
Novel Giant Phages vB_AerVM_332-Vera and vB_AerVM_332-Igor and Siphophage vB_AerVS_332-Yulya Infecting the Same Aeromonas veronii Strain


 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Phage Isolation
2.3. Phage Propagation
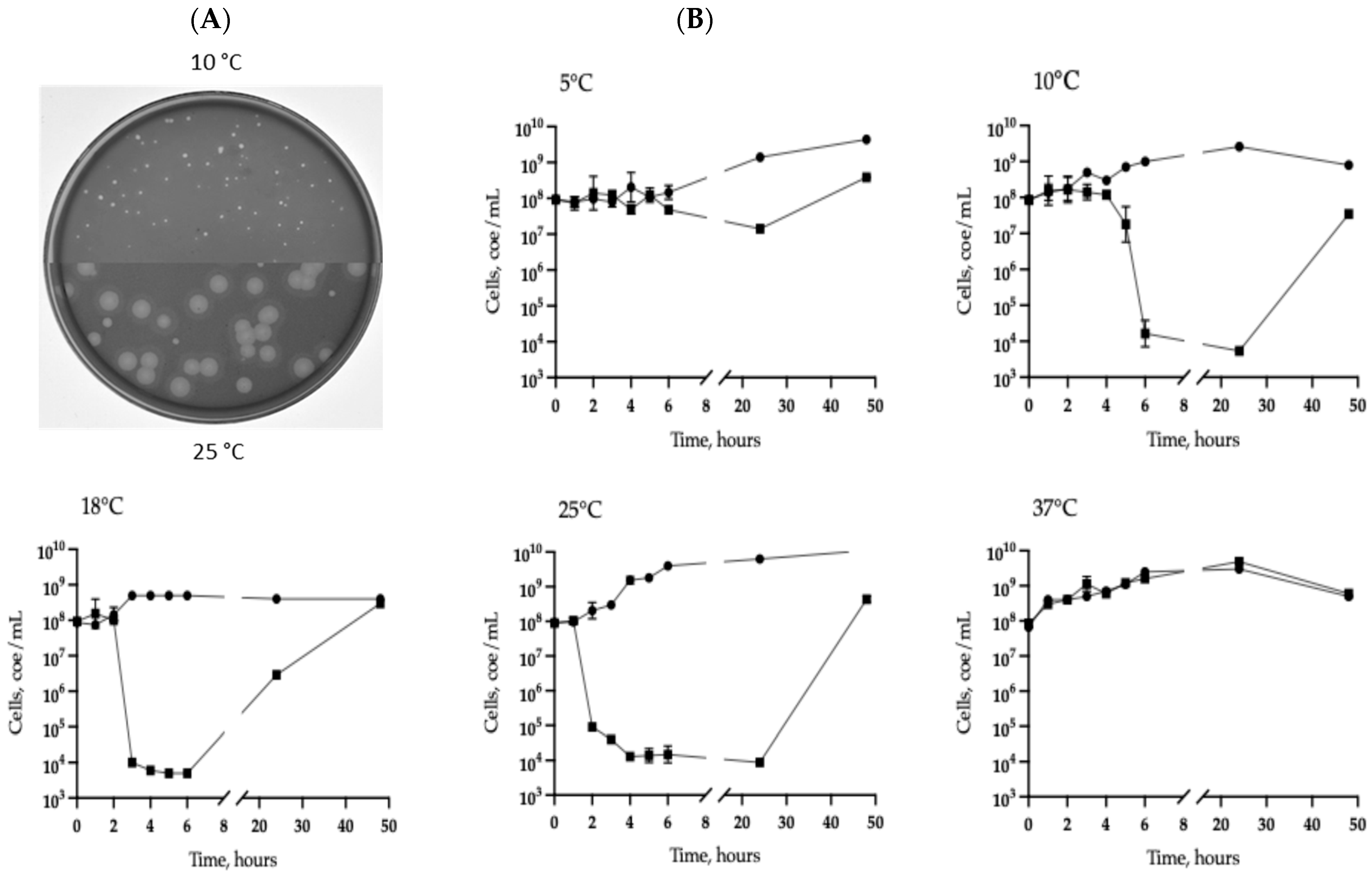
