
Tracking the Spatial and Functional Dispersion of Vaccine-Related Canine Distemper Virus Genotypes: Insights from a Global Scoping Review

 ,
, 
 , ,
, ,  and
and 
Abstract
1. Introduction
Assumptions and Objectives
2. Materials and Methods
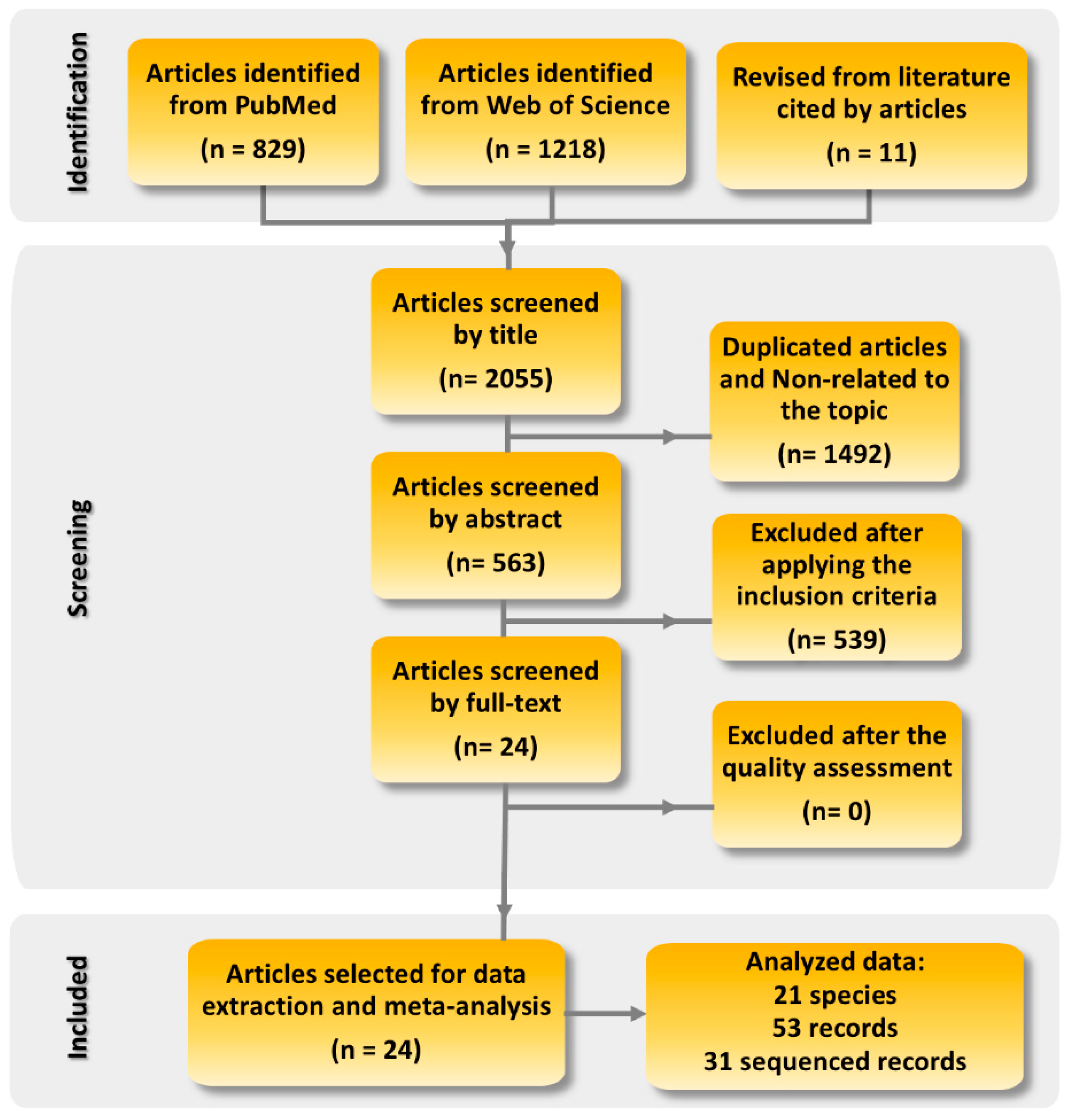
2.1. Scoping Review and Data Compilation
- Studies performed on domestic and/or wild carnivores.
- Studies that detect and sequence CDV classified within the America-1 and Rockborn-like genotypes.
- Articles written in Spanish, Portuguese, and English.
- -
- Articles: Total number of articles reviewed (Table S2, Supplementary File S2).
- -
- Species: Animal species analysed in the included articles.
- -
- Records: Categorisation that classifies the different individual studies analysed in the articles reviewed. To work with and identify these individual studies, we considered the following criteria: (i) all the different species analysed in each revised article, (ii) the existence of sequencing of the two vaccine-related genotypes (America-1 and Rockborn-like), (iii) all the different geographical areas from which the host species originate.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Category | Variable | Factor Label |
|---|---|---|
| Host species | Wild or domestic carnivore | Wild carnivore; domestic carnivore. |
| Suborder | Caniformia; Feliformia | |
| Family | Canidae, Mephitidae, Mustelidae, Phocidae, Procyonidae, Ursidae, Felidae, Herpestidae, Hyanidae, Viverridae. | |
