1. Introduction
The hepatitis C virus (HCV) is an enveloped RNA virus classified within the Hepacivirus genus of the
Flaviviridae family [
1]. Its genome is a positive-sense RNA molecule, approximately 9.6 kilobases in length, which is translated into a single polyprotein. This polyprotein is subsequently cleaved into three structural proteins (core protein, glycoproteins E1 and E2) and seven non-structural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A and NS5B) [
2].
It is estimated that more than 50 million people globally are suffering from hepatitis C infection. On average, 30% of infected individuals experience the acute form of viral hepatitis, which in most cases can be cleared spontaneously. In the remaining 70% of the cases, HCV infection leads to chronic viral hepatitis C (CHC). CHC is a significant health burden worldwide, with serious implications such as liver cirrhosis, hepatocellular carcinoma, increased risk of type 2 diabetes, or hypercholesterolemia [
3,
4,
5]. Current therapeutic approaches against HCV infection involve the use of third-generation direct-acting antivirals (DAAs), which have replaced the earlier interferon-alpha (IFN-α) and Ribavirin-based treatments [
6]. Nevertheless, due to economic inequity in low-income countries, the accessibility to DAAs often remains poor alongside social awareness [
7]. Therefore, the development of an effective HCV vaccine remains an ongoing challenge [
8].
Infection with the hepatitis C virus (HCV) was found to have significant effects on lipid metabolism and the rearrangement of intracellular membranes. Specifically, HCV manipulates the host cell’s endoplasmic reticulum (ER)-derived membranes to facilitate its own replication. HCV replication occurs in the replicase complexes on the cytoplasmic face of the ER [
9]. They consist of viral RNA, non-structural proteins and host cell factors. The precise localisation for the replication to happen is within specialised structures known as double-membrane vesicles (DMVs). Originating from the ER, these DMVs provide a suitable environment for viral RNA replication [
10]. HCV assembly occurs in close proximity to the ER, in regions known as detergent-resistant membranes (DRMs) or mitochondrial-associated ER membranes (MAMs) [
11]. These membrane regions have a distinctive lipid composition, being highly enriched in cholesterol and sphingolipids. A crucial link between HCV replication and assembly is established through cytosolic lipid droplets (cLDs) [
9]. cLDs serve as a connecting point between viral replication sites in DMVs and assembly sites in DRMs or MAMs. They play a pivotal role in facilitating the transfer of viral components between these compartments, supporting the efficient assembly of infectious HCV particles [
12]. Moreover, HCV virions are closely associated with various lipoproteins and apolipoproteins, resulting in the creation of lipoviroparticles (LVPs). Virion assembly, infectivity and entry strongly depend on the virus association with lipoproteins. The manipulation of lipid metabolism and the rearrangement of intracellular membranes by HCV are critical for its successful replication and assembly within the host cell [
13].
Hepatitis C virus infection is known to induce oxidative stress and to disrupt redox homeostasis, leading to cellular damage, altered cellular metabolism and impaired liver regeneration [
14,
15]. A key protective mechanism against oxidative stress is the Nrf2/Keap1 signalling pathway. Nuclear factor erythroid 2 related factor-2 (Nrf2) is a transcription factor that plays a crucial role in activating the expression of cytoprotective genes [
16]. Under normal physiological conditions, Nrf2 is bound to its inhibitor Kelch-like ECH-associated protein 1 (Keap1). However, upon activation, the Nrf2-Keap1 complex dissociates, allowing translocation of Nrf2 into the nucleus. Inside the nucleus, Nrf2 forms a heterodimer with small Maf proteins (sMafs) and binds to specific sequences known as antioxidant response elements (AREs) or electrophile response elements (EpREs). This binding activates the expression of cytoprotective genes [
17,
18,
19,
20]. In the context of HCV replication, impaired Nrf2/ARE signalling has been observed in the infected cells. This impairment is attributed to the withdrawal of sMaf proteins from the nucleus as they bind to NS3, an integral component of the HCV replicase complex, on the cytoplasmic face of the ER. Upon binding to NS3 at the replicase complexes, sMafs are withdrawn from the nucleus to the surface of the ER. The dislocation of sMaf from the nucleus to the surface of the ER has two consequences: (i) it leads to a lack of sMaf proteins in the nucleus, and (ii) it prevents Nrf2 from entry into the nucleus, as the released Nrf2 is trapped and sequestered by the ER-localised sMafs. This prevents the Nrf2-mediated induction of cytoprotective ARE-dependent genes. Consequently, the reactive oxygen species (ROS) within HCV-infected cells are not detoxified, and the ROS levels remain elevated [
21]. Apart from its relevance for HCV-associated pathogenesis, the elevated ROS level is crucial for the HCV life cycle by the activation of autophagy. On the one hand, autophagy represents a cellular defence mechanism; on the other hand, it is required for the MVB-dependent release of HCV in the form of exosomes [
19].
Another factor involved in maintaining the cellular redox homeostasis is the nuclear factor erythroid 2 related factor-1 (Nrf1; NFE2L1), a ubiquitously expressed transcription factor, belonging to the Cap′N′Collar family [
22]. In its unprocessed form, full-length Nrf1 is bound to the ER membranes. Upon stimulation, the inactive 120 kDa N-glycoprotein undergoes deglycosylation by N-glycanase 1 (NGLY1) and cleavage by the ubiquitin-directed endoprotease DNA damage inducible 1 homologue 2 (DDI2), resulting in the activation of the p95-Nrf1 isoform [
23,
24]. Subsequently, Nrf1 interacts with the endoplasmic reticulum-associated degradation (ERAD) machinery mediated by the proteasome [
25], leading to its cleavage [
22]. This releases Nrf1 from the ER, allowing its translocation to the nucleus. Upon translocation, Nrf1 undergoes further processing and cleavage, generating multiple truncated isoforms. These isoforms exhibit distinct activities and are responsible for regulating specific subsets of target genes controlled by antioxidant response elements (AREs). The versatile biological functions of the different Nrf1 isoforms rely on their structural domains, harbouring conserved motifs. Notably, Nrf1 contains two transactivation domains: AD1 (acidic domain 1) and AD2 (acidic domain 2). AD1 comprises amino acids 125–298 and functions as the major transactivation domain. AD2 (amino acids 403–455) further enhances Nrf1’s transactivation activity and is of particular significance for the truncated 65 kDa isoform. Moreover, Nrf1 encompasses the Nrf2–ECH homology 1-like region, spanning amino acids 581–730. Within this region, both CNC (Cap‘n’Collar) and bZIP (Basic-region leucine zipper) domains collectively function as the DBD (DNA-binding domain). The different Nrf1 isoforms play crucial roles in maintaining redox and proteasome homeostasis, protecting against oxidative stress and regulating hepatic fatty and amino acid metabolism [
26,
27]. In line with this, the deglycosylated and cleaved 85 kDa form of Nrf1 serves as the primary transcriptional activator, while the shorter fragments ranging from 36 to 25 kDa act as repressors [
28]. In addition to its role in redox homeostasis, Nrf1 is implicated in maintaining lipid and cholesterol homeostasis by acting as a cholesterol sensor, particularly in the liver. Here it binds to cholesterol via its N-terminal CRAC (cholesterol-recognition/amino acid consensus motif) domain, thereby modulating hepatic inflammatory signalling, adaptive metabolic responses and promoting cholesterol removal. This indicates that Nrf1 plays a crucial role in maintaining hepatic cholesterol homeostasis and protecting the liver from excessive cholesterol accumulation [
29].
Here, we investigate the HCV-Nrf1 crosstalk. We study the impact of HCV on Nrf1 expression and how modulation of Nrf1 expression interferes with HCV morphogenesis. Therefore, we take a close look at the viral replication and production of progeny virions upon Nrf1 overexpression and silenced Nrf1 expression. We found out that Nrf1 overexpression restricts multiple steps of the HCV life cycle. This effect can be reversed upon Nrf1 silencing. Moreover, our study revealed that the 85 kDa Nrf1 fragment can be withdrawn from the nucleus by association with the sMaf proteins. Therefore, the overall function of Nrf1 is weakened, which results in impaired activation of Nrf1/ARE-dependent genes as well as intracellular cholesterol accumulation, favouring HCV morphogenesis.
Taken together, our data indicate strong crosstalk between HCV and Nrf1 affecting the HCV life cycle and HCV-associated pathogenesis. In light of this, Nrf1 could serve as a target for interference with the viral morphogenesis.