2.4. Biological Properties and Host Range Assay
2.5. Phage DNA Isolation
2.6. Phage DNA Sequencing
2.7. PCR and Real-Time PCR for Phage Genomes Detection
2.8. Phage Genome Analysis
2.9. Phylogenetic Analysis
3. Results
3.1. Identification and Characterization of the Host
3.2. Sequencing of Phage Genomes
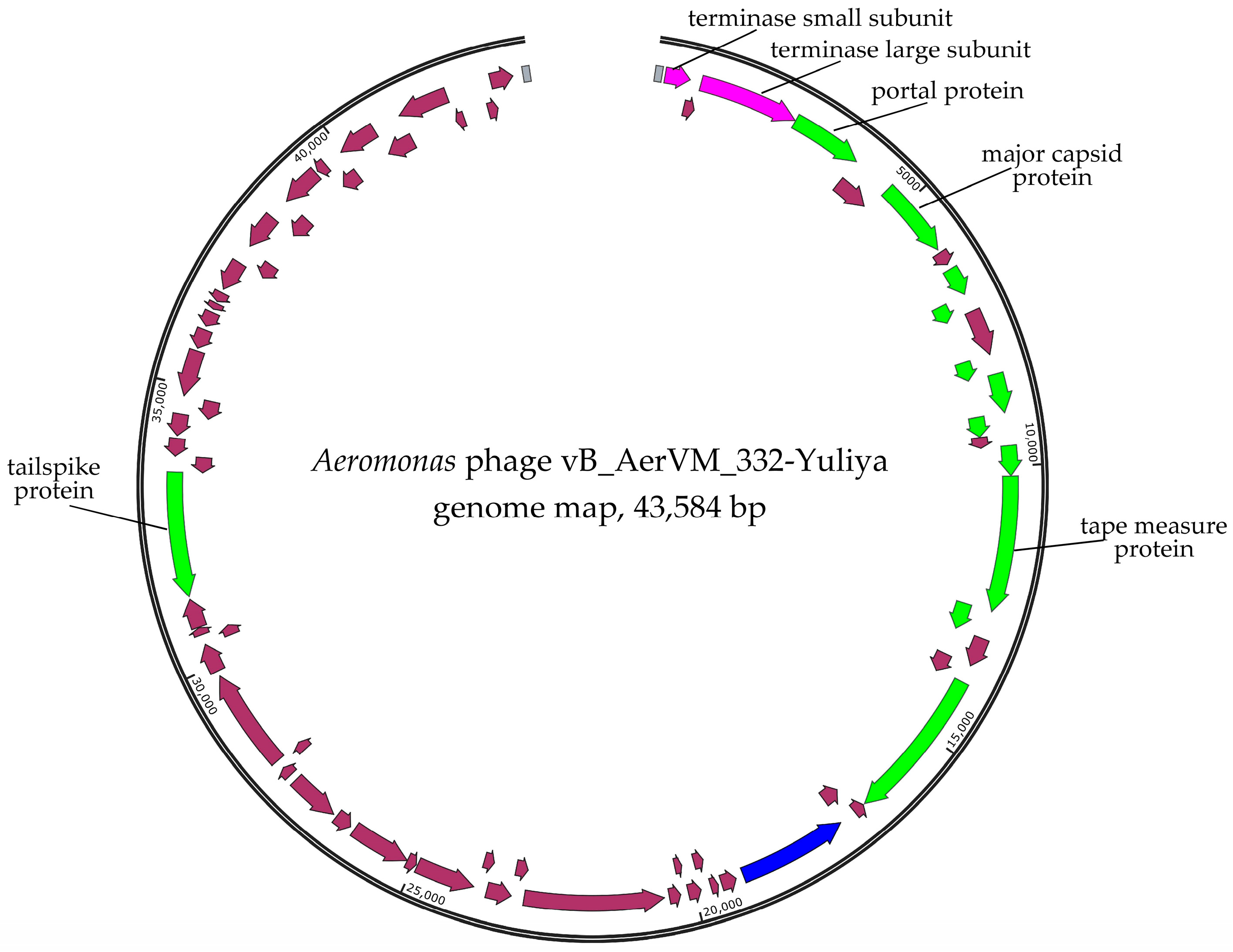
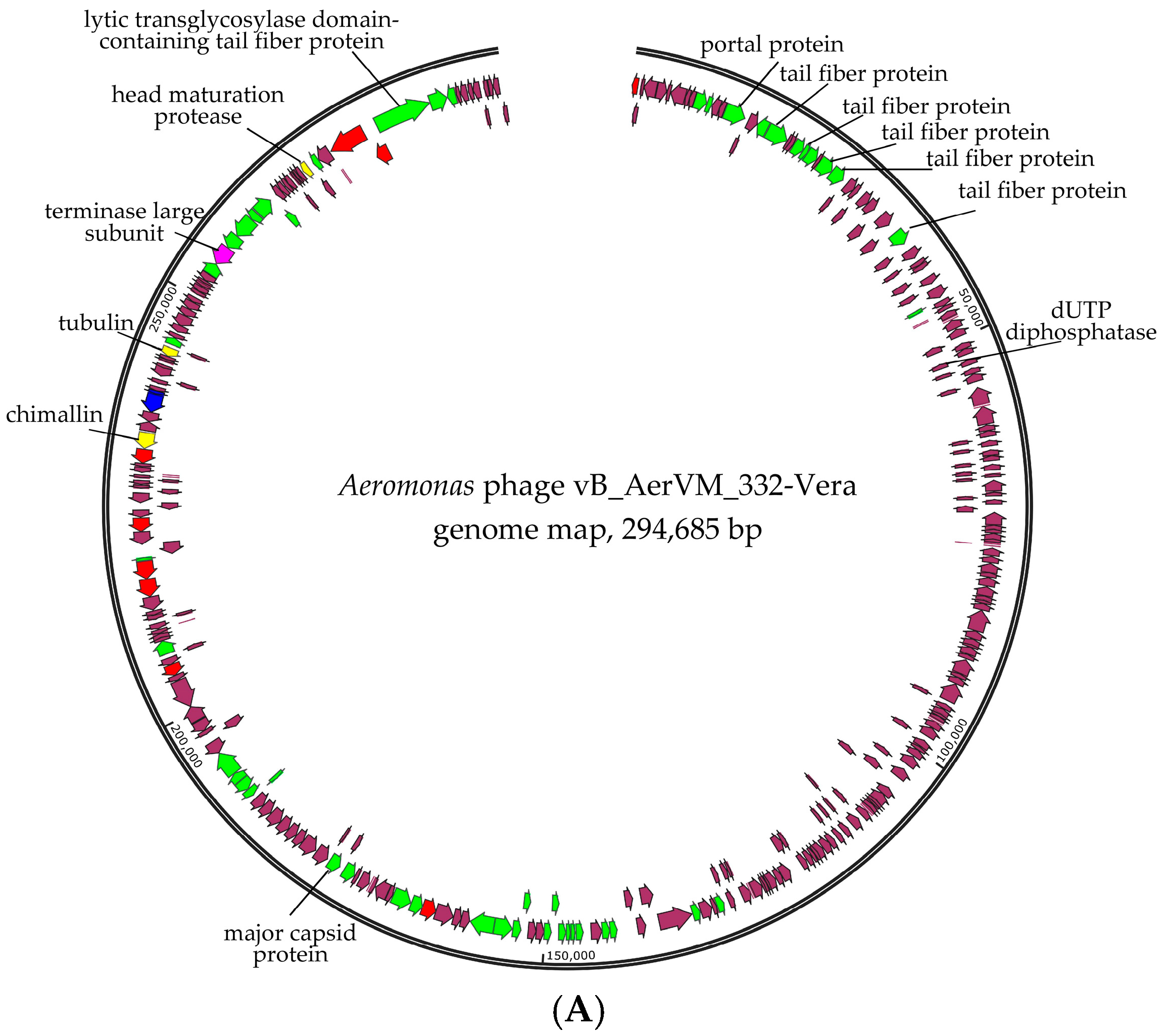
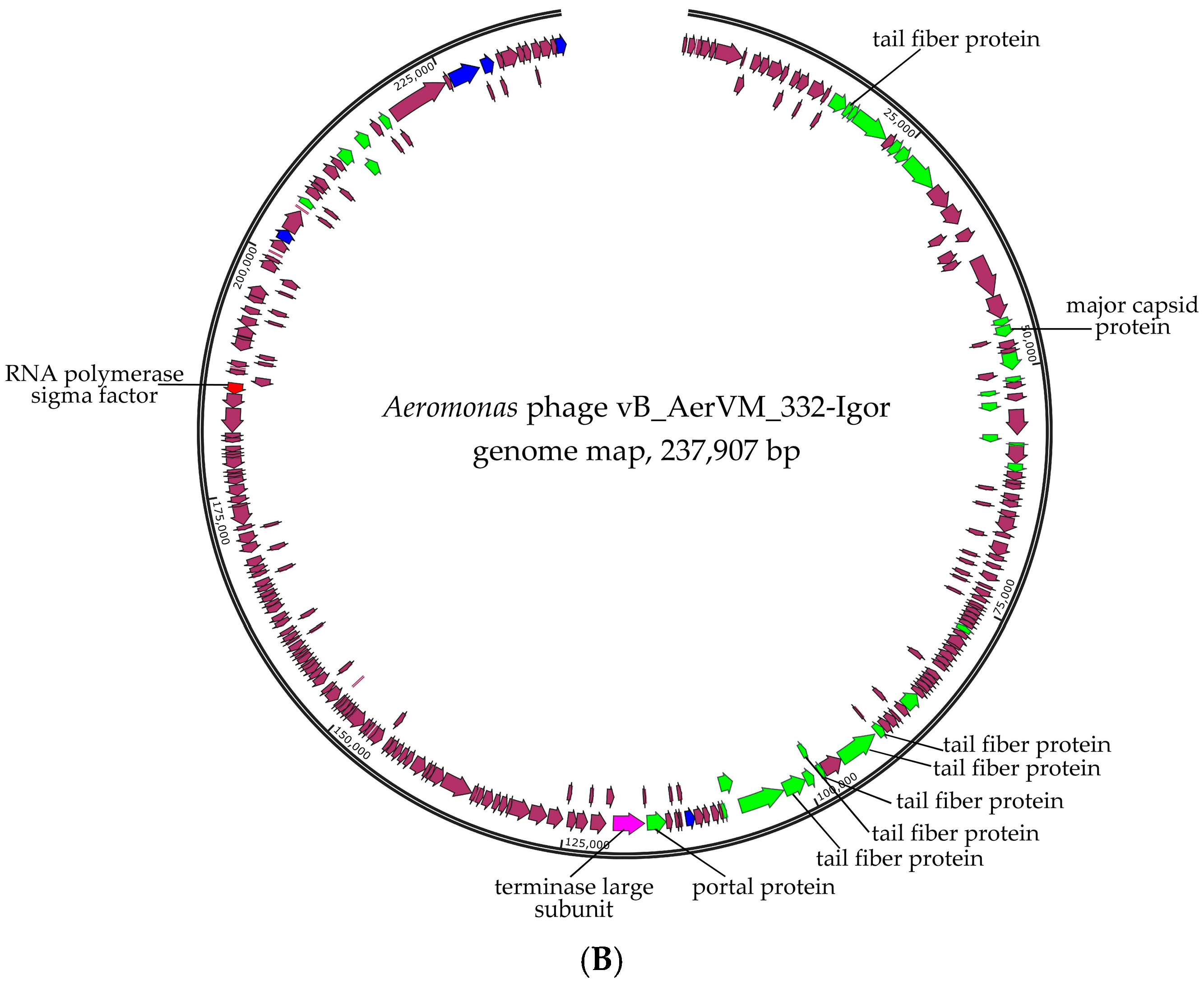
