Virus Infection Recognition and Early Innate Responses to Non-Enveloped Viral Vectors

{kind=link}
Abstract
:1. Introduction
2. Molecular Basis for Cellular Recognition of Virus Infection
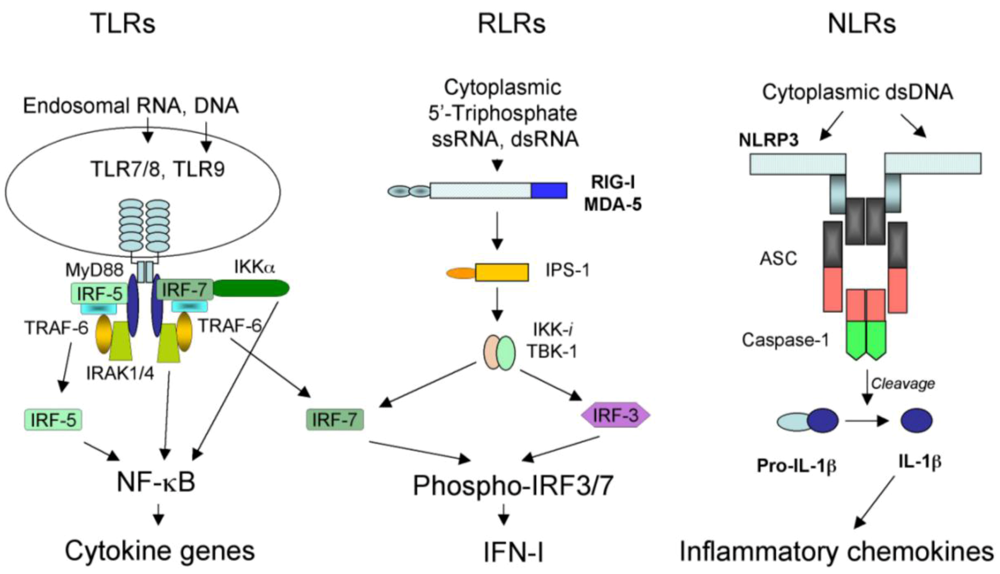
2.1. TLR-dependent recognition of virus infection
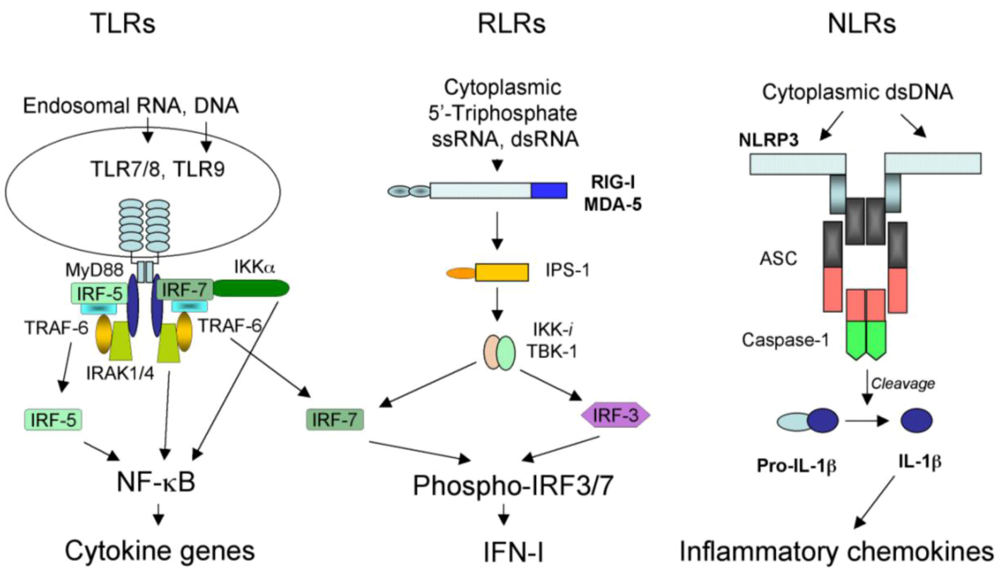
2.2. RLR-dependent recognition of virus infection
2.3. NLR-dependent recognition of virus infection
2.4. Recognition of cytoplasmic dsDNA by DAI and AIM2
3. Specifics of Innate Host Responses to Viral Gene Therapy Vectors
4. Innate Responses to Adenovirus Vectors
4.1. Molecular mediators of early innate immune responses to Ad vectors
4.2. Molecular sensors of Ad infection
5. Innate Responses to Adeno-Associated Virus Vectors
6. Conclusions
Acknowledgments
References
- Brunetti-Pierri, N.; Palmer, D.J.; Beaudet, A.L.; Carey, K.D.; Finegold, M.; Ng, P. Acute toxicity after high-dose systemic injection of helper-dependent adenoviral vectors into nonhuman primates. Hum. Gene Ther. 2004, 15, 35–46. [Google Scholar] [PubMed]
- Raper, S.E.; Chirmule, N.; Lee, F.S.; Wivel, N.A.; Bagg, A.; Gao, G.P.; Wilson, J.M.; Batshaw, M.L. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003, 80, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Raper, S.E.; Yudkoff, M.; Chirmule, N.; Gao, G.P.; Nunes, F.; Haskal, Z.J.; Furth, E.E.; Propert, K.J.; Robinson, M.B.; Magosin, S.; Simoes, H.; Speicher, L.; Hughes, J.; Tazelaar, J.; Wivel, N.A.; Wilson, J.M.; Batshaw, M.L. A pilot study of in vivo liver-directed gene transfer with an adenoviral vector in partial ornithine transcarbamylase deficiency. Hum. Gene Ther. 2002, 13, 163–175. [Google Scholar] [PubMed]
- Richman, D.D.; Whitley, R.; Hayden, F.G. Clinical Virology, 2nd ed. 2002; ASM Press: Washington, D.C., USA. [Google Scholar]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappa B by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; Akira, S. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Trinchieri, G.; Liu, Y.J. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 2004, 5, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. TLR signaling. Semin. Immunol. 2007, 19, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Hoebe, K.; Du, X.; Georgel, P.; Janssen, E.; Tabeta, K.; Kim, S.O.; Goode, J.; Lin, P.; Mann, N.; Mudd, S.; Crozat, K.; Sovath, S.; Han, J.; Beutler, B. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 2003, 424, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Matsumoto, M.; Funami, K.; Akazawa, T.; Seya, T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat. Immunol. 2003, 4, 161–167. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sato, S.; Mori, K.; Hoshino, K.; Takeuchi, O.; Takeda, K.; Akira, S. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J. Immunol. 2002, 169, 6668–6672. [Google Scholar] [PubMed]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; Akira, S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; tenOever, B.R.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Tabeta, K.; Georgel, P.; Janssen, E.; Du, X.; Hoebe, K.; Crozat, K.; Mudd, S.; Shamel, L.; Sovath, S.; Goode, J.; Alexopoulou, L.; Flavell, R. A.; Beutler, B. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. P. Natl. Acad. Sci. USA 2004, 101, 3516–3521. [Google Scholar] [CrossRef]
