1 Correlates Inversely with Viral Load in Plasma. J. Virol. 2002, 76, 2298–2305. [Google Scholar] [CrossRef] [PubMed]
- Kiepiela, P.; Ngumbela, K.; Thobakgale, C.; Ramduth, D.; Honeyborne, I.; Moodley, E.; Reddy, S.; de Pierres, C.; Mncube, Z.; Mkhwanazi, N.; Bishop, K.; van der Stok, M.; Nair, K.; Khan, N.; Crawford, H.; Payne, R.; Leslie, A.; Prado, J.; Prendergast, A.; Frater, J.; McCarthy, N.; Brander, C.; Learn, G.H.; Nickle, D.; Rousseau, C.; Coovadia, H.; Mullins, J.I.; Heckerman, D.; Walker, B.D.; Goulder, P. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 2007, 13, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, R.; Lucchetti, A.; Galvan, P.; Sanchez, S.; Sanchez, C.; Hernandez, A.; Sanchez, H.; Frahm, N.; Linde, C.H.; Hewitt, H.S.; Hildebrand, W.; Altfeld, M.; Allen, T.M.; Walker, B.D.; Korber, B.T.; Leitner, T.; Sanchez, J.; Brander, C. Relative Dominance of Gag p24-Specific Cytotoxic T Lymphocytes Is Associated with Human Immunodeficiency Virus Control. J. Virol. 2006, 80, 3122–3125. [Google Scholar] [CrossRef] [PubMed]
- Novitsky, V.; Gilbert, P.; Peter, T.; McLane, M.F.; Gaolekwe, S.; Rybak, N.; Thior, I.; Ndung’u, T.; Marlink, R.; Lee, T.H.; Essex, M. Association between virus-specific T-cell responses and plasma viral load in HIV-1 subtype C infection. J. Virol. 2003, 77, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Novitsky, V.A.; Gilbert, P.B.; Shea, K.; McLane, M.F.; Rybak, N.; Klein, I.; Thior, I.; Ndung'u, T.; Lee, T.H.; Essex, M.E. Interactive association of proviral load and IFN-gamma-secreting T cell responses in HIV-1C infection. Virology 2006, 349, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Betts, M.R.; Ambrozak, D.R.; Douek, D.C.; Bonhoeffer, S.; Brenchley, J.M.; Casazza, J.P.; Koup, R.A.; Picker, L.J. Analysis of Total Human Immunodeficiency Virus (HIV)-Specific CD4+ and CD8+ T-Cell Responses: Relationship to Viral Load in Untreated HIV Infection. J. Virol. 2001, 75, 11983–11991. [Google Scholar] [CrossRef] [PubMed]
- Masemola, A.; Mashishi, T.; Khoury, G.; Mohube, P.; Mokgotho, P.; Vardas, E.; Colvin, M.; Zijenah, L.; Katzenstein, D.; Musonda, R.; Allen, S.; Kumwenda, N.; Taha, T.; Gray, G.; McIntyre, J.; Karim, S.A.; Sheppard, H.W.; Gray, C.M. Hierarchical targeting of subtype C human immunodeficiency virus type 1 proteins by CD8+ T cells: correlation with viral load. J. Virol. 2004, 78, 3233–3243. [Google Scholar] [CrossRef] [PubMed]
- Serwanga, J.; Shafer, L.A.; Pimego, E.; Auma, B.; Watera, C.; Rowland, S.; Yirrell, D.; Pala, P.; Grosskurth, H.; Whitworth, J.; Gotch, F.; Kaleebu, P. Host HLA B*allele-associated multi-clade Gag T-cell recognition correlates with slow HIV-1 disease progression in antiretroviral therapy-naive Ugandans. PLoS ONE 2009, 4, e4188. [Google Scholar] [CrossRef] [PubMed]
- Geldmacher, C.; Currier, J.R.; Herrmann, E.; Haule, A.; Kuta, E.; McCutchan, F.; Njovu, L.; Geis, S.; Hoffmann, O.; Maboko, L.; Williamson, C.; Birx, D.; Meyerhans, A.; Cox, J.; Hoelscher, M. CD8 T-cell recognition of multiple epitopes within specific Gag regions is associated with maintenance of a low steady-state viremia in human immunodeficiency virus type 1-seropositive patients. J. Virol. 2007, 81, 2440–2448. [Google Scholar] [CrossRef] [PubMed]