| Species | Common name. | |
| Species | Scientific name. | |
| CDV linages | Genotype, following Panzera et al. [21] and Duque-Valencia et al. [12] | America-1, Rockborn-like (all vaccine strains classified within these genotypes). |
| Date and geographical procedence | Year of sampling | 1988–2025 |
| Continent | Africa; America; Asia; Europe; Oceania. | |
| Country | ||
| Geographical region | ||
| Latitude coordinates (decimal degrees) | ||
| Longitude coordinates (decimal degrees) | ||
| Accuracy | Accurate; Approximate; National. | |
| Host ecological and behavioral traits | Habitat type | Closed, open, both |
| Geographical distribution | Restricted, worldwide. | |
| Degree of ecological plasticity | Specialist, generalist. | |
| Social behaviour | Solitary, gregarious. | |
| Contact with human environments | Yes, No. | |
| Vaccination | Yes, No. | |
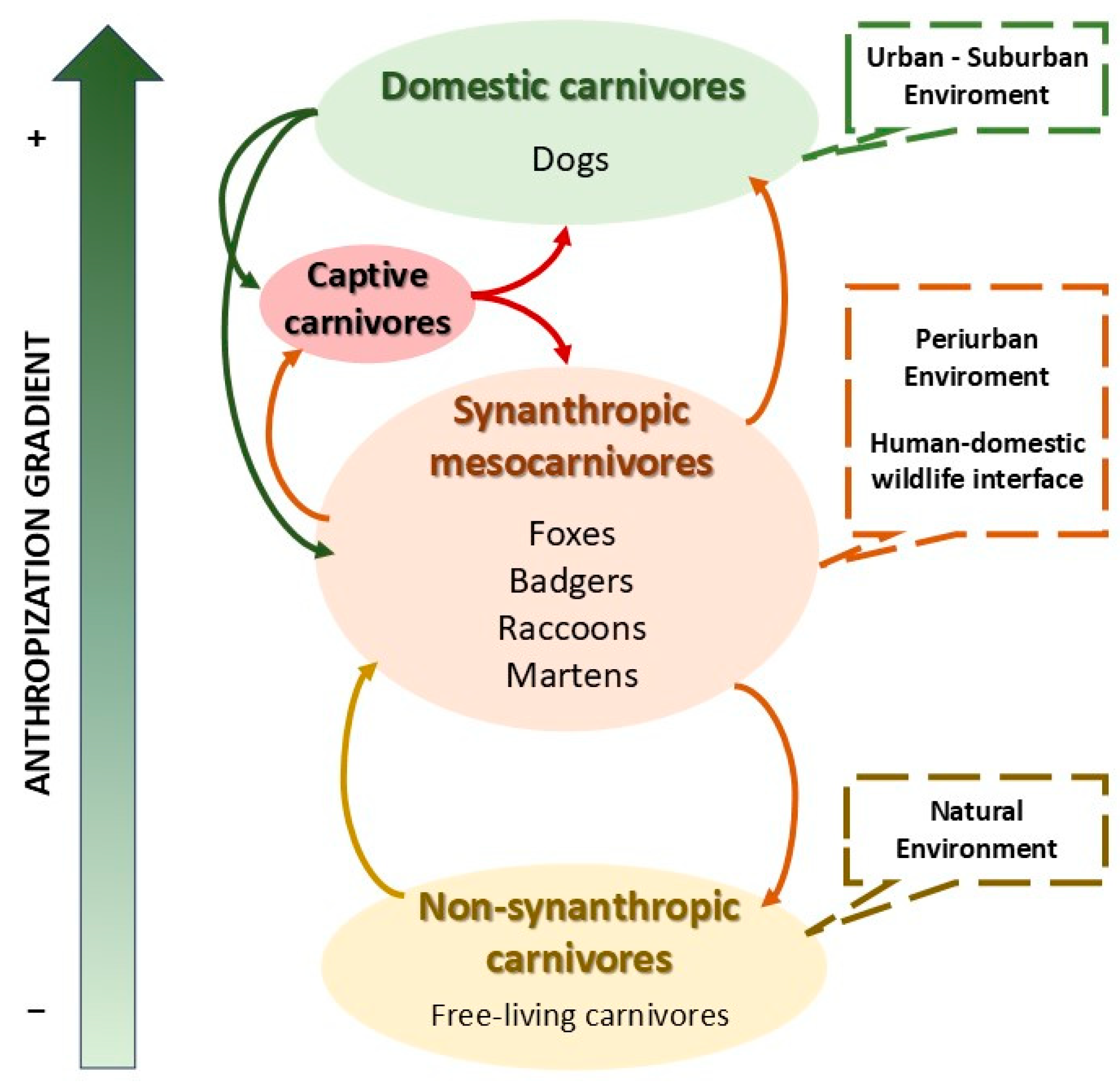
| Host functional group. Life status in relation with domestication (Figure 2) | Domestic dog | Canis lupus familiaris. |
| Captive carnivore | Zoo or farmed individuals. | |
| Synanthropic mesocarnivore | Species with periurban habits such as foxes, raccoons, and martens. | |
| Wild non-synanthropic carnivore | Those animals belonging Carnivora with limited or no human interface. |
2.2. Epidemiological Modelling of Vaccine-Related CDV Dispersion