- Takeuchi, O.; Takeda, K.; Hoshino, K.; Adachi, O.; Ogawa, T.; Akira, S. Cellular responses to bacterial cell wall components are mediated through MyD88-dependent signaling cascades. Int. Immunol. 2000, 12, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Mizutani, T.; Negishi, H.; Shimada, N.; Suzuki, N.; Ohba, Y.; Takaoka, A.; Yeh, W.C.; Taniguchi, T. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 15416–15421. [Google Scholar] [CrossRef]
- Kawai, T.; Sato, S.; Ishii, K.J.; Coban, C.; Hemmi, H.; Yamamoto, M.; Terai, K.; Matsuda, M.; Inoue, J.; Uematsu, S.; Takeuchi, O.; Akira, S. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 2004, 5, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.G.; Huang, X.P.; Yang, Y.P. Innate immune response to adenoviral vectors is mediated by both Toll-like receptor-dependent and -independent pathways. J. Virol. 2007, 81, 3170–3180. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.C.; Gopalkrishnan, R.V.; Wu, Q.; Jankowsky, E.; Pyle, A.M.; Fisher, P.B. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. USA 2002, 99, 637–642. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; Garcia-Sastre, A.; Katze, M.G.; Gale Jr., M. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008, 82, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale Jr., M. ; Akira, S.; Yonehara, S.; Kato, A.; Fujita, T. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [PubMed]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; Endres, S.; Hartmann, G. 5 '-triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Sousa, C.R.E. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5 '-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Schwerd, T.; Hamm, W.; Hellmuth, J.C.; Cui, S.; Wenzel, M.; Hoffmann, F.S.; Michallet, M.C.; Besch, R.; Hopfner, K.P.; Endres, S.; Rothenfusser, S. 5'-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc. Natl. Acad. Sci. USA 2009, 106, 12067–12072. [Google Scholar] [CrossRef]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, R. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.J.; Ea, C.K.; Chen, Z.J.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappa B and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.H.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Michallet, M.C.; Meylan, E.; Ermolaeva, M.A.; Vazquez, J.; Rebsamen, M.; Curran, J.; Poeck, H.; Bscheider, M.; Hartmann, G.; Konig, M.; Kalinke, U.; Pasparakis, M.; Tschopp, J. TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity 2008, 28, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Oganesyan, G.; Saha, S.K.; Guo, B.; He, J.Q.; Shahangian, A.; Zarnegar, B.; Perry, A.; Cheng, G. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 2006, 439, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Melchjorsen, J.; Jensen, S.B.; Malmgaard, L.; Rasmussen, S.B.; Weber, F.; Bowie, A.G.; Matikainen, S.; Paludan, S.R. Activation of innate defense against a paramyxovirus is mediated by RIG-I and TLR7 and TLR8 in a cell-type-specific manner. J. Virol. 2005, 79, 12944–12951. [Google Scholar] [CrossRef] [PubMed]