3.3. Phage Genomes Organization
3.4. Phage Genomes Detection by PCR and Real-Time PCR
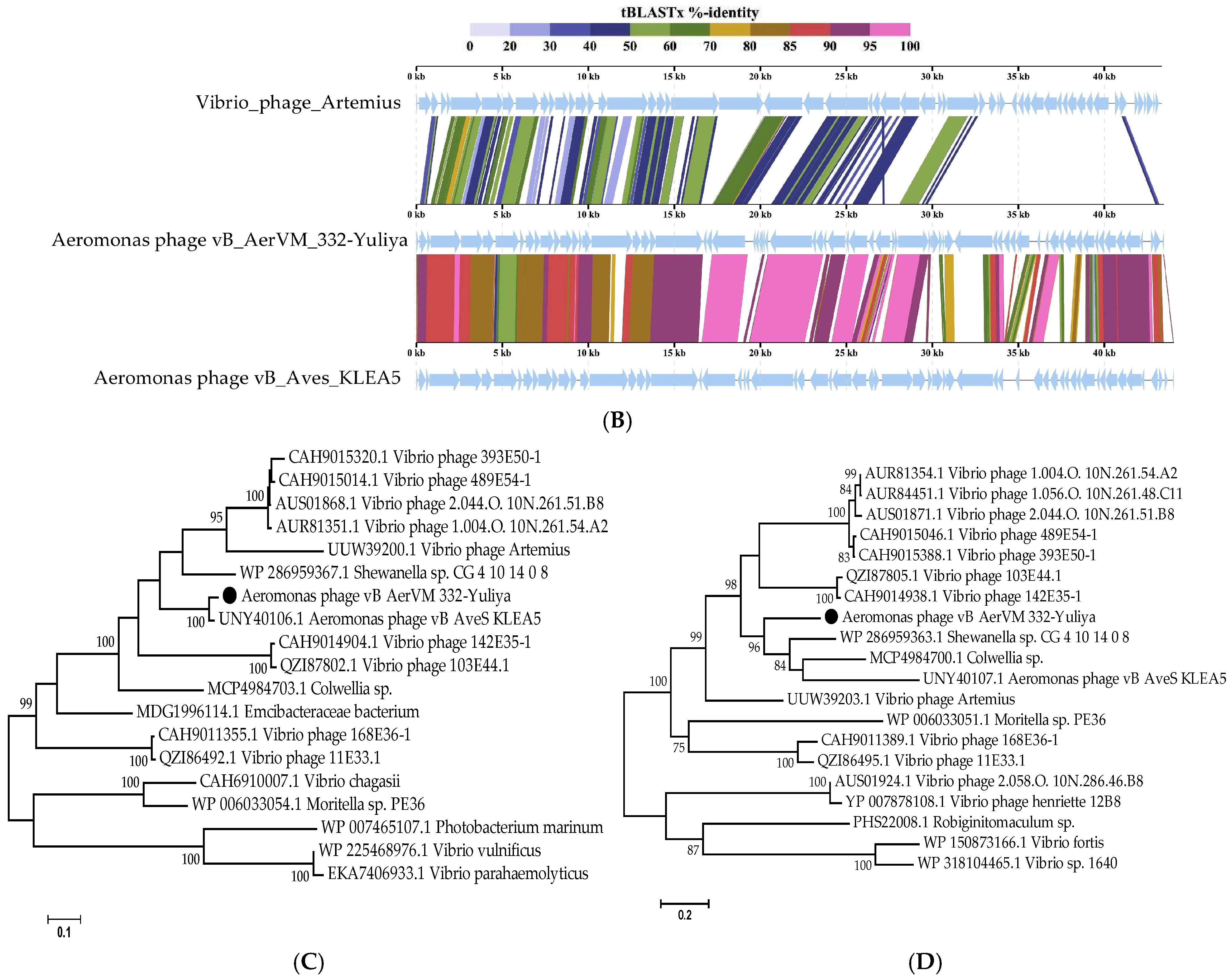
3.5. Comparative Analysis