2. Materials and Methods
2.1. Cell Culture
The Huh7-derived cell line Huh7.5 (RRID:CVCL_7927) [
30] was used for electroporation in all experiments due to its susceptibility to HCV infection and replication. The HCV genomic RNA was generated by in vitro run-off transcription of linearised plasmid DNA (pFK-pJFH1/GND and pFK-JFH1/J6) using T7 Scribe Standard RNA IVT Kit (Biozym, Hessisch Oldendorf. Germany) according to the manufacturer’s protocol. Huh7.5 cells’ electroporation with HCV RNAs was performed as described in [
31]. Cells were cultivated in Dulbecco’s modified Eagle’s medium (complete DMEM) supplemented with 2 mM L-glutamine (Bio&Sell, Feucht, Germany, K0283), 100 μg/mL streptomycin, 100 U/mL penicillin and 10%
v/
v foetal bovine serum; (FBS.S 0615, Bio & Sell GmbH, Feucht, Germany) at 37 °C with 95% relative humidity and 5% CO
2. HCV-replicating and non-replicating cells were passaged two to three times a week, no longer than passage six.
2.2. Plasmids
Plasmids encoding HCV DNA: pFK-pJFH1/GND (replication deficient) and pFK-JFH1/J6 have been previously described [
31,
32]. pFK-Luc-Jc1 was kindly provided by R. Bartenschlager, Heidelberg, Germany, and encodes a bicistronic reporter construct of the full-length Jc1 genome [
32]. Plasmid pGreenFire1-LXRE in LXR alpha was ordered from IBA Lifesciences. Luciferase reporter construct pNQO1luc harbouring the AREs from NAD(P)H-dependent quinone oxidoreductase 1 (pNQO1luc) was used as described in [
33]. Plasmid encoding EGFP (GenBank: AAB02572.1) was purchased from Clontech Laboratories Inc., Mountain View, CA, USA. The Δa1pEGFP-N1 (pJo23) vector, which is based on the pEGFP-N1 plasmid (GenBank: U55762.1) lacking the start codon, was purchased from Invitrogen, Carlsbad, CA, USA. Plasmids encoding 85 kDa or 25 kDa Nrf1 tagged with a C-terminal EGFP were constructed by inserting human Nrf1 DNA into the Δa1pEGFP-N1 vector. All the Nrf1 fragments were amplified via polymerase chain reaction using the Q5
® High-Fidelity DNA Polymerase (NEB #0491S), and the plasmid DNA Flag-Nrf1-HA (#34708; Addgene) was used as a template.
The following primers were used: 85 kDa-fwd (5′-AAAAAGCTTATGGTTCACCGAGACCCAGAGG-3′), 25 kDa-fwd (5′-AAAAAGCTTATGAGGCACCCAGTGCCCTG-3′) and rev (5′-AAAGGTACCCTTTCTCCGGTCCTTTGGCTTC-3′). Inserts were cloned into the backbone linearized with HindIII (NEB #R0104L) and KpnI (NEB #R0142M) restriction enzymes according to Gibson et al. [
34]. Plasmids encoding human sMafG protein (Gene ID: 4097) with nuclear export/localisation signal N-terminally tagged with mCherry (GenBank: AAV52164) (pEM_Z55_mCherry_sMaf-NES; pcDNA3.1(-) sMafG-NLS) were previously generated in our lab.
2.3. Transient Transfection
Huh7.5, GND and Jc1 electroporated cells were transfected using linear polyethyleneimine (PEI) (1 mg/mL) (23966-100; Polysciences, Warrington, PA, USA) or FuGENE® HD Transfection Reagent (E2311; Promega, Madison, WI, USA). The cells were seeded one day before the transfection at a density of 3 × 105 cells/well in 6-well plates. For CLSM analyses, the cells were seeded on coverslips (1.5 H) (Carl Roth, Karlsruhe, Germany) in 12-well plates at a density of 1.5 × 105 cells/well. A DNA:PEI mass ratio of 1:6 was used, followed by an 8 h incubation time and medium change. A DNA:FuGENE mass ratio of 1:3 was used, followed by a 24 h incubation and medium change. Cells were harvested 48 h after transfection, at the peak point of Nrf1 overexpression.
Nrf1 knockdown experiments were performed using the siPORTTM NeoFXTM Transfection Agent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol using 2 nM Nfe2l1 siRNA (SMARTPool M-019733-01-0010; Dharmacon, Lafayette, CO, USA) or scrambled siRNA (sc-37007, Santa Cruz Biotechnology, Dallas, TX, USA) as a control. The transfections were performed in 12-well plates using the overlay protocol. For one well, a Transfection mix of 0.2 µL Nfe2l1 siRNA (10 µM) or scrambled siRNA (10 µM) was prepared in 49.8 µL Opti-MEM® I (Thermo Fisher, Waltham, MA, USA) and 3 µL siPORTTM NeoFXTM Transfection Agent were mixed with 47 µL Opti-MEM® I (31985062, Thermo Fisher, Waltham, MA, USA). After combining the solutions, RNA was prepared in OptiMEM® medium (31985062; Thermo Fisher, Waltham, MA, USA) using the overlay protocol siPORT (AM4510; Thermo Fisher, Waltham, MA, USA) and incubated for 15 min at room temperature. The RNA/siPORTTM NeoFXTM Transfection Agent complexes were evenly dispersed in 12-well plates. A total of 100 µL per 12 wells of transfection mix was pipetted into the wells and dispersed evenly. Afterwards, 900 µL of 1 × 105 stably HCV-replicating Huh7.5 cells were laid over the transfection mix. Samples were analysed 96 h after the transfection via Western blot or the CLSM method, and silencing of the Nrf1 protein was confirmed using an antibody binding to the N-terminus of the protein (#8052, Cell Signaling, Danvers, MA, USA).
2.4. RT-qPCR
Total RNA was isolated using RNA-solv® Reagent (R6830-02; VWR) according to the manufacturer’s protocol. Equal amounts of total RNA (4 µg) were treated with RQ1 RNase-Free Dnase (M6101; WVR) and transcribed into cDNA using a random hexamer primer and RevertAid H Minus reverse transcriptase (EP0451; Thermo Fisher Scientific, Waltham, MA, USA). qPCR of 10-fold diluted samples was performed using the SYBR-green detection kit (K0251; Thermo Fisher Scientific, Waltham, MA, USA) and specific primers. Extracellular viral RNA was isolated from the cell culture supernatants via the QIAmp® Viral RNA Mini Kit (Qiagen, Hilden, Germany) and detected using LightMix Modular Hepatitis C Virus Kit (53-0557-96, TIB MOLBIOL, Berlin, Germany) in combination with LightCycler Multiplex RNA Virus Master (6754155001; Roche Diagnostics) according to the manufacturer’s protocol. All RT-qPCR experiments were performed using the LightCycler 480 Instrument II (Roche Diagnostics) and analysed using the LightCycler480 Software (v1.5.1; Roche Diagnostics).
2.5. Virus Titration
Virus titres were analysed based on limited dilution by determining the half-maximal tissue culture infectious dose (TCID
50) as described previously [
35]. Briefly, Huh7.5 cells were seeded in a 96-well plate at a density of 1 × 10
4 cells/well. For extracellular TCID
50, cells were infected using a serial dilution of cell culture supernatant (5 steps, 1:5 ratio) in 6 replicates. For intracellular TCID
50, cells were washed with PBS, trypsinised and pelleted. Pellets were then resuspended in 1 mL DMEM and subjected to 4–5 freeze/thaw cycles at −80 °C and 37 °C, respectively. Afterwards, Huh7.5 cells were infected with the obtained supernatant (5 steps, 1:5 ratio) in 6 replicates. Cells were cultivated for 72 h. Fixation was performed with 4% formaldehyde in PBS at RT. Cells were then washed with PBS and incubated overnight at 4 °C with the NS5A-specific antiserum to detect HCV-positive cells. Horseradish peroxidase–coupled donkey-α-rabbit IgG (NA934; GE Healthcare, Chicago, IL, USA) was used as a secondary antibody, and subsequent staining was performed using 3-amino-9-ethylcarbazol (30 mM Na-acetate, 12 mM acetic acid, 0.05%
w/
v 3-amino-9-ethylcarbazol, 0.01% H
2O
2) [
36]. The resulting TCID
50 was calculated based on the method of Spearman and Kärber [
37,
38].