- Sumpter Jr., R.; Loo, Y.M.; Foy, E.; Li, K.; Yoneyama, M.; Fujita, T.; Lemon, S.M.; Gale Jr., M. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I . J. Virol. 2005, 79, 2689–2699. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Uematsu, S.; Matsui, K.; Tsujimura, T.; Takeda, K.; Fujita, T.; Takeuchi, O.; Akira, S. Cell type-specific involvement of RIG-I in antiviral response. Immunity 2005, 23, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; Yamaguchi, O.; Otsu, K.; Tsujimura, T.; Koh, C.S.; Reis e Sousa, C.; Matsuura, Y.; Fujita, T.; Akira, S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Gitlin, L.; Barchet, W.; Gilfillan, S.; Cella, M.; Beutler, B.; Flavell, R.A.; Diamond, M.S.; Colonna, M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. U S A 2006, 103, 8459–8464. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Ting, J.P. NLR, the nucleotide-binding domain leucine-rich repeat containing gene family. Curr. Opin. Immunol. 2008, 20, 3–9. [Google Scholar] [PubMed]
- Ting, J.P.Y.; Willingham, S.B.; Bergstralh, D.T. NLRs at the intersection of cell death and immunity. Nat. Rev. Immunol. 2008, 8, 372–379. [Google Scholar] [CrossRef]
- Lich, J.D.; Ting, J.P. CATERPILLER (NLR) family members as positive and negative regulators of inflammatory responses. Proc. Am. Thorac. Soc. 2007, 4, 263–266. [Google Scholar] [CrossRef]
- Sutterwala, F.S.; Ogura, Y.; Flavell, R.A. The inflammasome in pathogen recognition and inflammation. J. Leukocyte Biol. 2007, 82, 259–264. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Kanneganti, T.D.; Franchi, L.; Nunez, G. Caspase-1 inflammasomes in infection and inflammation. J. Leukoc. Biol. 2007, 82, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed]
- Muruve, D.A.; Petrilli, V.; Zaiss, A.K.; White, L.R.; Clark, S.A.; Ross, P.J.; Parks, R.J.; Tschopp, J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008, 452, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.G.; Dash, P.; Aldridge Jr., J.R. ; Ellebedy, A.H.; Reynolds, C.; Funk, A.J.; Martin, W.J.; Lamkanfi, M.; Webby, R.J.; Boyd, K.L.; Doherty, P.C.; Kanneganti, T.D. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1 . Immunity 2009, 30, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; Ohba, Y.; Taniguchi, T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.J.; Coban, C.; Kato, H.; Takahashi, K.; Torii, Y.; Takeshita, F.; Ludwig, H.; Sutter, G.; Suzuki, K.; Hemmi, H.; Sato, S.; Yamamoto, M.; Uematsu, S.; Kawai, T.; Takeuchi, O.; Akira, S. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol. 2006, 7, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.L.; Idris, A.; Dunn, J.A.; Kelly, G.M.; Burnton, C.M.; Hodgson, S.; Hardy, L.L.; Garceau, V.; Sweet, M.J.; Ross, I.L.; Hume, D.A.; Stacey, K.J. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 2009, 323, 1057–1060. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC . Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Burckstummer, T.; Baumann, C.; Bluml, S.; Dixit, E.; Durnberger, G.; Jahn, H.; Planyavsky, M.; Bilban, M.; Colinge, J.; Bennett, K.L.; Superti-Furga, G. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 2009, 10, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Muruve, D.A.; Tschopp, J. Innate immunity: cytoplasmic DNA sensing by the AIM2 inflammasome . Curr. Biol. 2009, 19, R262–R265. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Benureau, Y.; Rijnbrand, R.; Yi, J.; Wang, T.; Warter, L.; Lanford, R.E.; Weinman, S.A.; Lemon, S.M.; Martin, A.; Li, K. GB virus B disrupts RIG-I signaling by NS3/4A-mediated cleavage of the adaptor protein MAVS. J. Virol. 2007, 81, 964–976. [Google Scholar] [CrossRef] [PubMed]
- Baril, M.; Racine, M.E.; Penin, F.; Lamarre, D. MAVS dimer is a crucial signaling component of innate immunity and the target of hepatitis C virus NS3/4A protease. J. Virol. 2009, 83, 1299–1311. [Google Scholar] [CrossRef] [PubMed]
- Hutchin, M.E.; Pickles, R.J.; Yarbrough, W.G. Efficiency of adenovirus-mediated gene transfer to oropharyngeal epithelial cells correlates with cellular differentiation and human coxsackie and adenovirus receptor expression. Hum. Gene Ther. 2000, 11, 2365–2375. [Google Scholar] [PubMed]
- Kobinger, G.P. Simian adenoviral vector expressing Ebola glycoprotein variants for safe establishment of a strong protective immunity . Mol. Ther. 2003, 7, S310. [Google Scholar]
- Tan Y, H.N.; Crystal, R.G. Rapid protective immunity evoked against anthrax lethal toxin following a single intramuscular administration of an adenovirus-based vaccine coding for humanized protective antigen . Mol. Ther. 2003, 7, S311. [Google Scholar]
- Morral, N.; O'Neal, W.K.; Rice, K.; Leland, M.M.; Piedra, P.A.; Aguilar-Cordova, E.; Carey, K.D.; Beaudet, A.L.; Langston, C. Lethal toxicity, severe endothelial injury, and a threshold effect with high doses of an adenoviral vector in baboons. Hum. Gene. Ther. 2002, 13, 143–154. [Google Scholar] [PubMed]
- Lozier, J.N.; Csako, G.; Mondoro, T.H.; Krizek, D.M.; Metzger, M.E.; Costello, R.; Vostal, J.G.; Rick, M.E.; Donahue, R.E.; Morgan, R.A. Toxicity of a first-generation adenoviral vector in rhesus macaques. Hum. Gene Ther. 2002, 13, 113–124. [Google Scholar] [PubMed]
- Alemany, R.; Suzuki, K.; Curiel, D.T. Blood clearance rates of adenovirus type 5 in mice. J. Gen. Virol. 2000, 81, 2605–2609. [Google Scholar] [PubMed]
- Kirn, D. Clinical research results with dl1520 (Onyx-015), a replication-selective adenovirus for the treatment of cancer: what have we learned? Gene Ther. 2001, 8, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D.; Martuza, R.L.; Zwiebel, J. Replication-selective virotherapy for cancer: Biological principles, risk management and future directions. Nat. Med. 2001, 7, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Tao, N.; Gao, G.P.; Parr, M.; Johnston, J.; Baradet, T.; Wilson, J.M.; Barsoum, J.; Fawell, S.E. Sequestration of adenoviral vector by Kupffer cells leads to a nonlinear dose response of transduction in liver. Mol. Ther. 2001, 3, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Worgall, S.; Wolff, G.; Falck-Pedersen, E.; Crystal, R.G. Innate immune mechanisms dominate elimination of adenoviral vectors following in vivo administration. Hum. Gene Ther. 1997, 8, 37–44. [Google Scholar] [CrossRef]