of the 332-Yuliya Genome
3.6. Comparative Analysis of the 332-Vera Genome
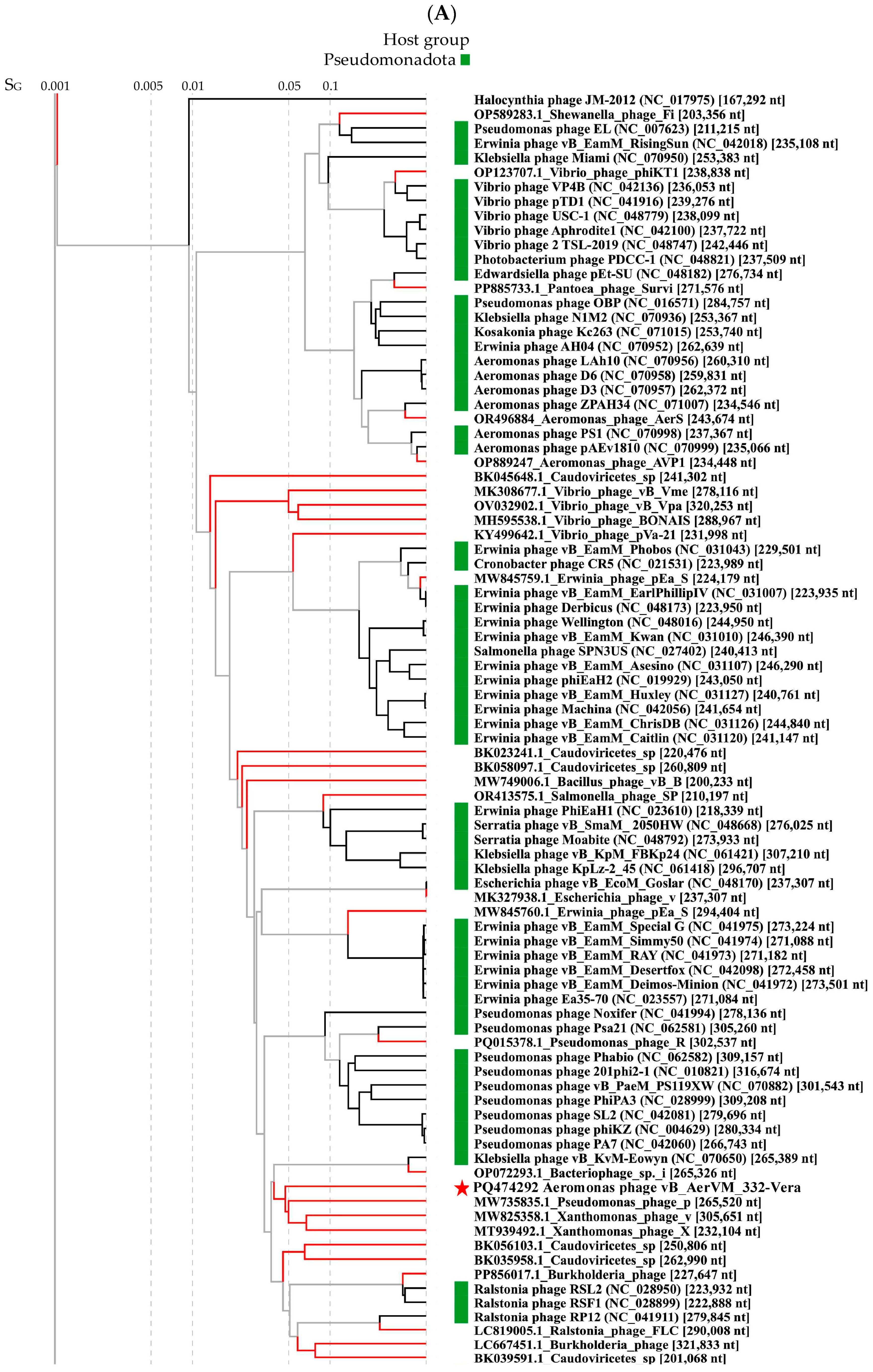
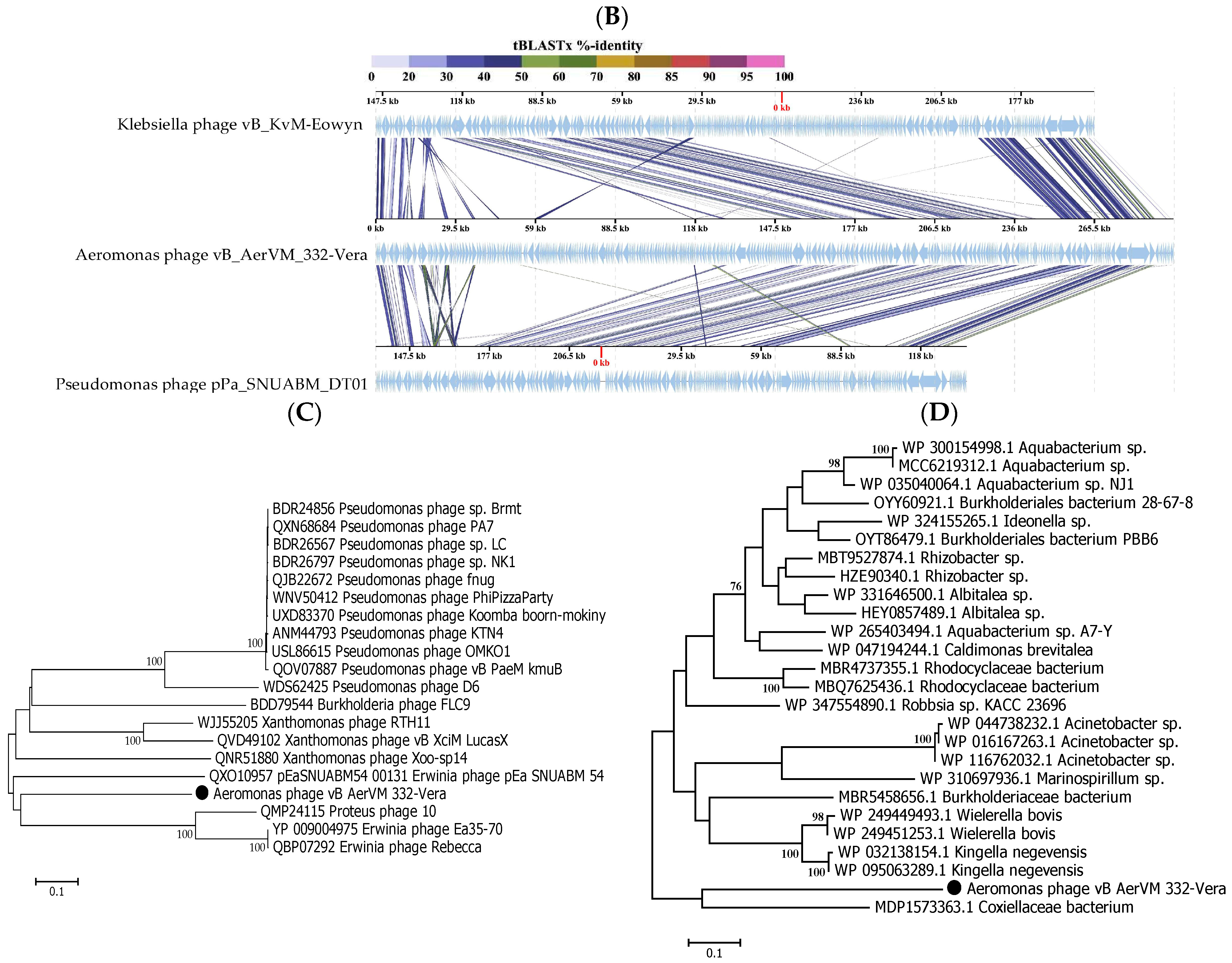