- Boaz, M.J.; Waters, A.; Murad, S.; Easterbrook, P.J.; Vyakarnam, A. Presence of HIV-1 Gag-Specific IFN-gamma(+)IL-2(+) and CD28(+)IL-2(+) CD4 T Cell Responses is Associated with Nonprogression in HIV-1 Infection. J. Immunol. 2002, 169, 6376–6385. [Google Scholar] [PubMed]
- Ramduth, D.; Chetty, P.; Mngquandaniso, N.C.; Nene, N.; Harlow, J.D.; Honeyborne, I.; Ntumba, N.; Gappoo, S.; Henry, C.; Jeena, P.; Addo, M.M.; Altfeld, M.; Brander, C.; Day, C.; Coovadia, H.; Kiepiela, P.; Goulder, P.; Walker, B. Differential immunogenicity of HIV-1 clade C proteins in eliciting CD8+ and CD4+ cell responses. J. Infect. Dis. 2005, 192, 1588–1596. [Google Scholar] [CrossRef] [PubMed]
- Ndongala, M.L.; Peretz, Y.; Boulet, S.; Doroudchi, M.; Yassine-Diab, B.; Boulassel, M.R.; Rouleau, D.; Tremblay, C.; Leblanc, R.; Routy, J.P.; Sekaly, R.P.; Bernard, N.F. HIV Gag p24 specific responses secreting IFN-gamma and/or IL-2 in treatment-naive individuals in acute infection early disease (AIED) are associated with low viral load. Clin. Immunol 2009. [Google Scholar]
- Rolland, M.; Heckerman, D.; Deng, W.; Rousseau, C.M.; Coovadia, H.; Bishop, K.; Goulder, P.J.; Walker, B.D.; Brander, C.; Mullins, J.I. Broad and Gag-biased HIV-1 epitope repertoires are associated with lower viral loads. PLoS ONE 2008, 3, e1424. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Skinner, P.J.; Ha, S.J.; Duan, L.; Mattila, T.L.; Hage, A.; White, C.; Barber, D.L.; O'Mara, L.; Southern, P.J.; Reilly, C.S.; Carlis, J.V.; Miller, C.J.; Ahmed, R.; Haase, A.T. Visualizing antigen-specific and infected cells in situ predicts outcomes in early viral infection. Science 2009, 323, 1726–1729. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, X.; Jin, X.; Liu, Z.; Huang, X.; Cao, Z.; Guo, C.; Dong, T.; Wu, H. Proliferation, but not interleukin 2 production, of Gag-specific CD8+ T cells is associated with low HIV viremia and high CD4 counts in HIV-1-infected Chinese individuals. J. Acquir. Immune. Defic. Syndr. 2009, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Altfeld, M.; Geer, S.C.; Kalife, E.T.; Moore, C.; O'Sullivan, K.M.; DeSouza, I.; Feeney, M.E.; Eldridge, R.L.; Maier, E.L.; Kaufmann, D.E.; Lahaie, M.P.; Reyor, L.; Tanzi, G.; Johnston, M.N.; Brander, C.; Draenert, R.; Rockstroh, J.K.; Jessen, H.; Rosenberg, E.S.; Mallal, S.A.; Walker, B.D. Selective Escape from CD8+ T-Cell Responses Represents a Major Driving Force of Human Immunodeficiency Virus Type 1 (HIV-1) Sequence Diversity and Reveals Constraints on HIV-1 Evolution. J. Virol. 2005, 79, 13239–13249. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Carrington, M.; Kaslow, R.A.; Gao, X.; Rinaldo, C.R.; Jacobson, L.P.; Margolick, J.B.; Phair, J.; O'Brien, S.J.; Detels, R. Association of polymorphisms in human leukocyte antigen class I and transporter associated with antigen processing genes with resistance to human immunodeficiency virus type 1 infection. J. Infect. Dis. 2003, 187, 1404–1410. [Google Scholar] [CrossRef] [PubMed]
- O'Connor, D.H.; McDermott, A.B.; Krebs, K.C.; Dodds, E.J.; Miller, J.E.; Gonzalez, E.J.; Jacoby, T.J.; Yant, L.; Piontkivska, H.; Pantophlet, R.; Burton, D.R.; Rehrauer, W.M.; Wilson, N.; Hughes, A.L.; Watkins, D.I. A Dominant Role for CD8+-T-Lymphocyte Selection in Simian Immunodeficiency Virus Sequence Variation. J. Virol. 2004, 78, 14012–14022. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.A.; Wei, X.; Flower, D.R.; Wong, M.; Michor, F.; Saag, M.S.; Hahn, B.H.; Nowak, M.A.; Shaw, G.M.; Borrow, P. Determinants of Human Immunodeficiency Virus Type 1 Escape from the Primary CD8+ Cytotoxic T Lymphocyte Response. J. Exp. Med. 2004, 200, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- Mullins, J.I.; Rolland, M.; Allen, T.M. Viral evolution and escape during primary human immunodeficiency virus-1 infection: implications for vaccine design. Curr. Opin. HIV AIDS 2008, 3, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Iversen, A.K.; Stewart-Jones, G.; Learn, G.H.; Christie, N.; Sylvester-Hviid, C.; Armitage, A.E.; Kaul, R.; Beattie, T.; Lee, J.K.; Li, Y.; Chotiyarnwong, P.; Dong, T.; Xu, X.; Luscher, M.A.; MacDonald, K.; Ullum, H.; Klarlund-Pedersen, B.; Skinhoj, P.; Fugger, L.; Buus, S.; Mullins, J.I.; Jones, E.Y.; van der Merwe, P.A.; McMichael, A.J. Conflicting selective forces affect T cell receptor contacts in an immunodominant human immunodeficiency virus epitope. Nat. Immunol. 2006, 7, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mullins, J.I.; Mittler, J.E. Waiting times for the appearance of cytotoxic T-lymphocyte escape mutants in chronic HIV-1 infection. Virology 2006, 347, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Novitsky, V.; Smith, U.R.; Gilbert, P.; McLane, M.F.; Chigwedere, P.; Williamson, C.; Ndung'u, T.; Klein, I.; Chang, S.Y.; Peter, T.; Thior, I.; Foley, B.T.; Gaolekwe, S.; Rybak, N.; Gaseitsiwe, S.; Vannberg, F.; Marlink, R.; Lee, T.H.; Essex, M. HIV-1 subtype C molecular phylogeny: consensus sequence for an AIDS vaccine design? J. Virol. 2002, 76, 5435–5451. [Google Scholar] [CrossRef] [PubMed]
- Abebe, A.; Pollakis, G.; Fontanet, A.L.; Fisseha, B.; Tegbaru, B.; Kliphuis, A.; Tesfaye, G.; Negassa, H.; Cornelissen, M.; Goudsmit, J.; Rinke de Wit, T.F. Identification of a genetic subcluster of HIV type 1 subtype C (C') widespread in Ethiopia. AIDS Res. Hum. Retroviruses 2000, 16, 1909–1914. [Google Scholar] [PubMed]
- Ayele, W.; Baar, M.P.; Goudsmit, J.; Kliphuis, A.; Tilahun, T.; Dorigo-Zetsma, W.; Wolday, D.; Abebe, A.; Mengistu, Y.; Pollakis, G. Surveillance technology for HIV-1 subtype C in Ethiopia: an env-based NASBA molecular beacon assay to discriminate between subcluster C and C'. J. Virol. Methods 2005, 130, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Ayele, W.; Pollakis, G.; Abebe, A.; Fisseha, B.; Tegbaru, B.; Tesfaye, G.; Mengistu, Y.; Wolday, D.; van Gemen, B.; Goudsmit, J.; Dorigo-Zetsma, W.; de Baar, M.P. Development of a nucleic acid sequence-based amplification assay that uses gag-based molecular beacons to distinguish between human immunodeficiency virus type 1 subtype C and C' infections in Ethiopia. J. Clin. Microbiol. 2004, 42, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Abebe, A.; Lukashov, V.V.; Rinke De Wit, T.F.; Fisseha, B.; Tegbaru, B.; Kliphuis, A.; Tesfaye, G.; Negassa, H.; Fontanet, A.L.; Goudsmit, J.; Pollakis, G. Timing of the introduction into Ethiopia of subcluster C' of HIV type 1 subtype C. AIDS Res. Hum. Retroviruses 2001, 17, 657–661. [Google Scholar] [PubMed]
- De Baar, M.P.; Abebe, A.; Kliphuis, A.; Tesfaye, G.; Goudsmit, J.; Pollakis, G. HIV type 1 C and C' subclusters based on long terminal repeat sequences in the Ethiopian type 1 subtype C epidemic. AIDS Res. Hum. Retroviruses 2003, 19, 917–922. [Google Scholar] [PubMed]