- Wolff, G.; Worgall, S.; van Rooijen, N.; Song, W.R.; Harvey, B.G.; Crystal, R.G. Enhancement of in vivo adenovirus-mediated gene transfer and expression by prior depletion of tissue macrophages in the target organ. J. Virol. 1997, 71, 624–629. [Google Scholar] [PubMed]
- Liu, Q.; Muruve, D.A. Molecular basis of the inflammatory response to adenovirus vectors. Gene Ther. 2003, 10, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Lieber, A.; He, C.Y.; Meuse, L.; Schowalter, D.; Kirillova, I.; Winther, B.; Kay, M.A. The role of Kupffer cell activation and viral gene expression in early liver toxicity after infusion of recombinant adenovirus vectors. J. Virol. 1997, 71, 8798–8807. [Google Scholar] [PubMed]
- Schnell, M.A.; Zhang, Y.; Tazelaar, J.; Gao, G.P.; Yu, Q.C.; Qian, R.; Chen, S.J.; Varnavski, A.N.; LeClair, C.; Raper, S.E.; Wilson, J.M. Activation of innate immunity in nonhuman primates following intraportal administration of adenoviral vectors. Mol. Ther. 2001, 3, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chirmule, N.; Gao, G.P.; Qian, R.; Croyle, M.; Joshi, B.; Tazelaar, J.; Wilson, J.M. Acute cytokine response to systemic adenoviral vectors in mice is mediated by dendritic cells and macrophages. Mol. Ther. 2001, 3, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Muruve, D.A.; Barnes, M.J.; Stillman, I.E.; Libermann, T.A. Adenoviral gene therapy leads to rapid induction of multiple chemokines and acute neutrophil-dependent hepatic injury in vivo. Hum. Gene Ther. 1999, 10, 965–976. [Google Scholar] [PubMed]
- Borgland, S.L.; Bowen, G.P.; Wong, N.C.; Libermann, T.A.; Muruve, D.A. Adenovirus vector-induced expression of the C-X-C chemokine IP-10 is mediated through capsid-dependent activation of NF-kappaB. J. Virol. 2000, 74, 3941–3947. [Google Scholar] [CrossRef] [PubMed]
- Bowen, G.P.; Borgland, S.L.; Lam, M.; Libermann, T.A.; Wong, N.C.; Muruve, D.A. Adenovirus vector-induced inflammation: capsid-dependent induction of the C-C chemokine RANTES requires NF-kappa B. Hum. Gene Ther. 2002, 13, 367–379. [Google Scholar] [PubMed]
- Bhat, N.R.; Fan, F. Adenovirus infection induces microglial activation: involvement of mitogen-activated protein kinase pathways. Brain Res. 2002, 948, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Reid, T.; Galanis, E.; Abbruzzese, J.; Sze, D.; Andrews, J.; Romel, L.; Hatfield, M.; Rubin, J.; Kirn, D. Intra-arterial administration of a replication-selective adenovirus (dl1520) in patients with colorectal carcinoma metastatic to the liver: a phase I trial. Gene Ther. 2001, 8, 1618–1626. [Google Scholar] [CrossRef]