2.6. Luciferase Reporter Gene Assay
Huh7.5 and HCV-replicating Huh7.5 cells were transfected using pGreenFire1-LXRE plasmid (encoding for an LXR promotor-driven firefly luciferase) or pNQO1luc (encoding for a NQO1 promotor-driven firefly luciferase). Then, 48 h after transfection, cells were lysed with 200 μL of luciferase lysis buffer (25 mM Tris-HCl, 0.1% Triton-X 100, 2 mM DTT, 2 mM EGTA, 10% glycerol, pH 7.5). The luciferase activity of lysates (50 µL) was analysed by the addition of luciferase substrate (25 mM Tris-HCl pH 7.8, 5 mM MgCl
2, 33.3 mM DTT, 0.1 mM EDTA, 470 μM Luciferin, 530 μM ATP) and subsequent detection in the Orion II microplate Luminometer (Titerek Berthold, Pforzheim, Germany). The samples were analysed as technical duplicates. Luciferase levels were referred to the protein concentration of the appropriate lysates, determined by Bradford assay [
36]. To monitor HCV replication, Huh7.5 cells were electroporated with the bicistronic replicon pFK-Luc-Jc1, and a luciferase assay was performed as described previously [
39].
2.7. Immunofluorescence Analysis
Huh7.5 and HCV-replicating Huh7.5 cells were grown in 12-well plates on coverslips (1.5H) (Carl Roth, Karlsruhe, Germany). Thereafter, cells were washed with PBS and fixed with 4% formaldehyde for 20 min at RT. These were then blocked for 15 min with 5% bovine serum (Carl Roth, Karlsruhe, Germany, T844.4) in TBS-T and incubated for 1h with primary and secondary antibodies in a humidity chamber, respectively. As a diluent, a blocking solution was used. After washing with TBS-T, coverslips were mounted on glass slides with Mowiol 40-88 (324590-100G; Sigma Aldrich, St. Louis, MO, USA). Nuclei were stained with DAPI (4.6-diamidino-2-phenylindole) (6335.1; Roth) solution (1 μg/μL). For lipid droplet visualisation, Nile Red (Cay30787-250; Biomol) solution (1 μM) was added during the secondary antibodies incubation. F-Actin was stained with phalloidin-Atto 633 (68825, Sigma Aldrich, St. Louis, MO, USA, 1:500). Filipin III (F4767-1MG; Sigma Aldrich, St. Louis, MO, USA) was used to stain total intracellular cholesterol. First, cells were fixed as mentioned above. Afterwards, Schiff-base from FA-fixation was quenched with TBS for 5 min at RT. Cells were then permeabilized with 0.05% TritonX-100 (T9284; Sigma Aldrich, St. Louis, MO, USA) and blocked in 5% bovine serum in TBS containing 0.05 mg/mL filipin. These were then incubated with the primary and secondary antibodies with 0.05 mg/mL filipin in a humidity chamber. After washing with TBS, coverslips were mounted as described above. Immunofluorescence staining was analysed using a confocal laser scanning microscope (Leica TCS SP8 System with a DMi8 microscope) and Las X Control Software (Leica, Wetzlar, Germany). The used objectives were 100×, numerical aperture 1.46. Total fluorescence per cell was calculated using ImageJ software [
40] and the following formula: corrected total cell fluorescence (CTCF) = integrated density—(area of selected cell × mean fluorescence of background readings). In total, a minimum of ten cells were measured.
2.8. Antibodies
Primary antibodies were raised against HCV core (MA1-080; Thermo Scientific, Waltham, MA, USA), NS3 (VIS-1847-100; Biozol/MyBioSource), β-actin (A5316; Sigma-Aldrich), C-terminal Nrf1 (PA590023; Thermo Scientific, Waltham, MA, USA and sc-28379; Santa Cruz), N-terminal Nrf1 (#8052; Cell Signaling, Danvers, MA, USA) and sMaf G/F/K (sc-22831; Santa Cruz). For NS5A detection, polyclonal rabbit-derived serum was used.
Secondary antibodies directed against mouse or rabbit were produced in donkey and were conjugated to Alexa Fluor 488 (28115; Invitrogen, Carlsbad, CA, USA), Alexa Fluor 546 (A10036; Invitrogen, Carlsbad, CA, USA or A10040; Thermo Fischer Scientific) and Alexa Fluor 633 (A-21052; Thermo Fischer Scientific, Waltham, MA, USA).
2.9. Immunohistochemistry
An immunostaining procedure was performed on paraffin-embedded human liver sections, obtained from patients chronically infected with HCV genotype 1a and healthy individuals as controls. Both infected and healthy individuals were male, aged between 45 and 58. The specimens were kindly provided by K. Klingel, Institute of Molecular Pathology, Universitätsklinikum Tübingen, Tübingen, Germany. Sample collection was supervised by the Ethics Committee at the Medical Faculty of the Eberhard Karls University and at the University Hospital of Tübingen. Samples were fixed with 4% formaldehyde in PBS. Then, 4 µm thick paraffin liver sections were deparaffinised for 15 min with xylene, 10 min with 99% ethanol, 10 min with 75% ethanol and 5 min in ddH2O. Antigen retrieval was performed by heating the sections in 10mM sodium citrate buffer, pH 6.0, at 95 °C for 30 min. These were then blocked for 60 min in 10% BSA with 0.1% Tween20. The primary antibodies used for staining were the anti-Nrf1 antibody, detecting the N-terminus of the protein (#8052; Cell Signalling, Danvers, MA, USA), and the anti-core antibody (MA1-080; Thermo Scientific, Waltham, MA, USA), detecting HCV-positive cells. Anti-rabbit Alexa Fluor 488-conjugated (28115; Invitrogen, Carlsbad, CA, USA) and anti-mouse Alexa Fluor 546-conjugated (A10036; Invitrogen, Carlsbad, CA, USA) were used as secondary antibodies. Nuclei were stained with DAPI (4.6-diamidino-2-phenylindole) (6335.1; Roth) solution (1 μg/μL). All the dilutions were performed in the blocking solution. After washing with TBS-T, liver sections were mounted on glass slides with Mowiol 40-88 (324590-100G; Sigma Aldrich, St. Louis, MO, USA). Staining was analysed using a confocal laser scanning microscope (Leica TCS SP8 System with a DMi8 microscope) and Las X Control Software (Leica, Wetzlar, Germany).
2.10. SDS-PAGE and Western Blot Analysis
Cells were lysed using RIPA buffer (50 mM Tris-HCl pH 7.2, 150 mM NaCl, 0.1% SDS (
w/
v), 1% sodium deoxycholate (
w/
v) and 1% Triton X-100) containing protease inhibitors, followed by sonication. An equal amount of protein was denatured in sample 1x SDS loading buffer, boiled for 10 min at 95 °C and separated by SDS-PAGE electrophoresis with 10%
v/
v acrylamide. Afterwards, proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane (P667.1, Roth) and blocked in 1× ROTI-Block solution (A151.2, Roth) [
36]. Detection of the chosen proteins was conducted using the following primary antibodies: anti-GFP (632592; Takara, San Jose, CA, USA), anti-mCherry (ab167453; Abcam, Cambridge, UK), anti-αTubulin (sc-5546; Santa Cruz) anti-NS3, anti-NS5A, anti-Nrf1 and anti-βactin as described above. Secondary antibodies directed against mouse or rabbit were coupled to IRDye680RD or IRDye800CW (926-68073, 926-32213; LI-COR Biosciences, San Jose, CA, USA) and horseradish peroxidase (NA934, NXA931; GE Healthcare). All the antibody dilutions were performed in the blocking reagent and incubated for 1h on the membrane. After washing with TBS-T, fluorescence was detected by the LI-COR Odyssey CLx scanner (LI-COR Biosciences, San Jose, CA, USA). After incubation with Luminata Forte Western HRP substrate (WBLUF0100; Merck Chemicals GmbH Millipore GmbH, Darmstadt, Germany), chemiluminescence was detected by the INTAS-Imaging System (Intas Pharmaceuticals Limited, Ahmedabad, India). Data was analysed using Image-Studio Lite v5.2.5 (LI-COR Biosciences, San Jose, CA, USA).
2.11. Determination of Half-Life
To inhibit protein synthesis, HCV-replicating and -negative cells were treated with 142 μM cycloheximide (C7698-5G; Sigma Aldrich, St. Louis, MO, USA). Cells were lysed using RIPA buffer (50 mM Tris-HCl pH 7.2, 150 mM NaCl, 0.1% SDS (w/v), 1% sodium deoxycholate (w/v), 1% Triton X-100) containing protease inhibitors, at different time points between 0 and 4 h after cycloheximide treatment, and analysed by Western blot. Western blot analysis of cellular lysates derived from HCV-positive and HCV-negative cells was performed using an antibody binding to the N-terminus of the protein (#8052, Cell Signaling, Danvers, MA, USA), detecting 140-120 kDa Nrf1, representing full-length protein. The protein amount of Nrf1 was normalised to beta-actin, and the half-life of Nrf1 was determined by exponential regression.