- Pollakis, G.; Abebe, A.; Kliphuis, A.; De Wit, T.F.; Fisseha, B.; Tegbaru, B.; Tesfaye, G.; Negassa, H.; Mengistu, Y.; Fontanet, A.L.; Cornelissen, M.; Goudsmit, J. Recombination of HIV type 1C (C'/C") in Ethiopia: possible link of EthHIV-1C' to subtype C sequences from the high-prevalence epidemics in India and Southern Africa. AIDS Res. Hum. Retroviruses 2003, 19, 999–1008. [Google Scholar] [PubMed]
- Moulton, V.; Huber, K.T. Split networks. A tool for exploring complex evolutionary relationships in molecular data, 2nd; In The Phylogenetic Handbood. A Practical Approach to Phylogenetic Analysis and Hypothesis TestingLemey, P., Salemi, M., Vandamme, A.M., Eds.; University Press: Cambridge, UK, 2009. [Google Scholar]
- Huson, D.H. SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics 1998, 14, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.R.; Slatkin, M.; Maddison, W.P. Estimation of levels of gene flow from DNA sequence data. Genetics 1992, 132, 583–589. [Google Scholar] [PubMed]
- Slatkin, M. Isolation by distance in equilibrium and non-equilibrium populations. Evolution 1993, 47. [Google Scholar] [CrossRef]
- Hudson, R.R.; Boos, D.D.; Kaplan, N.L. A statistical test for detecting geographic subdivision. Mol. Biol. Evol. 1992, 9, 138–151. [Google Scholar] [PubMed]
- Pond, S.L.; Frost, S.D.; Muse, S.V. HyPhy: hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol.Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Frater, A.J.; Brown, H.; Oxenius, A.; Gunthard, H.F.; Hirschel, B.; Robinson, N.; Leslie, A.J.; Payne, R.; Crawford, H.; Prendergast, A.; Brander, C.; Kiepiela, P.; Walker, B.D.; Goulder, P.J.; McLean, A.; Phillips, R.E. Effective T-cell responses select human immunodeficiency virus mutants and slow disease progression. J. Virol. 2007, 81, 6742–6751. [Google Scholar] [CrossRef] [PubMed]
- Brumme, Z.L.; Brumme, C.J.; Carlson, J.; Streeck, H.; John, M.; Eichbaum, Q.; Block, B.L.; Baker, B.; Kadie, C.; Markowitz, M.; Jessen, H.; Kelleher, A.D.; Rosenberg, E.; Kaldor, J.; Yuki, Y.; Carrington, M.; Allen, T.M.; Mallal, S.; Altfeld, M.; Heckerman, D.; Walker, B.D. Marked epitope- and allele-specific differences in rates of mutation in human immunodeficiency type 1 (HIV-1) Gag, Pol, and Nef cytotoxic T-lymphocyte epitopes in acute/early HIV-1 infection. J. Virol. 2008, 82, 9216–9227. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, Y.; Pfafferott, K.; Frater, J.; Matthews, P.; Payne, R.; Addo, M.; Gatanaga, H.; Fujiwara, M.; Hachiya, A.; Koizumi, H.; Kuse, N.; Oka, S.; Duda, A.; Prendergast, A.; Crawford, H.; Leslie, A.; Brumme, Z.; Brumme, C.; Allen, T.; Brander, C.; Kaslow, R.; Tang, J.; Hunter, E.; Allen, S.; Mulenga, J.; Branch, S.; Roach, T.; John, M.; Mallal, S.; Ogwu, A.; Shapiro, R.; Prado, J.G.; Fidler, S.; Weber, J.; Pybus, O.G.; Klenerman, P.; Ndung'u, T.; Phillips, R.; Heckerman, D.; Harrigan, P.R.; Walker, B.D.; Takiguchi, M.; Goulder, P. Adaptation of HIV-1 to human leukocyte antigen class I. Nature 2009. [Google Scholar]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Ho, S.Y.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol 2006, 4, e88. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Gemmel, M.; Teuwen, D.E.; Haselkorn, T.; Kunstman, K.; Bunce, M.; Muyembe, J.J.; Kabongo, J.M.; Kalengayi, R.M.; Van Marck, E.; Gilbert, M.T.; Wolinsky, S.M. Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960. Nature 2008, 455, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Keele, B.F.; Giorgi, E.E.; Salazar-Gonzalez, J.F.; Decker, J.M.; Pham, K.T.; Salazar, M.G.; Sun, C.; Grayson, T.; Wang, S.; Li, H.; Wei, X.; Jiang, C.; Kirchherr, J.L.; Gao, F.; Anderson, J.A.; Ping, L.-H.; Swanstrom, R.; Tomaras, G.D.; Blattner, W.A.; Goepfert, P.A.; Kilby, J.M.; Saag, M.S.; Delwart, E.L.; Busch, M.P.; Cohen, M.S.; Montefiori, D.C.; Haynes, B.F.; Gaschen, B.; Athreya, G.S.; Lee, H.Y.; Wood, N.; Seoighe, C.; Perelson, A.S.; Bhattacharya, T.; Korber, B.T.; Hahn, B.H.; Shaw, G.M. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. P. Natl. Acad. Sci. USA 2008, 105, 7552–7557. [Google Scholar] [CrossRef]
- Salazar-Gonzalez, J.F.; Salazar, M.G.; Keele, B.F.; Learn, G.H.; Giorgi, E.E.; Li, H.; Decker, J.M.; Wang, S.; Baalwa, J.; Kraus, M.H.; Parrish, N.F.; Shaw, K.S.; Guffey, M.B.; Bar, K.J.; Davis, K.L.; Ochsenbauer-Jambor, C.; Kappes, J.C.; Saag, M.S.; Cohen, M.S.; Mulenga, J.; Derdeyn, C.A.; Allen, S.; Hunter, E.; Markowitz, M.; Hraber, P.; Perelson, A.S.; Bhattacharya, T.; Haynes, B.F.; Korber, B.T.; Hahn, B.H.; Shaw, G.M. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 2009. [Google Scholar]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Weiss, R.E.; Sinsheimer, J.S. Bayesian selection of continuous-time Markov chain evolutionary models. Mol. Biol. Evol. 2001, 18, 1001–1013. [Google Scholar] [PubMed]
- Hahn, B.H.; Shaw, G.M.; De Cock, K.M.; Sharp, P.M. AIDS as a zoonosis: scientific and public health implications. Science 2000, 287, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Hahn, B.H. AIDS: prehistory of HIV-1. Nature 2008, 455, 605–606. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Muldoon, M.; Theiler, J.; Gao, F.; Gupta, R.; Lapedes, A.; Hahn, B.H.; Wolinsky, S.; Bhattacharya, T. Timing the ancestor of the HIV-1 pandemic strains. Science 2000, 288, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.T.; Rambaut, A.; Wlasiuk, G.; Spira, T.J.; Pitchenik, A.E.; Worobey, M. The emergence of HIV/AIDS in the Americas and beyond. Proc. Natl. Acad. Sci. USA 2007, 104, 18566–18570. [Google Scholar] [CrossRef]
- Elena, S.F.; Sanjuán, R. Virus Evolution: Insights from an Experimental Approach. Annu. Rev. Ecol. Evol. Syst. 2007, 38, 27–52. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Posada, D. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4; 2003; Sinauer Associates: Sunderland, Massachusetts, USA; pp. 47–52. [Google Scholar]
- Rambaut, A. FigTree v1.1.2. Available online: http://tree.bio.ed.ac.uk/software/figtree/. [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Poon, A.; Frost, S.D.W. How does SLAC infer selection? Available online: http://www.datamonkey.org/help/SLAC.php. [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Novitsky, V.; Wang, R.; Lagakos, S.; Essex, M. HIV-1 Subtype C Phylodynamics in the Global Epidemic. Viruses 2010, 2, 33-54. https://doi.org/10.3390/v2010033
Novitsky V, Wang R, Lagakos S, Essex M. HIV-1 Subtype C Phylodynamics in the Global Epidemic. Viruses. 2010; 2(1):33-54. https://doi.org/10.3390/v2010033
Chicago/Turabian StyleNovitsky, Vlad, Rui Wang, Stephen Lagakos, and Max Essex. 2010. "HIV-1 Subtype C Phylodynamics in the Global Epidemic" Viruses 2, no. 1: 33-54. https://doi.org/10.3390/v2010033
APA StyleNovitsky, V., Wang, R., Lagakos, S., & Essex, M. (2010). HIV-1 Subtype C Phylodynamics in the Global Epidemic. Viruses, 2(1), 33-54. https://doi.org/10.3390/v2010033