2.2.1. Conceptual Model of Transmission Pathways
- Dogs, as the main host of CDV and a domestic species closely associated with humans, play a significant epidemiological role. They are highly susceptible to CDV and can become infected by any host species, regardless of the degree of interaction with human environments (anthropisation). Moreover, their high colonisation potential, generalist ecology, and the common practice of domestic dogs traveling extended distances with their owners allow them to disseminate the infection to areas far from its original focus.
- Wild carnivores are highly susceptible to CDV infection. In addition, the proliferation of synanthropic species, including mesocarnivores as the red fox, raccoon, skunk, and badger at the human–domestic–wildlife interface, boosts both intra- and interspecies contact rates. Increased densities of these species in anthropised areas facilitate the spread of directly transmitted viruses, such as CDV. In contrast, elusive carnivores with synanthropic habits often act as connectors between domestic and non-synanthropic wild reservoirs in their respective habitats.
- Flow of CDV from domestic dogs to any carnivore species (both captive and wild), anywhere in the world.
- Transmission from any carnivore species (both captive and wild) globally to domestic dogs.
- Transmission among synanthropic mesocarnivores (specifically fox, raccoon, skunk and badger) and towards any other carnivore species (both captive and wild) within their shared geographical range.
- Transmission from any other elusive carnivore species that is neither generalist nor synanthropic (both captive and wild) towards synanthropic mesocarnivores (fox, raccoon, skunk and badger), within their shared geographical range.
2.2.2. Generation and Filtering of Temporal Connection Data
2.2.3. Exploratory Data Analysis
2.3. Statistical and Spatial Assessment of Transmission Patterns
2.3.1. Statistical Analysis of Dispersion Plausibility
2.3.2. Spatial Processing and Vector Generation
3. Results
3.1. Reported Cases of Post-Vaccination CD or Detection of CDV Vaccine Strains in Domestic and Wild Carnivores
3.2. Descriptive Epidemiology of CD Due to Strains of Vaccine Origin
3.3. Functional Epidemiological Profiles of Vaccine-Related Genotypes
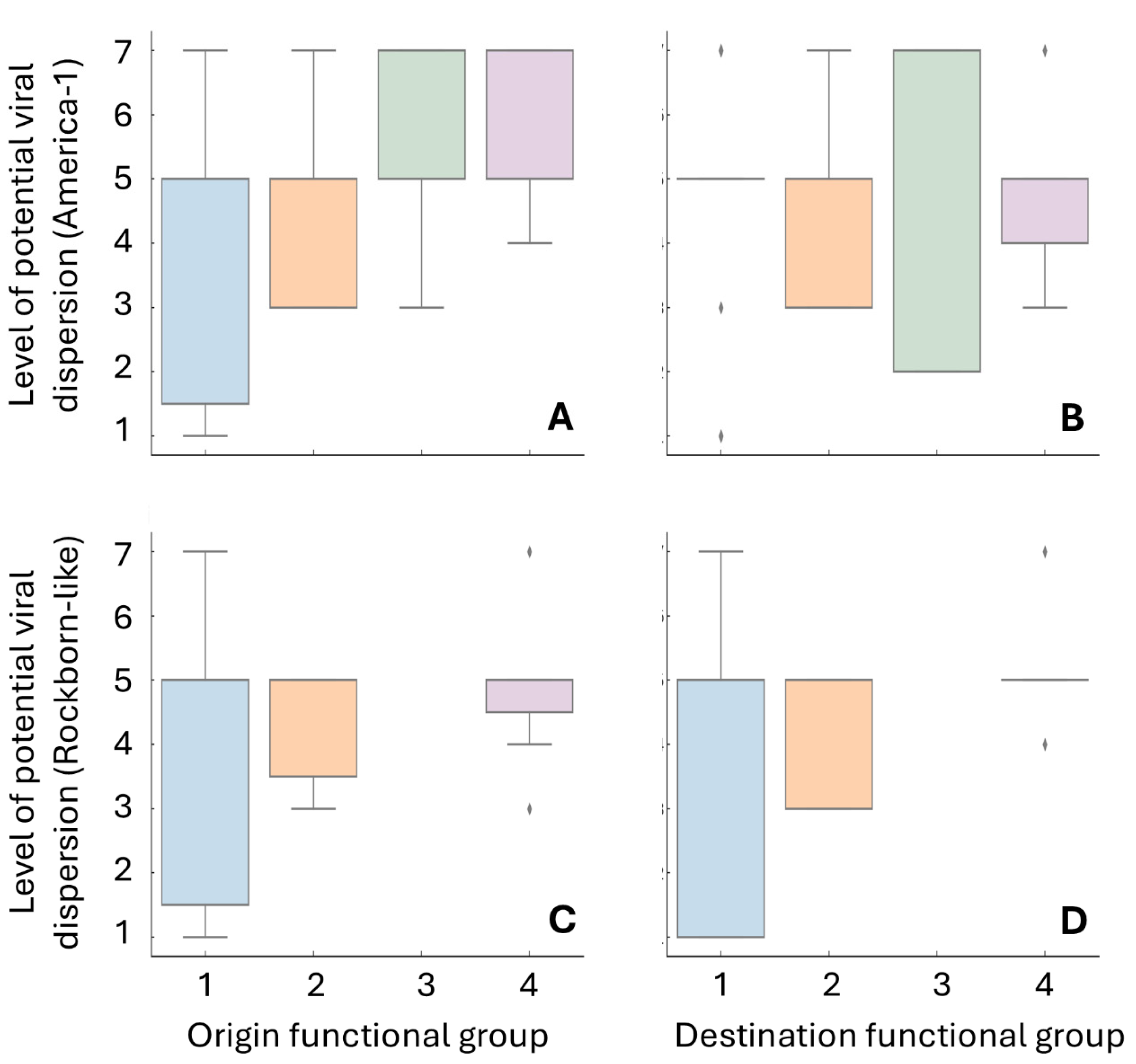
3.4. Statistical Assessment of Dispersion Patterns
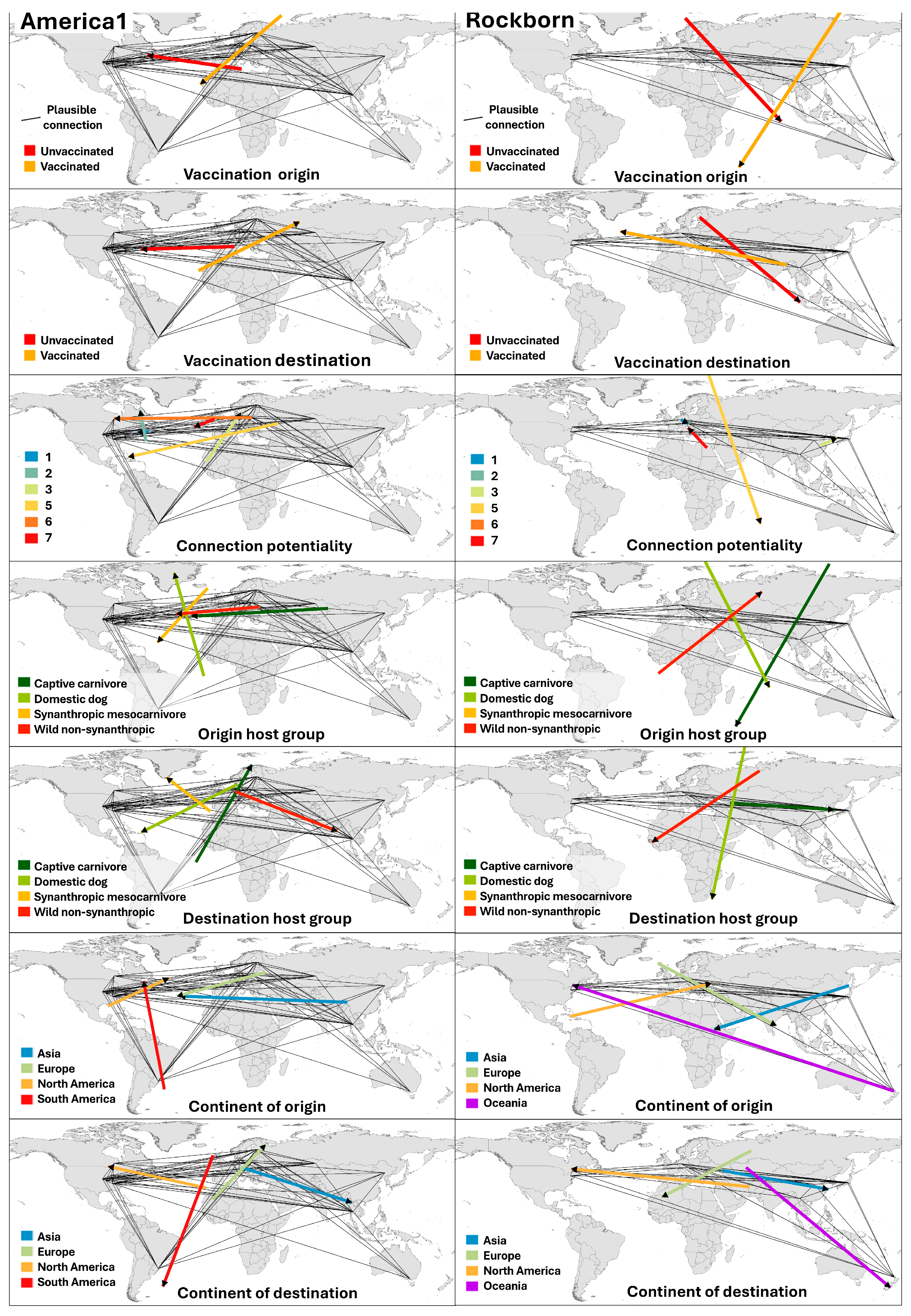
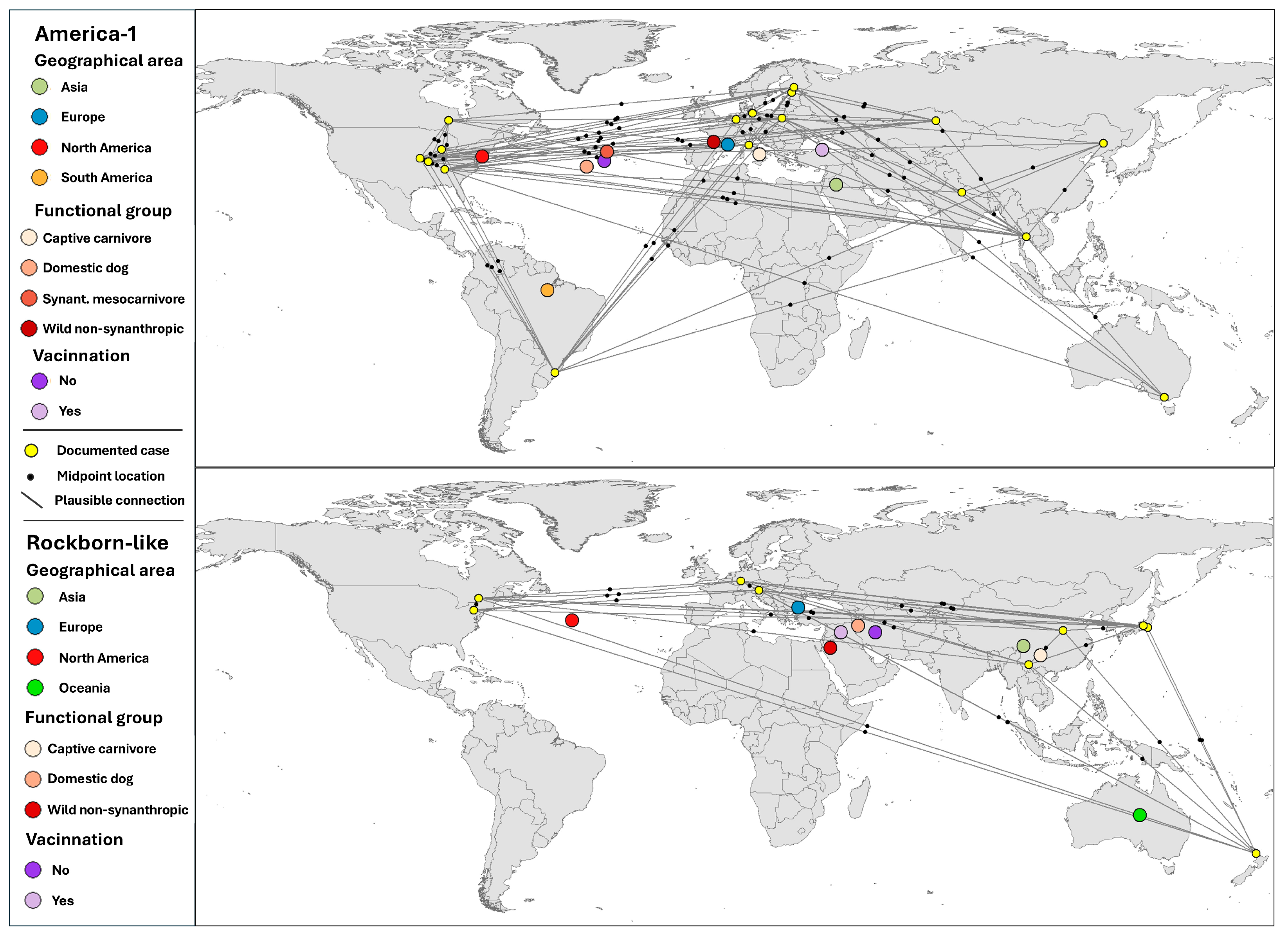
3.5. Spatial Patterns of Plausible Viral Connections: Directional Structure and Functional Centres
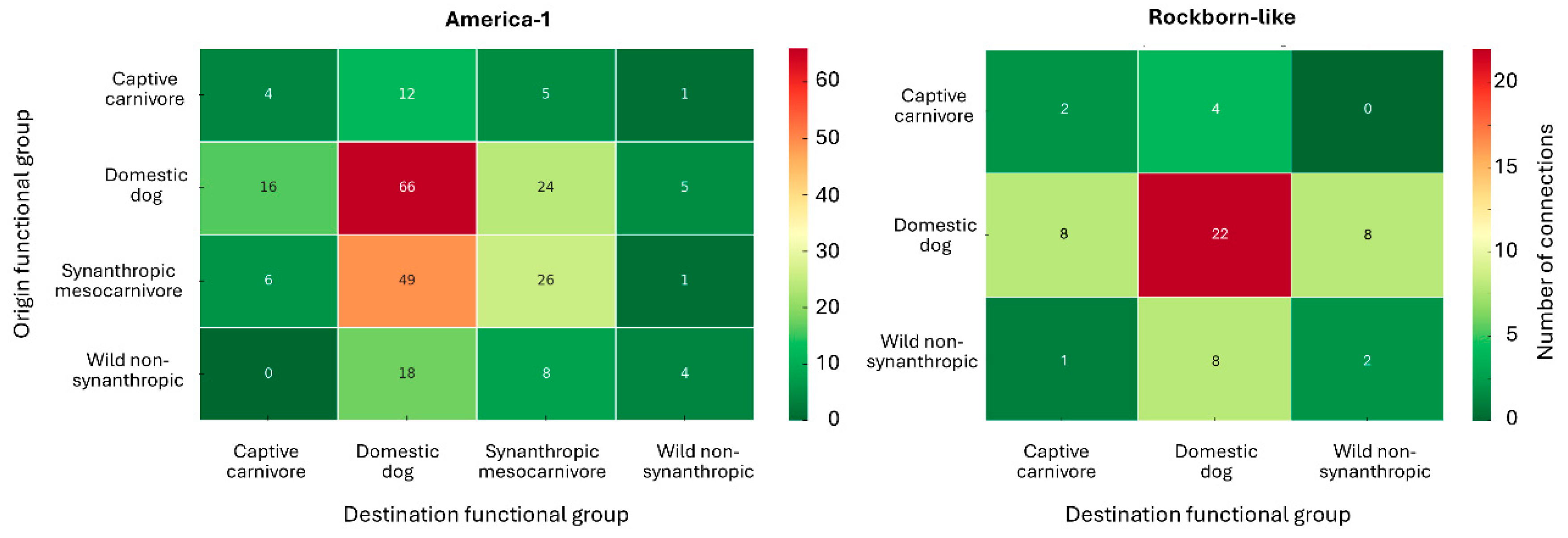
3.5.1. Spatial Structure of Plausible Connections: Comparative Analysis by Genotype
3.5.2. Spatial Aggregation of Plausible Pathways by Host and Epidemiological Attributes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://ictv.global/taxonomy (accessed on 10 May 2024).