2.12. Kinome Analysis
Huh7.5 cells stably electroporated with HCV RNAs were seeded in a 6-well plate with a density of 3 × 10
5 cells/well and cultivated in fully supplemented DMEM medium. After cells adhered, transfection of plasmid DNA encoding Nrf1 constructs was carried out using FuGENE
® HD Transfection Reagent as described earlier. Medium exchange was performed 24 h post-transfection and cells were lysed 72 h post-transfection using M-PER™ Mammalian Protein Extraction Reagent (78503, Thermo Scientific, Waltham, MA, USA) supplemented with Halt™ Protease Inhibitor Cocktail (87785, Thermo Scientific, Waltham, MA, USA) and Halt™ Phosphatase Inhibitor Cocktail (78420, Thermo Scientific, Waltham, MA, USA). Similar protein amounts, as assessed via the Pierce™ BCA Protein Assay Kit (23225, Thermo Scientific, Waltham, MA, USA), of cleared lysates were then used to determine relative changes in peptide phosphorylation and subsequent analyses of upstream kinases. Measurements were carried out on peptide arrays comprising distinct sets of peptides being targets of certain kinases using PamChips
® and respective reagent kits (32516, 32112, 32501, 32201, PamGene International, Hertogenbosch, The Netherlands) in combination with the PamStation
®12 instrument operated with Evolve2 software. Active kinases in lysates phosphorylate the immobilised peptides, which were visualised via fluorescently conjugated antibodies and a CCD camera. Relative changes in peptide phosphorylation, along with statistics, were then computed using the BioNavigator v6.3.67 software. These were used to predict the activity of upstream kinases, making use of distinct peptides as substrates, with the help of public databases (PhosphoNet, published in vitro or in vivo experiments or Kinexus), as described elsewhere [
41]. Changes in peptide phosphorylation were considered significant below a
p-value of 0.05, whereas upstream kinases were considered relevant above a final score of 2 (combinatory score of kinase specificity, the extent of relative change in activity and the corresponding statistics). Finally, significantly deregulated kinases were screened for their presence in certain gene ontology terms (GO terms) retrieved from the EMBL-EBI database as of 4 December 2022 (
http://purl.obolibrary.org/obo/go/releases/2022-12-04/go.owl). GO terms included covered inflammatory response (GO:0006954), innate immune response (GO:0045087), cholesterol biosynthetic process (GO:0006695) and lipid biosynthetic process (GO:0008610).
2.13. Lipid Droplet Analysis
LD analysis was performed using the particle analysis feature in Fiji (Image J) open-source analysis software [
40]. The size of the particle was set as 0.01-infinity (inch^2). Circularity was set as 0.00–1.00. The total count and perimeter were measured. The total volume of lipid droplets was calculated based on the average number of LDs multiplied by the average volume of LDs. Additional equations used in the calculations include
where
V is the volume,
r is the radius,
d is the diameter, and
P is the perimeter.
2.14. Statistical Analysis
Similar conditions were applied for all experiments. Each figure legend shows the number of independent experiments that correspond to that figure. Prism v9.2 software (GraphPad Prism version 9.2 for Windows, GraphPad Software, Boston, Massachusetts, USA,
www.graphpad.com) was used to perform statistical analysis and plot the graphical representation of the data. The results are described as the mean ± standard error of the mean (SEM). For statistical comparisons, a normality test was performed using the Shapiro–Wilk conditions. For normally distributed data, statistical significance was calculated by the unpaired
t-test. Data not showing a Gaussian distribution were tested using the Mann–Whitney test. Statistical significance is displayed as stated: ns—not significant, *
p < 0.05, **
p < 0.01, ***
p < 0.001, ****
p < 0.0001. The threshold for the
p-value was set using the Holm–Šídák method.
4. Discussion
The life cycle of HCV is tightly associated with lipid metabolism. HCV replication is associated with an intense rearrangement of the ER and the ‘membranous web’ formation. The ‘membranous web’ serves as the replication site and comprises ER-derived double-membrane vesicles (DMVs), multiple host factors and viral RNA and proteins [
48]. HCV assembly, however, takes place in close proximity—in detergent-resistant membranes (DRMs) of the ER or mitochondrial-associated ER membranes (MAMs) [
11]. A central place for HCV morphogenesis is cytosolic lipid droplets, as they serve as a platform that links the HCV replication and assembly sites [
9]. The late steps of the viral life cycle are also connected with lipid metabolism, as the release pathway of HCV is described to share many steps with lipoprotein release and uptake [
49]. Moreover, the mature viral particle is referred to as a lipoviroparticle due to its envelope that includes very-low-density lipoprotein (VLDL) components [
50,
51]. Nrf1, a transcription factor, plays a vital role in cellular homeostasis not only by responding to oxidative stress, but also as a cholesterol sensor and modulator of intracellular lipid levels [
26,
29]. HCV was described to modulate Nrf2/ARE-dependent gene expression by an unprecedented mechanism: in HCV-positive cells, sMaf proteins are translocated from the nucleus to the replicase complex on the cytoplasmic face of the ER by binding to NS3 protein. This traps Nrf2 at the replicase complexes and thereby prevents the entry of Nrf2 into the nucleus. Moreover, the lack of sMafs in the nucleus prevents their heterodimerysation with Nrf2 and in turn inhibits the activation of Nrf2/ARE-dependent cytoprotective genes [
33]. A recent study reports that both Nrf1 and Nrf2 transcription factors co-regulate genes that protect against hepatic stress induced by excess cholesterol accumulation [
52].
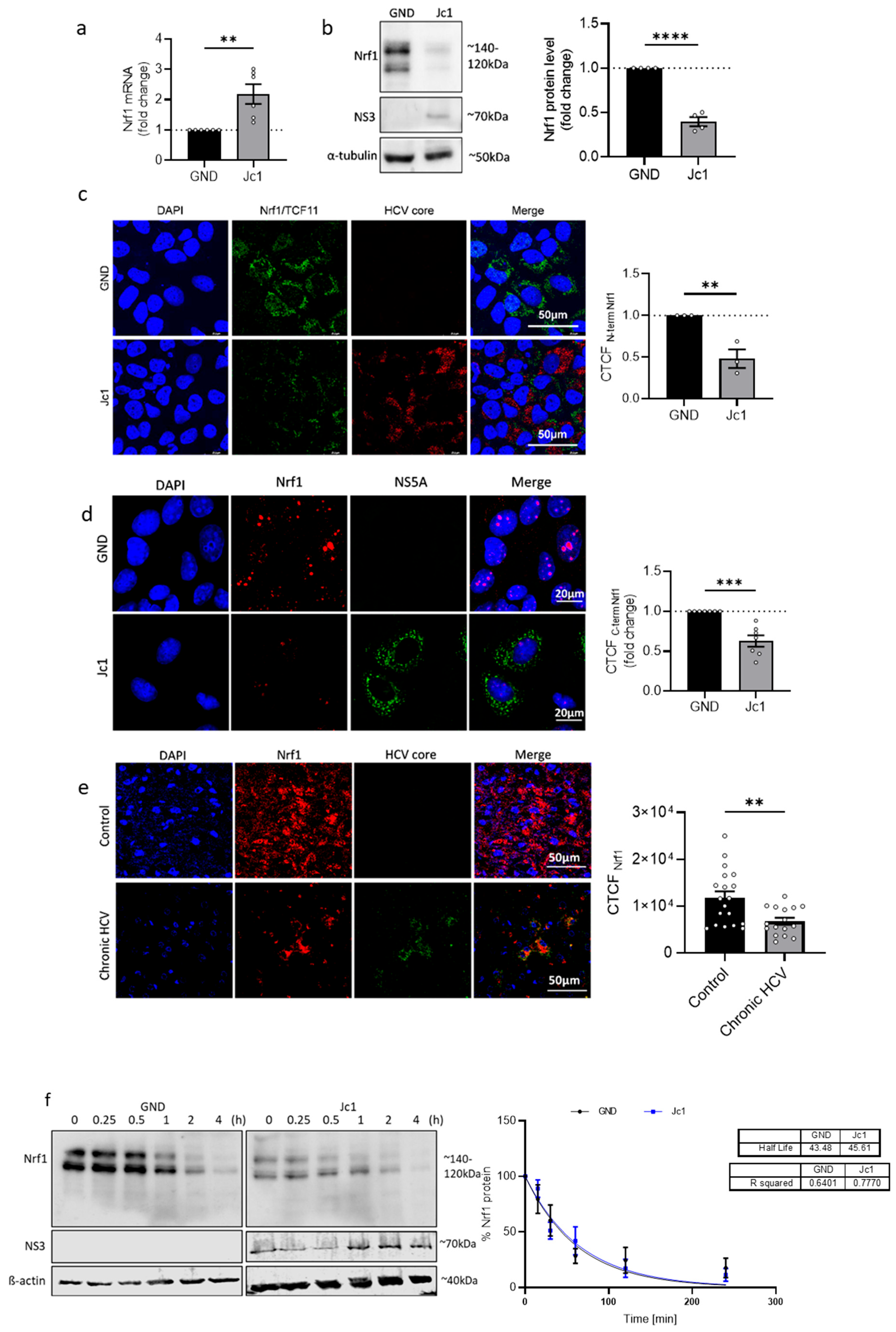
In light of the essential role of Nrf1 in the control of hepatic cholesterol levels and the intimate connection of the HCV life cycle with lipid metabolism, it was tempting to investigate the impact of HCV on Nrf1 expression, turnover, and localisation. Nrf1 protein amount was significantly decreased in HCV-positive cells and in liver tissues derived from a patient suffering from chronic HCV. In contrast, Nrf1 mRNA levels remained significantly increased. As we could not observe any difference in the half-life of the full-length Nrf1 protein in HCV-positive and HCV-negative cells, we assume that HCV modulates mRNA translation, as recently observed in ZIKV- or DNEV-infected cells. Singh et al. recently identified 19 or 7 repressed mRNAs in ZIKV- or DENV-infected cells, respectively. This finding is even more significant in the context of our research, as one of the translationally repressed genes in DENV-infected cells is NRF1. The regulation presumably occurs via the recruitment of various RNA-binding proteins [
53].