- Crystal, R.G.; Harvey, B.G.; Wisnivesky, J.P.; O'Donoghue, K.A.; Chu, K.W.; Maroni, J.; Muscat, J.C.; Pippo, A.L.; Wright, C.E.; Kaner, R.J.; Leopold, P.L.; Kessler, P.D.; Rasmussen, H.S.; Rosengart, T.K.; Hollmann, C. Analysis of risk factors for local delivery of low- and intermediate-dose adenovirus gene transfer vectors to individuals with a spectrum of comorbid conditions. Hum. Gene Ther. 2002, 13, 65–100. [Google Scholar] [PubMed]
- Ben-Gary, H.; McKinney, R.L.; Rosengart, T.; Lesser, M.L.; Crystal, R.G. Systemic interleukin-6 responses following administration of adenovirus gene transfer vectors to humans by different routes. Mol. Ther. 2002, 6, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Mickelson, C.A. Department of Health and Human Services National Institutes of Health Recombinant DNA Advisory Committee. Minutes of meeting March 8-10, 2000. Hum. Gene Ther. 2000, 11, 2159–2192. [Google Scholar] [PubMed]
- McCoy, R.D.; Davidson, B.L.; Roessler, B.J.; Huffnagle, G.B.; Janich, S.L.; Laing, T.J.; Simon, R.H. Pulmonary inflammation induced by incomplete or inactivated adenoviral particles. Hum. Gene Ther. 1995, 6, 1553–1560. [Google Scholar] [CrossRef]
- Fejer, G.; Drechsel, L.; Liese, J.; Schleicher, U.; Ruzsics, Z.; Imelli, N.; Greber, U.F.; Keck, S.; Hildenbrand, B.; Krug, A.; Bogdan, C.; Freudenberg, M.A. Key Role of Splenic Myeloid DCs in the IFN-alpha beta Response to Adenoviruses In Vivo. Plos Pathog. 2008, 4, e1000208. [Google Scholar] [CrossRef] [PubMed]
- Nociari, M.; Ocheretina, O.; Schoggins, J.W.; Falck-Pedersen, E. Sensing infection by adenovirus: Toll-like receptor-independent viral DNA recognition signals activation of the interferon regulatory factor 3 master regulator. J. Virol. 2007, 81, 4145–4157. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.G.; Huang, X.P.; Yang, Y.P. Type IIFN signaling on both B and CD4 T cells is required for protective antibody response to adenovirus. J. Immunol. 2007, 178, 3505–3510. [Google Scholar] [PubMed]
- Hartman, Z.C.; Kiang, A.; Everett, R.S.; Serra, D.; Yang, X.Y.; Clay, T.M.; Amalfitano, A. Adenovirus infection triggers a rapid, MyD88-regulated transcriptome response critical to acute-phase and adaptive immune responses in vivo. J. Virol. 2007, 81, 1796–1812. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.C.; Black, E.P.; Amalfitano, A. Adenoviral infection induces a multi-faceted innate cellular immune response that is mediated by the toll-like receptor pathway in A549 cells. Virology 2007, 358, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Iacobelli-Martinez, M.; Nemerow, G.R. Preferential activation of toll-like receptor nine by CD46-utilizing adenoviruses. J. Virol. 2007, 81, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Basner-Tschakarjan, E.; Gaffal, E.; O'Keeffe, M.; Tormo, D.; Limmer, A.; Wagner, H.; Hochrein, H.; Tuting, T. Adenovirus efficiently transduces plasmacytoid dendritic cells resulting in TLR9-dependent maturation and IFN-alpha production. J. Gene Med. 2006, 8, 1300–1306. [Google Scholar] [CrossRef]