- Krakowka, S.; Higgins, R.J.; Koestner, A. Canine distemper virus: Review of structural and functional modulations in lymphoid tissues. Am. J. Vet. Res. 1980, 41, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Beineke, A.; Puff, C.; Seehusen, F.; Baumgärtner, W. Pathogenesis and immunopathology of systemic and nervous canine distemper. Vet. Immunol. Immunopathol. 2009, 127, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pope, J.P.; Miller, D.L.; Riley, M.C.; Anis, E.; Wilkes, R.P. Characterization of a novel canine distemper virus causing disease in wildlife. J. Vet. Diagn. Investig. 2016, 28, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Loots, A.K.; Mitchell, E.; Dalton, D.L.; Kotz’e, A.; Venter, E.H. Advances in canine distemper virus pathogenesis research: A wildlife perspective. J. Gen. Virol. 2017, 98, 311–321. [Google Scholar] [CrossRef]
- Brown, A.T.; McAloose, D.; Calle, P.P.; Auer, A.; Posautz, A.; Slavinski, S.; Brennan, R.; Walzer, C.; Seimon, T.A. Development and validation of a portable, point-of-care canine distemper virus qPCR test. PLoS ONE 2020, 15, e0232044. [Google Scholar] [CrossRef]
- Appel, M.; Robson, D.S. A microneutralization test for canine distemper virus. Am. J. Vet. Res. 1973, 34, 1459–1463. [Google Scholar] [CrossRef]
- Barret, T. Rinderpest and distemper viruses. In Desk Encyclopedia of Animal and Bacterial Virology; Mahy, B.W., Van Regenmortel, M.H., Eds.; Elsevier: San Diego, CA, USA, 2010. [Google Scholar]
- van Moll, P.; Alldinger, S.; Baumgärtner, W.; Adami, M. Distemper in wild carnivores: An epidemiological, histological and immunocytochemical study. Vet. Microbiol. 1995, 44, 193–199. [Google Scholar] [CrossRef]
- Wipf, A.; Perez-Cutillas, P.; Ortega, N.; Huertas-López, A.; Martínez-Carrasco, C.; Candela, M.G. Geographical Distribution of Carnivore Hosts and Genotypes of Canine Distemper Virus (CDV) Worldwide: A Scoping Review and Spatial Meta-Analysis. Transbound. Emerg. Dis. 2025, 2025, 6632068. [Google Scholar] [CrossRef]
- Deem, S.L.; Spelman, L.H.; Yates, R.A.; Montali, R.J. Canine distemper in terrestrial carnivores: A review. J. Zoo Wildl. Med. 2000, 31, 441–451. [Google Scholar] [CrossRef]
- Duque-Valencia, J.; Sarute, N.; Olarte-Castillo, X.A.; Ruíz-Sáenz, J. Evolution and interspecies transmission of canine distemper virus—An outlook of the diverse evolutionary landscapes of a multi-host virus. Viruses 2019, 11, 582. [Google Scholar] [CrossRef]
- Kapil, S.; Yeary, T.J. Canine Distemper Spillover in Domestic Dogs from Urban Wildlife. Vet. Clin. North Am. Small Anim. Pr. 2011, 41, 1069–1086. [Google Scholar] [CrossRef]
- Wilkes, R.P. Canine Distemper Virus in Endangered Species: Species Jump, Clinical Variations, and Vaccination. Pathogens 2022, 12, 57. [Google Scholar] [CrossRef]
- Rendon-Marin, S.; Higuita-Gutiérrez, L.F.; Ruiz-Saenz, J. Safety and Immunogenicity of Morbillivirus canis Vaccines for Domestic and Wild Animals: A Scoping Review. Viruses 2024, 16, 1078. [Google Scholar] [CrossRef] [PubMed]
- Martella, V.; Blixenkrone-Møller, M.; Elia, G.; Lucente, M.S.; Cirone, F.; Decaro, N.; Nielsen, L.; Bányai, K.; Carmichael, L.E.; Buonavoglia, C. Lights and shades on a historical vaccine canine distemper virus, the Rockborn strain. Vaccine 2011, 29, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Harder, T.C.; Osterhaus, A.D. Canine distemper virus-a morbillivirus in search of new hosts? Trends Microbiol. 1997, 5, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Buczkowski, H.; Muniraju, M.; Parida, S.; Banyard, A.C. Morbillivirus vaccines: Recent successes and future hopes. Vaccine 2014, 32, 3155–3161. [Google Scholar] [CrossRef]
- Noyce, R.S.; Delpeut, S.; Richardson, C.D. Dog nectin-4 is an epithelial cell receptor for canine distemper virus that facilitates virus entry and syncytia formation. Virology 2013, 436, 210–220. [Google Scholar] [CrossRef]
- Summers, B.A.; Appel, M.J. Aspects of canine distemper virus and measles virus encephalomyelitis. Neuropathol. Appl. Neurobiol. 1994, 20, 525–534. [Google Scholar] [CrossRef]
- Panzera, Y.; Sarute, N.; Iraola, G.; Hernández, M.; Pérez, R. Molecular phylogeography of canine distemper virus: Geographic origin and global spreading. Mol. Phylogenet Evol. 2015, 92, 147–154. [Google Scholar] [CrossRef]
- George, A.M.; Wille, M.; Wang, J.; Anderson, K.; Cohen, S.; Moselen, J.; Lee, L.Y.Y.; Suen, W.W.; Bingham, J.; Dalziel, A.E.; et al. A novel and highly divergent Canine Distemper Virus lineage causing distemper in ferrets in Australia. Virology 2022, 576, 117–126. [Google Scholar] [CrossRef]
- Rockborn, G. An attenuated strain of canine distemper virus in tissue culture. Nature 1959, 184 (Suppl. 11), 822. [Google Scholar] [CrossRef]
- Zhang, H.; Shan, F.; Zhou, X.; Li, B.; Zhai, J.Q.; Zou, S.Z.; Wu, M.F.; Chen, W.; Zhai, S.L.; Luo, M.L. Outbreak and genotyping of canine distemper virus in captive Siberian tigers and red pandas. Sci. Rep. 2017, 7, 8132. [Google Scholar] [CrossRef]
- Shi, N.; Zhang, L.; Yu, X.; Zhu, X.; Zhang, S.; Zhang, D.; Duan, M. Insight into an Outbreak of Canine Distemper Virus Infection in Masked Palm Civets in China. Front. Vet. Sci. 2021, 8, 728238. [Google Scholar] [CrossRef]
- Wang, J.; Liu, L.; Zong, X.; Wang, C.; Zhu, G.; Yang, G.; Jiang, Y.; Yang, W.; Huang, H.; Shi, C.; et al. Immunogenicity and protective efficacy of a novel bacterium-like particle-based vaccine displaying canine distemper virus antigens in mice and dogs. Microbiol. Spectr. 2024, 12, e03477-23. [Google Scholar] [CrossRef]
- Wimsatt, J.; Biggins, D.; Innes, K.; Taylor, B.; Garell, D. Evaluation of oral and subcutaneous delivery of an experimental canarypox recombinant canine distemper vaccine in the Siberian polecat (Mustela eversmanni). J. Zoo Wildl. Med. 2003, 34, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.; Rubio, S.; Salvador, L.C.M.; Nemeth, N.M.; Fishburn, J.D.; Gottdenker, N.L. Canine distemper virus phylogenetic structure and ecological correlates of infection in mesocarnivores across anthropogenic land use gradients. Microbiol. Spectr. 2025, 13, e0122524. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, D.M.; Dunn, A.M.; Smith, M.J.; Telfer, S. Wildlife diseases: From individuals to ecosystems. J. Anim. Ecol. 2011, 80, 19–38. [Google Scholar] [CrossRef] [PubMed]