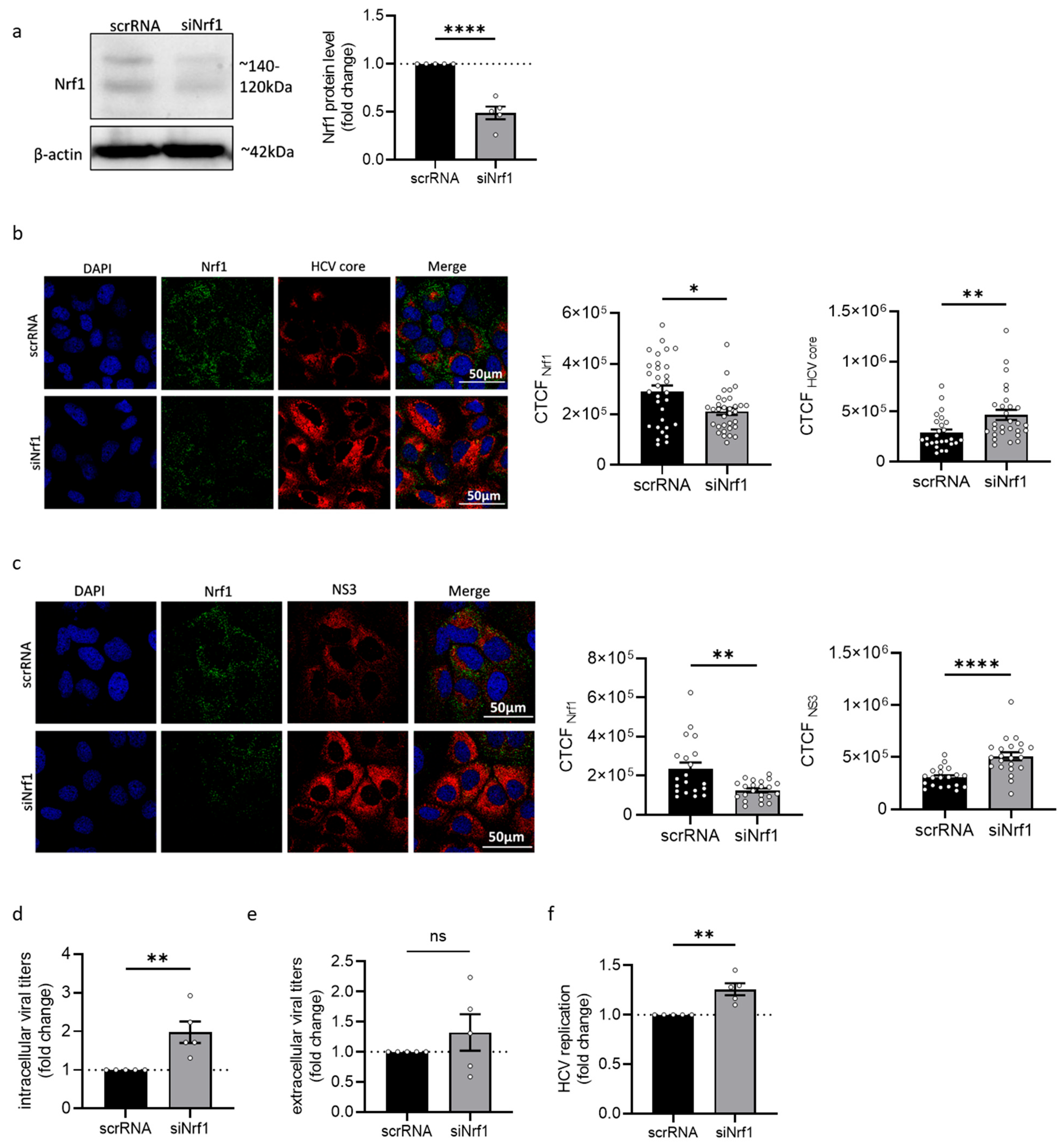
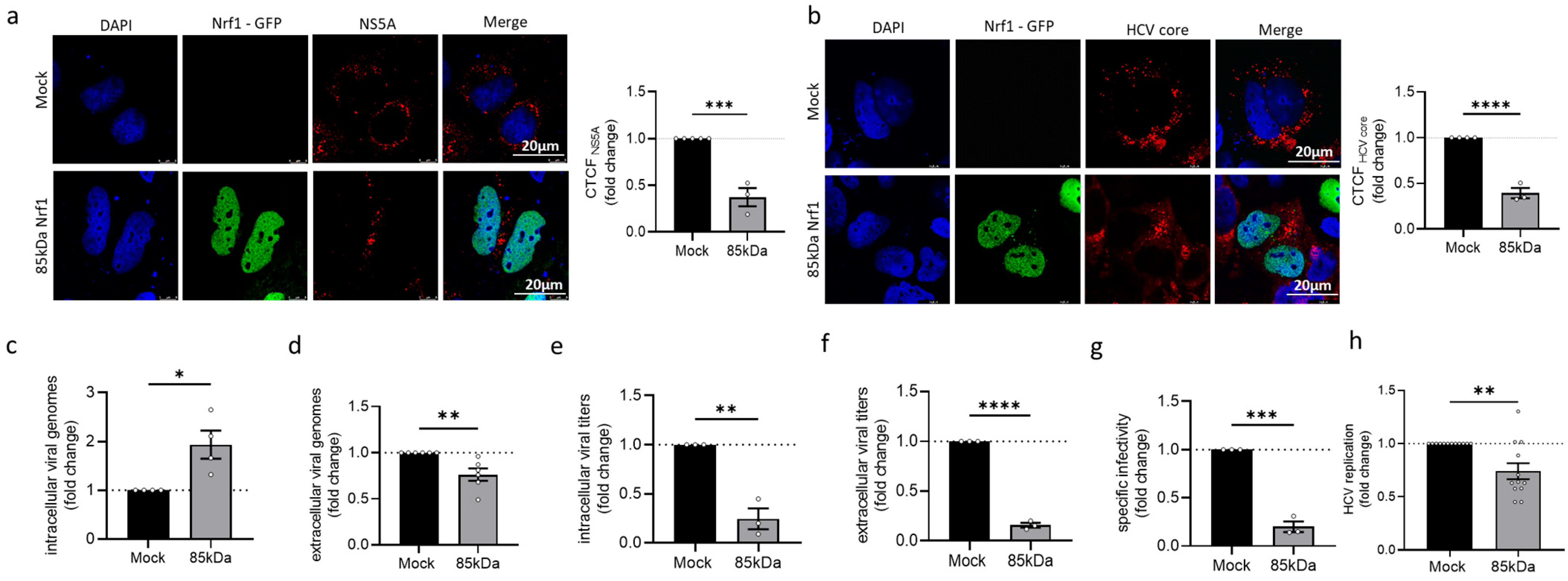
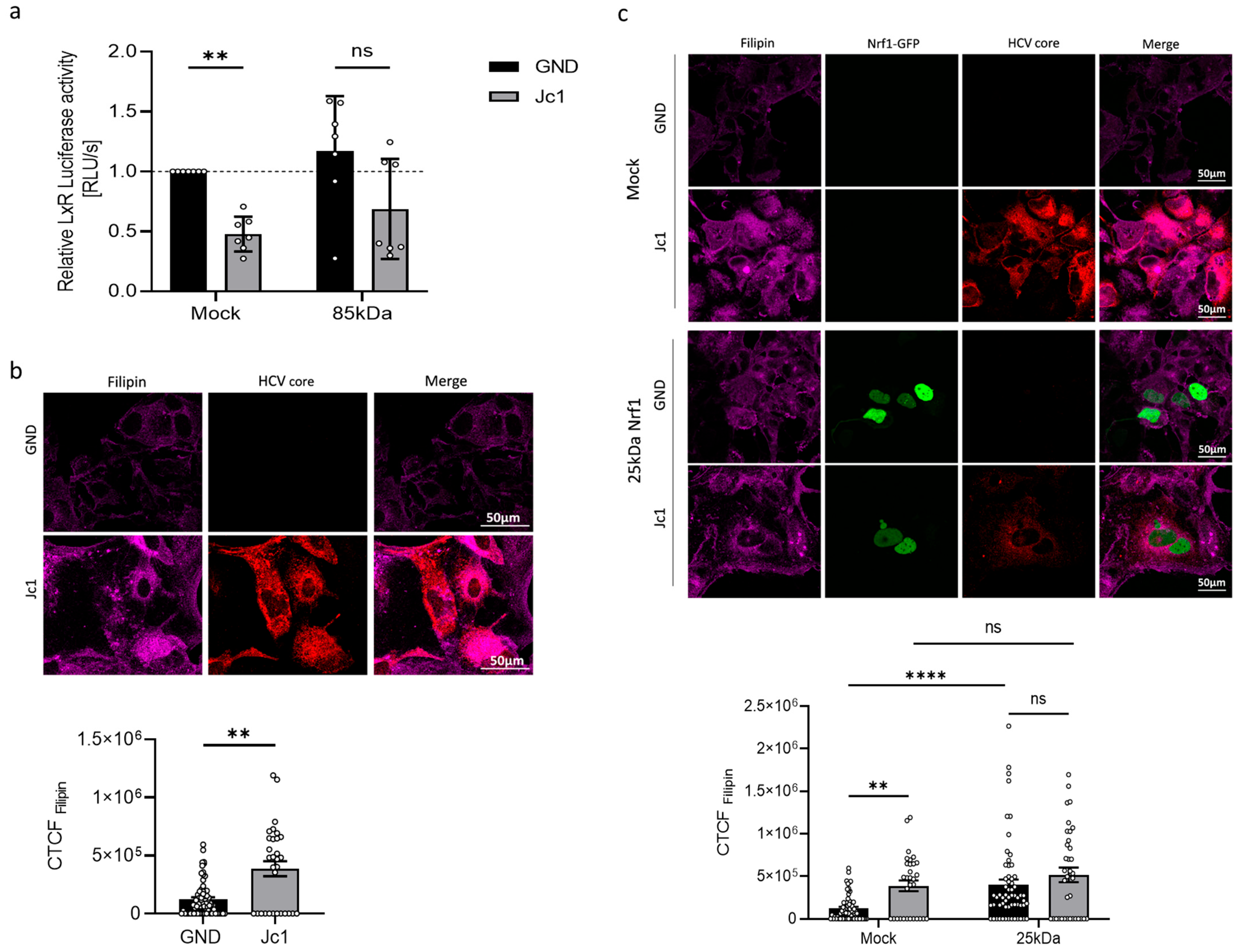
Since the Nrf1 protein amount is decreased in HCV-positive cells, we hypothesise that the reduced levels of Nrf1 favour HCV replication. Indeed, silencing of Nrf1 was found to increase HCV replication. In contrast, overexpression of the transcriptionally active 85 kDa Nrf1 was found to decrease HCV replication. Impaired Nrf1 activity is associated with enhanced intracellular lipid levels and formation of enlarged lipid droplets. Moreover, as a cholesterol sensor, Nrf1 has recently been linked to the regulation of LxR, one of the key players in the detoxification of cells from excess cholesterol [
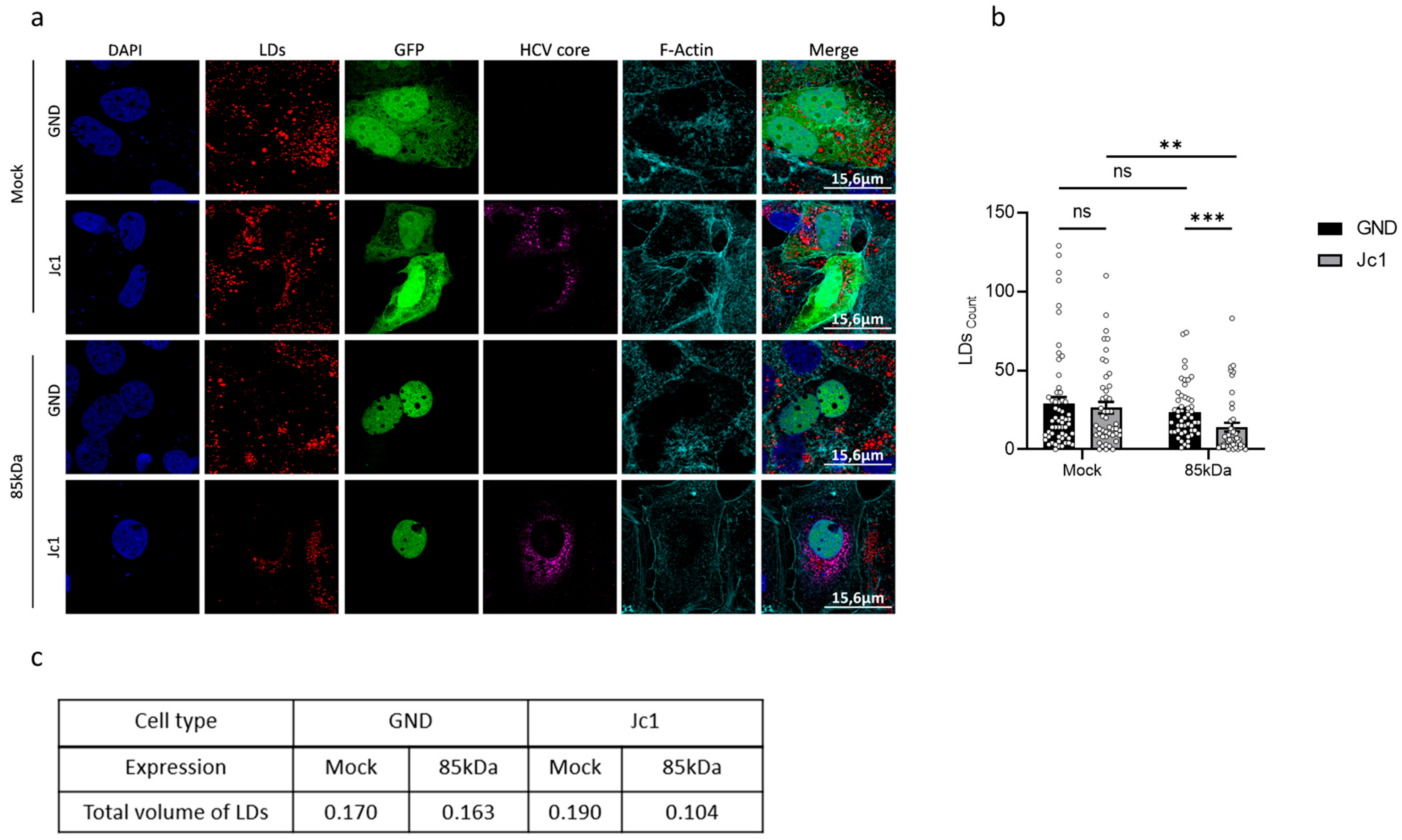
29]. In line with this, we observe that in HCV-positive cells, the activation of the LXR-promoter is impaired. Our data suggest that impaired Nrf1 functionality contributes to an elevated intracellular cholesterol level, observed in HCV-positive cells. In principle, the impaired Nrf1 functionality in HCV-positive cells could be a factor that contributes to reduced LXR activation and expression. This in turn could be a mechanism that, by defective cholesterol export, leads to elevated intracellular cholesterol levels observed in HCV-positive cells or HCV-negative cells with impaired Nrf1 functionality. However, it cannot be excluded that the observed effects represent a more indirect crosstalk or a coincidence and do not reflect a direct correlation. Moreover, HCV replication is associated with intracellular cholesterol accumulation and an impact on lipid droplets. Rescue of Nrf1 functionality by overexpression of the 85 kDa Nrf1 reduces the number of lipid droplets in HCV-positive cells. This effect is much more pronounced in HCV-positive cells compared to HCV-negative cells. Likewise, overexpression of the 85 kDa form triggered a much more pronounced reduction in the lipid droplet volume in HCV-positive cells compared to HCV-negative cells. This might reflect the fact that in the HCV-negative cells, there is an intact Nrf1 activity regulating the number and volume of lipid droplets. In contrast to this, in HCV-positive cells, there is an impaired Nrf1 functionality. Under these conditions, the rescue of Nrf1 functionality by overexpression of the 85 kDa form exerts a much stronger effect on these parameters.