- Shayakhmetov, D.M.; Li, Z.Y.; Ternovoi, V.; Gaggar, A.; Gharwan, H.; Lieber, A. The interaction between the fiber knob domain and the cellular attachment receptor determines the intracellular trafficking route of adenoviruses. J. Virol. 2003, 77, 3712–3723. [Google Scholar] [CrossRef] [PubMed]
- Cerullo, V.; Seiler, M.P.; Mane, V.; Brunetti-Pierri, N.; Clarke, C.; Bertin, T.K.; Rodgers, J.R.; Lee, B. Toll-like receptor 9 triggers an innate immune response to helper-dependent adenoviral vectors. Mol. Ther. 2007, 15, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, N.C.; Miao, E.A.; Iwakura, Y.; Murali-Krishna, K.; Aderem, A.; Flavell, R.A.; Papayannopoulou, T.; Shayakhmetov, D.M. Virus binding to a plasma membrane receptor triggers interleukin-1 alpha-mediated proinflammatory macrophage response in vivo. Immunity 2009, 31, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Wickham, T.J.; Mathias, P.; Cheresh, D.A.; Nemerow, G.R. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 1993, 73, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F.; Willetts, M.; Webster, P.; Helenius, A. Stepwise dismantling of adenovirus 2 during entry into cells. Cell 1993, 75, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Kono, H.; Golenbock, D.; Reed, G.; Akira, S.; Rock, K.L. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med. 2007, 13, 851–856. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, A.P.; Fawcett, P.; Nakai, H.; McCaffrey, R.L.; Ehrhardt, A.; Pham, T.T.; Pandey, K.; Xu, H.; Feuss, S.; Storm, T.A.; Kay, M.A. The host response to adenovirus, helper-dependent adenovirus, and adeno-associated virus in mouse liver. Mol. Ther. 2008, 16, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Hasbrouck, N.C.; High, K.A. AAV-mediated gene transfer for the treatment of hemophilia B: problems and prospects. Gene Ther. 2008, 15, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K.A. Immune responses to AAV in clinical trials. Curr. Gene Ther. 2007, 7, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, A.K.; Liu, Q.; Bowen, G.P.; Wong, N.C.; Bartlett, J.S.; Muruve, D.A. Differential activation of innate immune responses by adenovirus and adeno-associated virus vectors. J. Virol. 2002, 76, 4580–4590. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Huang, X.; Yang, Y. The TLR9-MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J. Clin. Invest. 2009, 119, 2388–2398. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Shayakhmetov, D.M. Virus Infection Recognition and Early Innate Responses to Non-Enveloped Viral Vectors. Viruses 2010, 2, 244-261. https://doi.org/10.3390/v2010244
Shayakhmetov DM. Virus Infection Recognition and Early Innate Responses to Non-Enveloped Viral Vectors. Viruses. 2010; 2(1):244-261. https://doi.org/10.3390/v2010244
Chicago/Turabian StyleShayakhmetov, Dmitry M. 2010. "Virus Infection Recognition and Early Innate Responses to Non-Enveloped Viral Vectors" Viruses 2, no. 1: 244-261. https://doi.org/10.3390/v2010244
APA StyleShayakhmetov, D. M. (2010). Virus Infection Recognition and Early Innate Responses to Non-Enveloped Viral Vectors. Viruses, 2(1), 244-261. https://doi.org/10.3390/v2010244