- Martella, V.; Elia, G.; Buonavoglia, C. Canine distemper virus. Vet. Clin. N. Am. Small Anim. Pract. 2008, 38, 787–797. [Google Scholar] [CrossRef]
- Bradley, C.A.; Altizer, S. Urbanization and the ecology of wildlife diseases. Trends Ecol. Evol. 2007, 22, 95–102. [Google Scholar] [CrossRef]
- Candela, M.G.; Fanelli, A.; Carvalho, J.; Serrano, E.; Domenech, G.; Alonso, F.; Martínez-Carrasco, C. Urban landscape and infection risk in free-roaming cats. Zoonoses Public Health 2022, 69, 295–311. [Google Scholar] [CrossRef]
- Appel, M.J.G. Reversion to virulence of attenuated canine distemper virus in vivo and in vitro. J. Gen. Virol. 1978, 41, 385–393. [Google Scholar] [CrossRef]
- Ke, G.M.; Ho, C.H.; Chiang, M.J.; Sanno-Duanda, B.; Chung, C.S.; Lin, M.Y.; Shi, Y.Y.; Yang, M.H.; Tyan, Y.C.; Liao, P.C.; et al. Phylodynamic analysis of the canine distemper virus hemagglutinin gene. BMC Vet. Res. 2015, 11, 164. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.; Akl, E.A.; Brennan, S.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Fairley, R.; Knesl, O.; Pesavento, P.; Elias, B. Post-vaccinal distemper encephalitis in two Border Collie cross littermates. N. Zeal. Vet. J. 2015, 63, 117–120. [Google Scholar] [CrossRef]
- Vandenberghe, H.; Escauriaza, L.; Nye, G.; Teague, M.; Granger, N. Postvaccination encephalomyelitis in German pinschers. Vet. Rec. 2021, 188, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.M.; Hartley, W.J.; Hodgkinson, N.L. An outbreak of post-vaccinal suspected distemper-like encephalitis in farmed ferrets (Mustela putorius furo). N. Zeal. Vet. J. 1988, 36, 173–176. [Google Scholar] [CrossRef]
- Haig, D.A. Canine distemper-immunisation with avianised virus Onderstepoort. J. Vet. Res. 1956, 27, 19–53. [Google Scholar]
- Krakowka, S.; Olsen, R.G.; Axthelm, M.K.; Rice, J.; Winters, K. Canine parvovirus infection potentiates canine distemper encephalitis attributable to modified live-virus vaccine. J. Am. Vet. Med. Assoc. 1982, 180, 137–139. [Google Scholar] [CrossRef] [PubMed]
- McCandlish, I.A.; Cornwell, H.J.; Thompson, H.; Nash, A.S.; Lowe, C.M. Distemper encephalitis in pups after vaccination of the dam. Vet. Rec. 1992, 130, 27–30. [Google Scholar] [CrossRef]
- Rzeutka, A.; Mizak, B. Sequence analysis of the fragment of the phosphoprotein gene of Polish distemper virus isolates. Arch. Virol. 2003, 148, 1623–1631. [Google Scholar] [CrossRef]
- Keawcharoen, J.; Theamboonlers, A.; Jantaradsamee, P.; Rungsipipat, A.; Poovorawan, Y.; Oraveerakul, K. Nucleotide sequence analysis of nucleocapsid protein gene of canine distemper virus isolates in Thailand. Vet. Microbiol. 2005, 105, 137–142. [Google Scholar] [CrossRef]
- Fischer, C.D.; Ikuta, N.; Canal, C.W.; Makiejczuk, A.; da Costa Allgayer, M.; Cardoso, C.H.; Lehmann, F.K.; Fonseca, A.S.; Lunge, V.R. Detection and differentiation of field and vaccine strains of canine distemper virus using reverse transcription followed by nested real-time PCR (RT-nqPCR) and RFLP analysis. J. Virol. Methods 2013, 194, 39–45. [Google Scholar] [CrossRef]
- Wilkes, R.P.; Sanchez, E.; Riley, M.C.; Kennedy, M.A. Real-time reverse transcription polymerase chain reaction method for detection of Canine distemper virus modified live vaccine shedding for differentiation from infection with wild-type strains. J. Vet. Diagn. Invest. 2014, 26, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Anis, E.; Newell, T.K.; Dyer, N.; Wilkes, R.P. Phylogenetic analysis of the wild-type strains of canine distemper virus circulating in the United States. Virol. J. 2018, 15, 118. [Google Scholar] [CrossRef] [PubMed]
- Ricci, I.; Cersini, A.; Manna, G.; Marcario, G.A.; Conti, R.; Brocherel, G.; Grifoni, G.; Eleni, C.; Scicluna, M.T. A Canine Distemper Virus Retrospective Study Conducted from 2011 to 2019 in Central Italy (Latium and Tuscany Regions). Viruses 2021, 13, 272. [Google Scholar] [CrossRef] [PubMed]
- Pekkarinen, H.M.; Karkamo, V.K.; Vainio-Siukola, K.J.; Hautaniemi, M.K.; Kinnunen, P.M.; Gadd, T.K.; Holopainen, R.H. Post-vaccinal distemper-like disease in two dog litters with confirmed infection of vaccine virus strain. Comp. Immunol. Microbiol. Infect. Dis. 2024, 105, 102114. [Google Scholar] [CrossRef]
- Frölich, K.; Czupalla, O.; Haas, L.; Hentschke, J.; Dedek, J.; Fickel, J. Epizootiological investigations of canine distemper virus in free-ranging carnivores from Germany. Vet. Microbiol. 2000, 74, 283–292. [Google Scholar] [CrossRef]
- Lednicky, J.A.; Dubach, J.; Kinsel, M.J.; Meehan, T.P.; Bocchetta, M.; Hungerford, L.L.; Sarich, N.A.; Witecki, K.E.; Braid, M.D.; Pedrak, C.; et al. Genetically distant American Canine distemper virus lineages have recently caused epizootics with somewhat different characteristics in raccoons living around a large suburban zoo in the USA. Virol. J. 2004, 1, 2. [Google Scholar] [CrossRef]
- Martella, V.; Elia, G.; Lucente, M.S.; Decaro, N.; Lorusso, E.; Banyai, K.; Blixenkrone-Møller, M.; Lan, N.T.; Yamaguchi, R.; Cirone, F.; et al. Genotyping canine distemper virus (CDV) by a hemi-nested multiplex PCR provides a rapid approach for investigation of CDV outbreaks. Vet. Microbiol. 2007, 122, 32–42. [Google Scholar] [CrossRef]
- Giacinti, J.A.; Pearl, D.L.; Ojkic, D.; Campbell, G.D.; Jardine, C.M. Genetic characterization of canine distemper virus from wild and domestic animal submissions to diagnostic facilities in Canada. Prev. Vet. Med. 2022, 198, 105535. [Google Scholar] [CrossRef]
- Ek-Kommonen, C.; Rudbäck, E.; Anttila, M.; Aho, M.; Huovilainen, A. Canine distemper of vaccine origin in European mink, Mustela lutreola—A case report. Vet. Microbiol. 2003, 92, 289–293. [Google Scholar] [CrossRef]
- Kadam, R.G.; Karikalan, M.; Siddappa, C.M.; Mahendran, K.; Srivastava, G.; Rajak, K.K.; Bhardwaj, Y.; Varshney, R.; War, Z.A.; Singh, R.; et al. Molecular and pathological screening of canine distemper virus in Asiatic lions, tigers, leopards, snow leopards, clouded leopards, leopard cats, jungle cats, civet cats, fishing cat, and jaguar of different states, India. Infect. Genet. Evol. 2022, 98, 105211. [Google Scholar] [CrossRef]