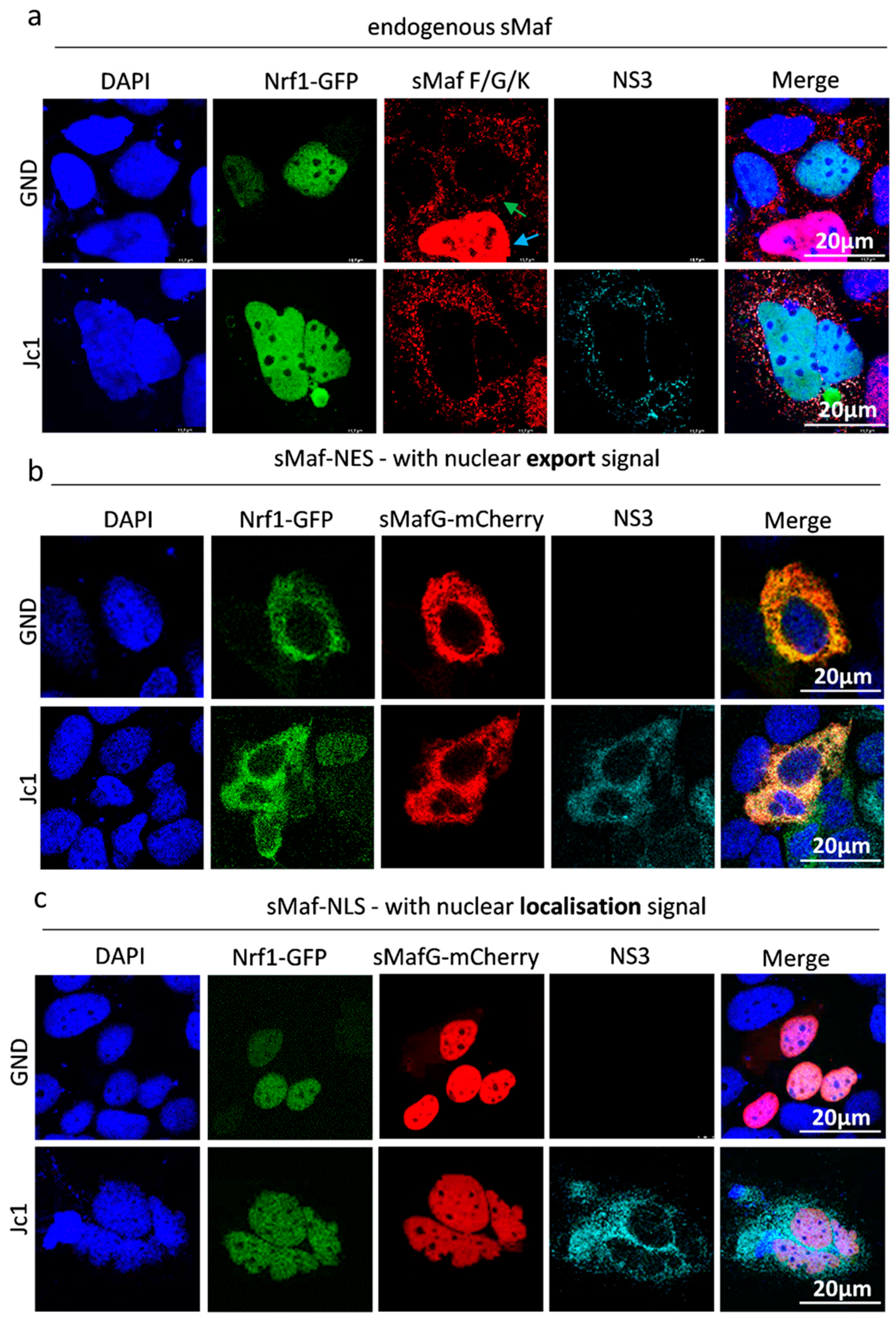
Apart from the decreased Nrf1 levels, the Nrf1/ARE-dependent gene expression is impaired in HCV-replicating cells. It is known that members of the Cap’N’Collar family, including Nrf1 and Nrf2, form heterodimers with sMaf proteins [
54]. Our previous work showed that in HCV-replicating cells, due to the NS3-mediated delocalisation of sMafs to the replicase complex on the cytoplasmic face of the ER, Nrf2 was incapable of entering the nucleus [
33]. Therefore, we wondered whether a similar phenomenon occurs in the case of Nrf1. Indeed, we observed significantly lower amounts of cleavage products of endogenous full-length Nrf1 in the nucleus of HCV-positive cells. However, overexpression of the GFP-tagged 85 kDa Nrf1, which is no longer associated with the ER but still has the capacity to bind to sMaf, revealed a strong nuclear localisation in HCV-positive cells. Yet, it was unclear whether the observed nuclear localisation of Nrf1 reflects its inability to bind to sMaf proteins or if the strong overexpression of Nrf1 fragments leads to the saturation of ER-localised sMaf proteins. The saturation of extranuclear sMaf with Nrf proteins (endogenous Nrf1 or Nrf2 and overexpressed Nrf1) could prevent the retention of the large excess of Nrf1-GFP fusion proteins outside the nucleus. The observation that co-expression of recombinant sMaf constructs harbouring a nuclear localisation (NLS) or a nuclear export signal (NES) clearly demonstrates the capacity of sMaf proteins to bind to Nrf1 and modulate its localisation. Moreover, these experiments show that Nrf1/ARE-dependent gene expression can be modulated by delocalised sMaf proteins. Apart from the impact on the HCV life cycle, sMaf-NES fusion proteins could represent a novel and very specific tool to modulate Nrf1 and/or Nrf2 activity.
The impaired activation of Nrf2 and the resulting elevated ROS levels were found to be essential for the HCV life cycle. Increased ROS levels further result in the induction of the autophagic pathway, which plays an essential role in the MVB-dependent release of HCV particles. Nevertheless, the oxidative stress further contributes to the HCV-associated pathogenesis, as elevated ROS levels induce DNA damage, which in turn leads to genetic mutations of the host genome. In addition, impaired Nrf2-functionality has been reported to interfere with liver regeneration based on a ROS-mediated inhibition of the insulin/insulin growth factor 1 (IGF1) signalling cascade [
45,
55]. Another factor involved in redox homeostasis is the transcription factor Nrf1. However, besides its role in redox homeostasis, Nrf1 is implicated in maintaining lipid and cholesterol homeostasis and modulates inflammatory processes. In this study, we were curious if the inhibition of Nrf1-dependent transcriptional activation, caused by HCV, could affect lipid metabolism and thereby impact viral replication, as the HCV life cycle is tightly connected to lipid metabolism. One important factor involved in the removal of intracellular cholesterol is the liver X receptor alpha (LXR-α). Notably, Nrf1 interferes with LXR activities to support cholesterol homeostasis [
29]. In the context of HCV, the LXR agonists GW3965 and T0901317 or the natural LXR ligand 24(S),25-epoxycholesterol have been reported to inhibit HCV infection [
56]. In accordance with these reports, we could observe that in HCV-positive cells, the activation of the LXRα promoter is impaired. It can be assumed that the defect in the Nrf1-LXRα-axis in HCV-positive cells, along with other factors, further results in elevated intracellular cholesterol levels. Yet, this phenotype can be partially rescued by co-expression of the 85 kDa Nrf1 isoform, corroborating the essential role of Nrf1 in maintaining lipid homeostasis. Vice versa, inhibition of Nrf1 activity by co-expression of the 25 kDa form of Nrf1 in HCV-negative cells leads to elevated intracellular cholesterol levels. Interestingly, a recent study described the interplay of Nrf1 and Nrf2 in the modulation against excess liver cholesterol [
52].