- Hartley, W.J. A post-vaccinal inclusion body encephalitis in dogs. Vet. Pathol. 1974, 11, 301–312. [Google Scholar] [CrossRef]
- Bestetti, G.; Fatzer, R.; Frankhauser, R. Encephalitis following vaccination against distemper and infectious hepatitis in the dog. An optical and ultrastructural study. Acta Neuropathol. 1978, 43, 69–75. [Google Scholar] [CrossRef]
- Cornwell, H.J.; Thompson, H.; McCandlish, I.A.; Macartney, L.; Nash, A.S. Encephalitis in dogs associated with a batch of canine distemper (Rockborn) vaccine. Vet. Rec. 1988, 122, 54–59. [Google Scholar] [CrossRef]
- Frisk, A.L.; König, M.; Moritz, A.; Baumgärtner, W. Detection of canine distemper virus nucleoprotein RNA by reverse transcription-PCR using serum, whole blood, and cerebrospinal fluid from dogs with distemper. J. Clin. Microbiol. 1999, 37, 3634–3643. [Google Scholar] [CrossRef]
- Rätsep, E.; Ojkic, D. Canine distemper virus infection of vaccinal origin in a 14-week-old puppy. J. Vet. Diagn. Invest. 2024, 36, 287–290. [Google Scholar] [CrossRef]
- Pardo, I.D.; Johnson, G.C.; Kleiboeker, S.B. Phylogenetic characterization of canine distemper viruses detected in naturally infected dogs in North America. J. Clin. Microbiol. 2005, 43, 5009–5017. [Google Scholar] [CrossRef] [PubMed]
- Uema, M.; Ohashi, K.; Wakasa, C.; Kai, C. Phylogenetic and restriction fragment length polymorphism analyses of hemagglutinin (H) protein of canine distemper virus isolates from domestic dogs in Japan. Virus Res. 2005, 109, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Gulliver, E.; Taylor, H.; Eames, M.; Chernyavtseva, A.; Jauregui, R.; Wilson, A.; Bestbier, M.; O’Connell, J.; Buckle, K.; Castillo-Alcala, F. Investigation of post-vaccinal canine distemper involving the Rockborn-like strain in nine puppies in New Zealand. N. Zeal. Vet. J. 2025, 73, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Bush, M.; Montali, R.J.; Brownstein, D.; James, A.E., Jr.; Appel, M.J. Vaccine-induced canine distemper in a lesser panda. J. Am. Vet. Med. Assoc. 1976, 169, 959–960. [Google Scholar] [CrossRef]
- Kennedy, J.M.; Earle, J.A.P.; Omar, S.; Abdullah, H.; Nielsen, O.; Roelke-Parker, M.E.; Cosby, S.L. Canine and Phocine Distemper Viruses: Global Spread and Genetic Basis of Jumping Species Barriers. Viruses 2019, 11, 944. [Google Scholar] [CrossRef]
- Carpenter, J.W.; Appel, M.J.; Erickson, R.C.; Novilla, M.N. Fatal vaccine-induced canine distemper virus infection in black-footed ferrets. J. Am. Vet. Med. Assoc. 1976, 169, 961–964. [Google Scholar] [CrossRef]
- Kazacos, K.R.; Thacker, H.L.; Shivaprasad, H.L.; Burger, P.P. Vaccine-induced distemper in kinkajous. J. Am. Vet. Med. Assoc. 1981, 179, 1166–1168. [Google Scholar] [CrossRef]
- Halbrooks, R.D.; Swango, L.J.; Schnurrenberger, P.R.; Mitchell, F.E.; Hill, E.P. Response of gray foxes to modified live-virus canine distemper vaccines. J. Am. Vet. Med. Assoc. 1981, 179, 1170–1174. [Google Scholar] [CrossRef]
- Thomas-Baker, B. Vaccination-induced distemper in maned wolves, vaccination-induced corneal opacity in a maned wolf. In Proceedings of the American Association of Zoo Veterinarians, Scottsdale, AZ, USA, 5–10 October 1985. [Google Scholar]
- Vergara-Wilson, V.; Hidalgo-Hermoso, E.; Sanchez, C.R.; Abarca, M.J.; Navarro, C.; Celis-Diez, S.; Soto-Guerrero, P.; Diaz-Ayala, N.; Zordan, M.; Cifuentes-Ramos, F.; et al. Canine Distemper Outbreak by Natural Infection in a Group of Vaccinated Maned Wolves in Captivity. Pathogens 2021, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Montali, R.J.; Bartz, C.R.; Bush, M. Canine distemper virus. In Virus Infections of Carnivores; Appel, M.J., Ed.; Elsevier Science Publishers: Amsterdam, The Netherlands, 1987; 441p. [Google Scholar]
- Tamukai, K.; Minami, S.; Kurihara, R.; Shimoda, H.; Mitsui, I.; Maeda, K.; Une, Y. Molecular evidence for vaccine-induced canine distemper virus and canine adenovirus 2 coinfection in a fennec fox. J. Vet. Diagn. Invest. 2020, 32, 598–603. [Google Scholar] [CrossRef] [PubMed]
- McCormick, A.E. Canine distemper in African Cape hunting dogs (Lycaon pictus). Possibly vaccine induced. J. Zoo. Anim. Med. 1983, 14, 66–71. [Google Scholar] [CrossRef]
- Van Heerden, J.; Bainbridge, N.; Burroughs, R.E.J.; Kriek, N.P.J. Distemper-like disease and encephalitozoonosis in wild dogs (Lycaon pictus). J. Wildl. Dis. 1989, 25, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Woodroffe, R. Modified live distemper vaccines carry low mortality risk for captive African wild dogs, Lycaon pictus. J. Zoo Wildl. Med. 2021, 52, 176–184. [Google Scholar] [CrossRef]
- McInnes, E.F.; Burroughs, R.E.; Duncan, N.M. Possible vaccine-induced canine distemper in a South American bush dog (Speothos venaticus). J. Wildl. Dis. 1992, 28, 614–617. [Google Scholar] [CrossRef]
- Sutherland-Smith, M.R.; Rideout, B.A.; Mikolon, A.B.; Appel, M.J.; Morris, P.J.; Shima, A.L.; Janssen, D.J. Vaccine-induced canine distemper in European mink, Mustela lutreola. J. Zoo Wildl. Med. 1997, 28, 312–318. [Google Scholar]
- Cottrell, W.O.; Keel, M.K.; Brooks, J.W.; Mead, D.G.; Phillips, J.E. First report of clinical disease associated with canine distemper virus infection in a wild black bear (Ursus americanus). J. Wildl. Dis. 2013, 49, 1024–1027. [Google Scholar] [CrossRef] [PubMed]
- Loots, A.K.; Mokgokong, P.S.; Mitchell, E.; Venter, E.H.; Kotze, A.; Dalton, D.L. Phylogenetic analysis of canine distemper virus in South African wildlife. PLoS ONE 2018, 13, e0199993. [Google Scholar] [CrossRef] [PubMed]
- Lunardi, M.; Darold, G.M.; Amude, A.M.; Headley, S.A.; Sonne, L.; Yamauchi, K.C.I.; Boabaid, F.M.; Alfieri, A.F.; Alfieri, A.A. Canine distemper virus active infection in order Pilosa, family Myrmecophagidae, species Tamandua tetradactyla. Vet. Microbiol. 2018, 220, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Karki, M.; Rajak, K.K.; Singh, R.P. Canine morbillivirus (CDV): A review on current status, emergence and the diagnostics. Virus Dis. 2022, 33, 309–321. [Google Scholar] [CrossRef]
- Kennedy, S. Morbillivirus infections in aquatic mammals. J. Comp. Pathol. 1998, 119, 201–225. [Google Scholar] [CrossRef]
- Sakai, K.; Nagata, N.; Ami, Y.; Seki, F.; Suzaki, Y.; Iwata-Yoshikawa, N.; Suzuki, T.; Fukushi, S.; Mizutani, T.; Yoshikawa, T.; et al. Lethal canine distemper virus outbreak in cynomolgus monkeys in Japan in 2008. J. Virol. 2013, 87, 1105–1114. [Google Scholar] [CrossRef]
- Sun, Z.; Li, A.; Ye, H.; Shi, Y.; Hu, Z.; Zeng, L. Natural infection with canine distemper virus in hand-feeding Rhesus monkeys in China. Vet. Microbiol. 2010, 141, 374–378. [Google Scholar] [CrossRef]
- Mares-Guia, M.A.M.M.; Furtado, M.C.; Chalhoub, F.L.L.; Portugal, M.D.; de Oliveira Coelho, J.M.C.; de Filippis, A.M.B.; Naveca, F.G. Coinfection with Canine Distemper Virus and Yellow Fever Virus in a Neotropical Primate in Brazil. Viruses 2024, 16, 1670. [Google Scholar] [CrossRef]
- Debesa Belizário Granjeiro, M.; Lima Kavasaki, M.; Morgado, T.O.; Avelino Dandolini Pavelegini, L.; Alves de Barros, M.; Fontana, C.; de Assis Bianchini, M.; de Oliveira Souza, A.; Gonçalves Lima Oliveira Santos, A.R.; Lunardi, M.; et al. First report of a canine morbillivirus infection in a giant anteater (Myrmecophaga tridactyla) in Brazil. Vet. Med. Sci. 2020, 6, 606–611. [Google Scholar] [CrossRef]
- Wang, T.; Du, H.; Feng, N.; Liu, Y.; Xu, Y.; Sun, H.; Peng, P.; Qin, S.; Zhang, X.; Liu, Y.; et al. First complete genomic sequence analysis of canine distemper virus in wild boar. Virol. Sin. 2024, 39, 702–704. [Google Scholar] [CrossRef]
- Martínez-Gutierrez, M.; Ruiz-Saenz, J. Diversity of susceptible hosts in canine distemper virus infection: A systematic review and data synthesis. BMC Vet. Res. 2016, 12, 78. [Google Scholar] [CrossRef]
- Devenish-Nelson, E.S.; Harris, S.; Soulsbury, C.D.; Richards, S.A.; Stephens, P.A. Demography of a carnivore, the red fox, Vulpes vulpes: What have we learnt from 70 years of published studies? Oikos 2013, 122, 705–716. [Google Scholar] [CrossRef]
- Prange, S.; Gehrt, S.D.; Wiggers, E.P. Demographic factors contributing to high raccoon densities in urban landscapes. J. Wildl. Manag. 2003, 67, 324. [Google Scholar] [CrossRef]
- Gras, P.; Knuth, S.; Börner, K.; Marescot, L.; Benhaiem, S.; Aue, A.; Wittstatt, U.; Kleinschmit, B.; Kramer-Schadt, S. Landscape Structures Affect Risk of Canine Distemper in Urban Wildlife. Front. Ecol. Evol. 2018, 6, 136. [Google Scholar] [CrossRef]
- Deuel, N.R.; Conner, L.M.; Miller, K.V.; Chamberlain, M.J.; Cherry, M.J.; Tannenbaum, L.V. Habitat selection and diurnal refugia of gray foxes in southwestern Georgia, USA. PLoS ONE 2017, 12, e0186402. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wu, X.; Li, L.; Zou, Y.; Liu, S.; Wang, Z. Evolutionary characteristics of morbilliviruses during serial passages in vitro: Gradual attenuation of virus virulence. Comp. Immunol. Microbiol. Infect. Dis. 2016, 47, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.J.; Streicker, D.G.; Altizer, S. Linking anthropogenic resources to wildlife-pathogen dynamics: A review and meta-analysis. Ecol. Lett. 2015, 18, 483–495. [Google Scholar] [CrossRef]
- Ditchkoff, S.S.; Saalfeld, S.T.; Gibson, C.J. Animal behavior in urban ecosystems: Modifications due to human-induced stress. Urban Ecosyst. 2006, 9, 5–12. [Google Scholar] [CrossRef]
- Bozek, C.K.; Prange, S.; Gehrt, S.D. The influence of anthropogenic resources on multi-scale habitat selection by raccoons. Urban Ecosyst. 2007, 10, 413–425. [Google Scholar] [CrossRef]
- Lednicky, J.A.; Meehan, T.P.; Kinsel, M.J.; Dubach, J.; Hungerford, L.L.; Sarich, N.A.; Witecki, K.E.; Braid, M.D.; Pedrak, C.; Houde, C.M. Effective primary isolation of wild-type canine distemper virus in MDCK, MV1 Lu and Vero cells without nucleotide sequence changes within the entire haemagglutinin protein gene and in subgenomic sections of the fusion and phospho protein genes. J. Virol. Methods 2004, 118, 147–157. [Google Scholar] [CrossRef]
- Yuan, C.; Liu, W.; Wang, Y.; Hou, J.; Zhang, L.; Wang, G. Homologous recombination is a force in the evolution of canine distemper virus. PLoS ONE 2017, 12, e0175416. [Google Scholar] [CrossRef] [PubMed]
- Demeter, Z.; Palade, E.A.; Hornyák, A.; Rusvai, M. Controversial results of the genetic analysis of a canine distemper vaccine strain. Vet. Microbiol. 2010, 142, 420–426. [Google Scholar] [CrossRef]
- Williams, E.S.; Thorne, E.T.; Appel, M.J.; Belitsky, D.W. Canine distemper in black-footed ferrets (Mustela nigripes) from Wyoming. J. Wildl. Dis. 1988, 24, 385–398. [Google Scholar] [CrossRef]
- Tricco, A.C.; Lillie, E.; Zarin, W.; O’Brien, K.K.; Colquhoun, H.; Levac, D.; Moher, D.; Peters, M.D.; Horsley, T.; Weeks, L.; et al. PRISMA Extension for Scoping Reviews (PRISMAScR): Checklist and Explanation. Ann. Intern. Med. 2018, 169, 467–473. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Candela, M.G.; Wipf, A.; Ortega, N.; Huertas-López, A.; Martínez-Carrasco, C.; Perez-Cutillas, P. Tracking the Spatial and Functional Dispersion of Vaccine-Related Canine Distemper Virus Genotypes: Insights from a Global Scoping Review. Viruses 2025, 17, 1045. https://doi.org/10.3390/v17081045
Candela MG, Wipf A, Ortega N, Huertas-López A, Martínez-Carrasco C, Perez-Cutillas P. Tracking the Spatial and Functional Dispersion of Vaccine-Related Canine Distemper Virus Genotypes: Insights from a Global Scoping Review. Viruses. 2025; 17(8):1045. https://doi.org/10.3390/v17081045
Chicago/Turabian StyleCandela, Mónica G., Adrian Wipf, Nieves Ortega, Ana Huertas-López, Carlos Martínez-Carrasco, and Pedro Perez-Cutillas. 2025. "Tracking the Spatial and Functional Dispersion of Vaccine-Related Canine Distemper Virus Genotypes: Insights from a Global Scoping Review" Viruses 17, no. 8: 1045. https://doi.org/10.3390/v17081045
APA StyleCandela, M. G., Wipf, A., Ortega, N., Huertas-López, A., Martínez-Carrasco, C., & Perez-Cutillas, P. (2025). Tracking the Spatial and Functional Dispersion of Vaccine-Related Canine Distemper Virus Genotypes: Insights from a Global Scoping Review. Viruses, 17(8), 1045. https://doi.org/10.3390/v17081045