Cholesterol-rich membrane domains play a central role in the release and infectivity of progeny virus, virus entry, and replication, i.e., affecting the ‘membranous web’ formation [
57,
58,
59]. In healthy adipocytes, lipid droplets serve as the main storage organelle for free cholesterol and its esterified derivatives [
60,
61]. In the case of HCV infection, intracellular accumulation of lipids in the form of lipid droplets is a prerequisite for viral replication. LDs play an important role as the site of viral morphogenesis and as a central part of the ‘membranous web’ [
50]. Furthermore, in HCV-positive cells, activation of LXRα is impaired, preventing the induction of an effective cholesterol removal programme. This leads to elevated intracellular cholesterol levels, which favour HCV replication and can contribute to HCV-associated pathogenesis as steatosis [
62]. Apart from these direct effects of Nrf1 on lipid metabolism and thereby on the HCV life cycle, there might be a variety of indirect factors involved. Several kinome-based studies performed by our group yielded a fundamental insight into the host–virus interplay [
63,
64,
65,
66]. Therefore, we decided to utilise this tool in the context of HCV-Nrf1 interplay. We aimed to identify a general trend in the deregulation of kinases that could potentially serve as drug targets against viral infection. We were able to pinpoint a set of kinases involved in inflammatory processes and, most importantly, cholesterol metabolism. The most prominent example is PTK6, as it is the kinase that appears in both HCV-positive cells and HCV-negative cells expressing the 25 kDa Nrf1 isoform. PTK6 is described to modulate autophagy pathways; thus, it potentially plays a role in HCV pathogenesis, as HCV is strongly dependent on autophagy [
67]. Moreover, the analysis revealed that overexpression of the Nrf1-85kDa fragment triggers an activation of AMP-activated kinase (AMPK). It has recently been described that NS5A has the capacity to inactivate AMPK. This leads to a diminished nuclear import of the RNA-binding protein HuR through the inhibition of AMPK-mediated phosphorylation and acetylation of importin-α1. Cytoplasmic HuR is crucial for HCV replication. It is involved in the assembly of the replication complex on \ viral-3′UTR, and the loss of cytoplasmic HuR hampers viral replication. The activation of AMPK decreases cytoplasmic HuR and thereby reduces HCV replication [
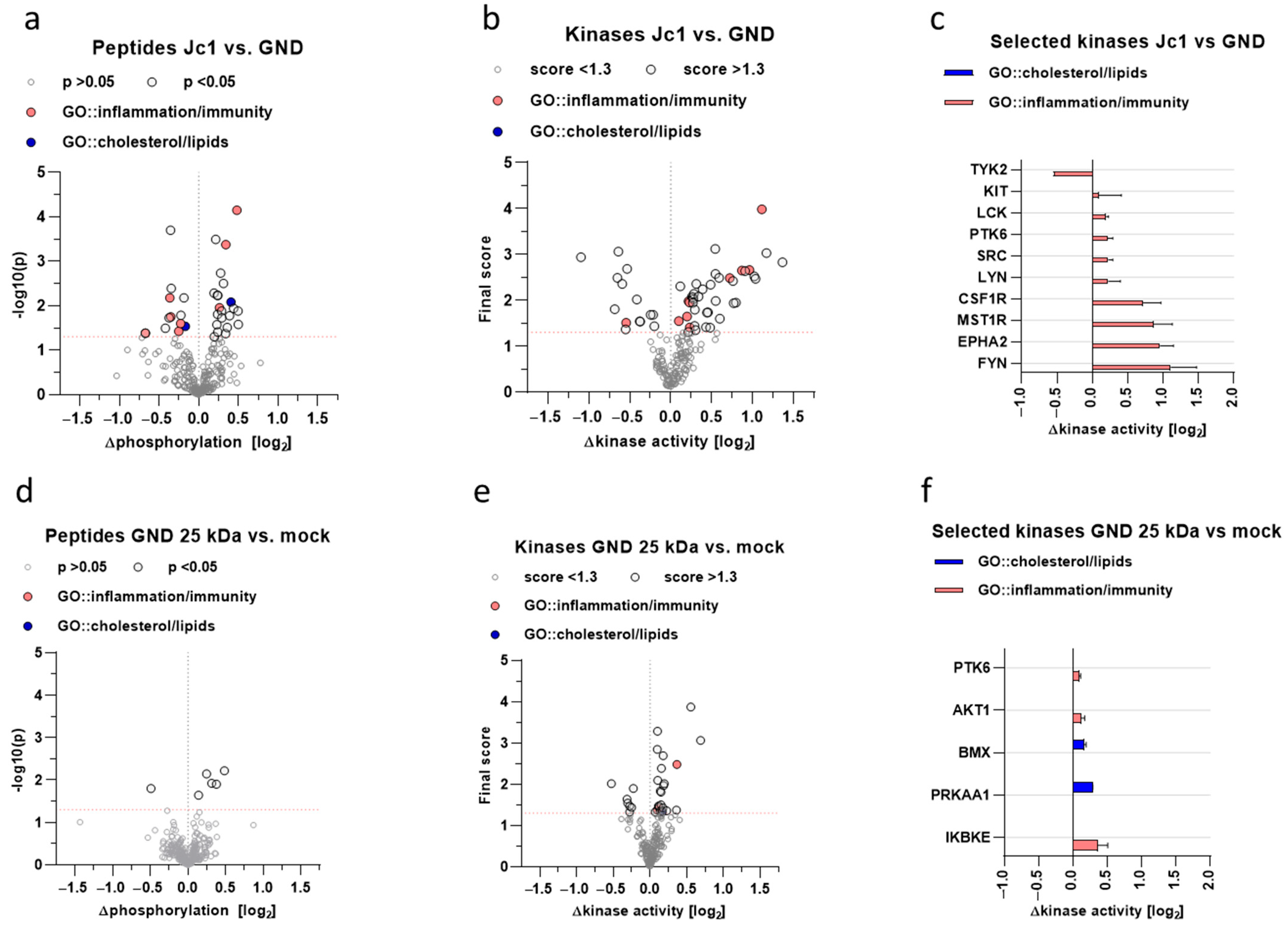
68]. Moreover, the strong impact of trans-dominant-negative Nrf1 (25 kDa form) in GND cells on the inhibitor of the nuclear factor kappa B kinase subunit epsilon (IKBKE) reflects a potent role in the regulation of inflammatory processes and underlines the potential relevance for HCV-associated pathogenesis. This indicates that, in addition to the direct impact of Nrf1 on the intracellular lipid content, a variety of further factors are relevant for both HCV replication and HCV-associated pathogenesis. The kinome analysis and kinases that we were able to identify could serve as an anchoring point for more extended and detailed studies in the future, identifying therapeutic targets in real-life clinical settings.
Taken together, these data describe crosstalk between HCV and Nrf1. In light of Nrf1’s prominent role in controlling intracellular ROS levels, expressing cytoprotective genes, and modulating lipid metabolism and inflammatory processes, as well as the impact of deregulated ROS levels and lipids on the HCV life cycle and HCV-associated pathogenesis, our data might contribute to our understanding of HCV-associated pathogenesis and the identification of therapeutic intervention targets.
There is a correlation between the HCV-dependent deregulation of Nrf1-driven transcriptional activation and the HCV life cycle. One of the underlying mechanisms is the modulation of the Nrf1-sMaf interaction, which affects, inter alia, the expression of ARE-regulated genes. Moreover, disrupted Nrf1 function impairs lipid removal programmes, leads to elevated intracellular cholesterol content, and influences the number and volume of lipid droplets, thus favouring the HCV life cycle. Apart from the impact on the HCV life cycle, the inflammatory processes are deregulated once again, reflecting the relevance of Nrf1 for HCV-associated pathogenesis. It is tempting to speculate if reconstitution of Nrf1 activity could represent a new therapeutic target by initiating a cholesterol removal programme affecting HCV replication and, in consequence, HCV-associated pathogenesis. Future studies will further characterise the role of various Nrf1 isoforms in HCV-associated infection. Particular emphasis will be put on lipid metabolism, as it is a central factor in viral morphogenesis, and the full-length Nrf1, as it encompasses the cholesterol-binding domain.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}