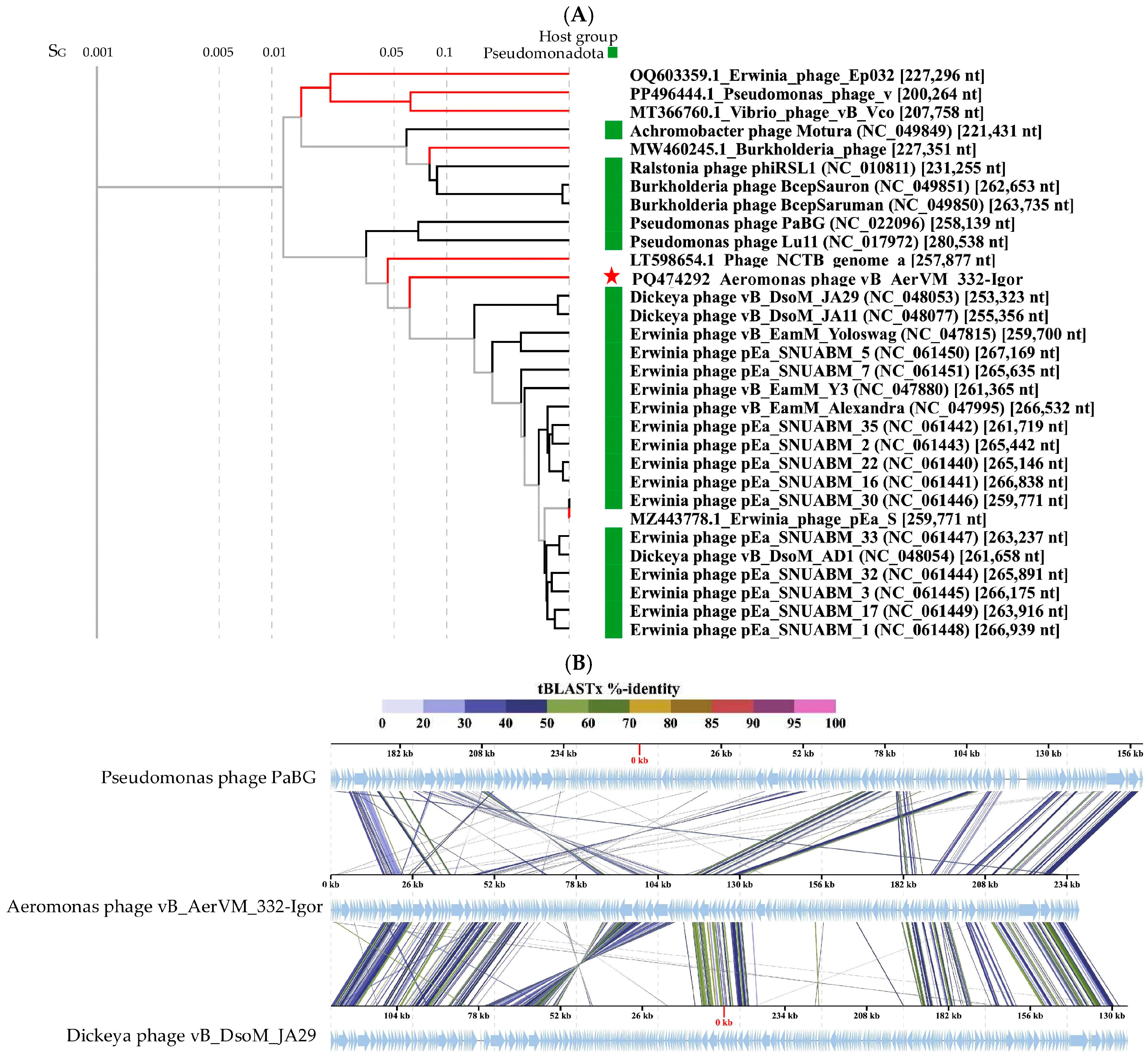
3.7. Comparative Analysis of the 332-Igor Genome
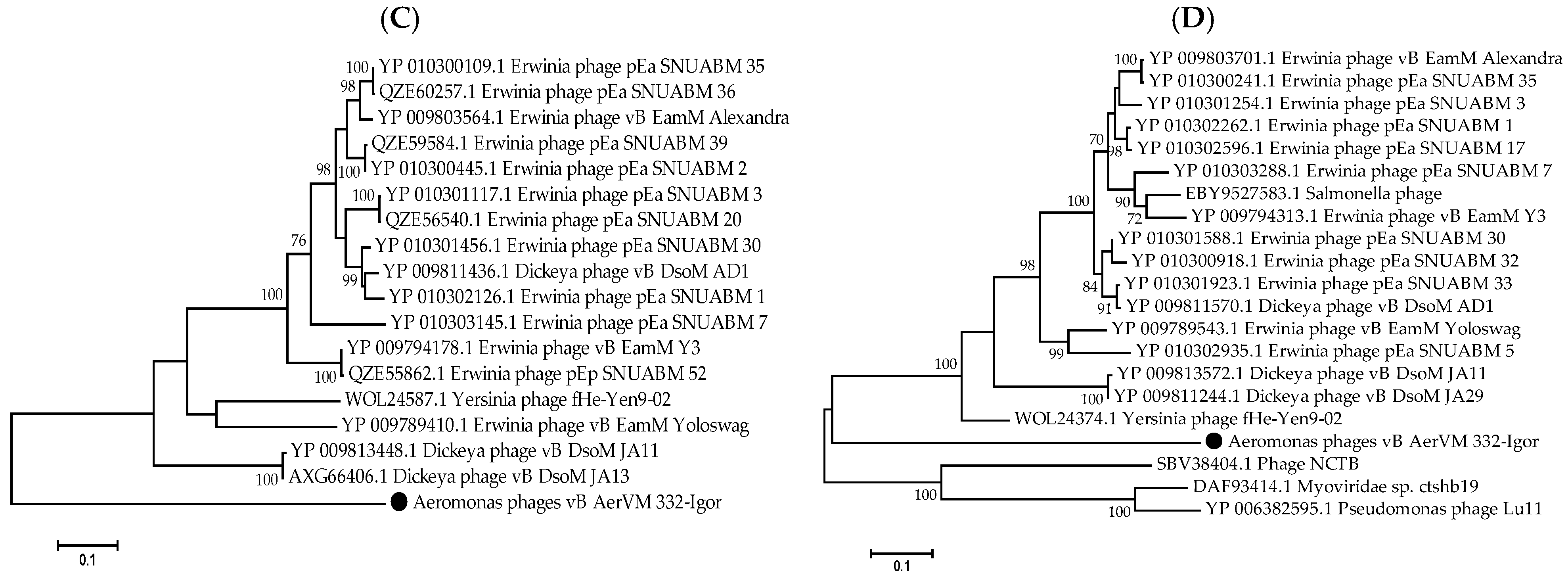
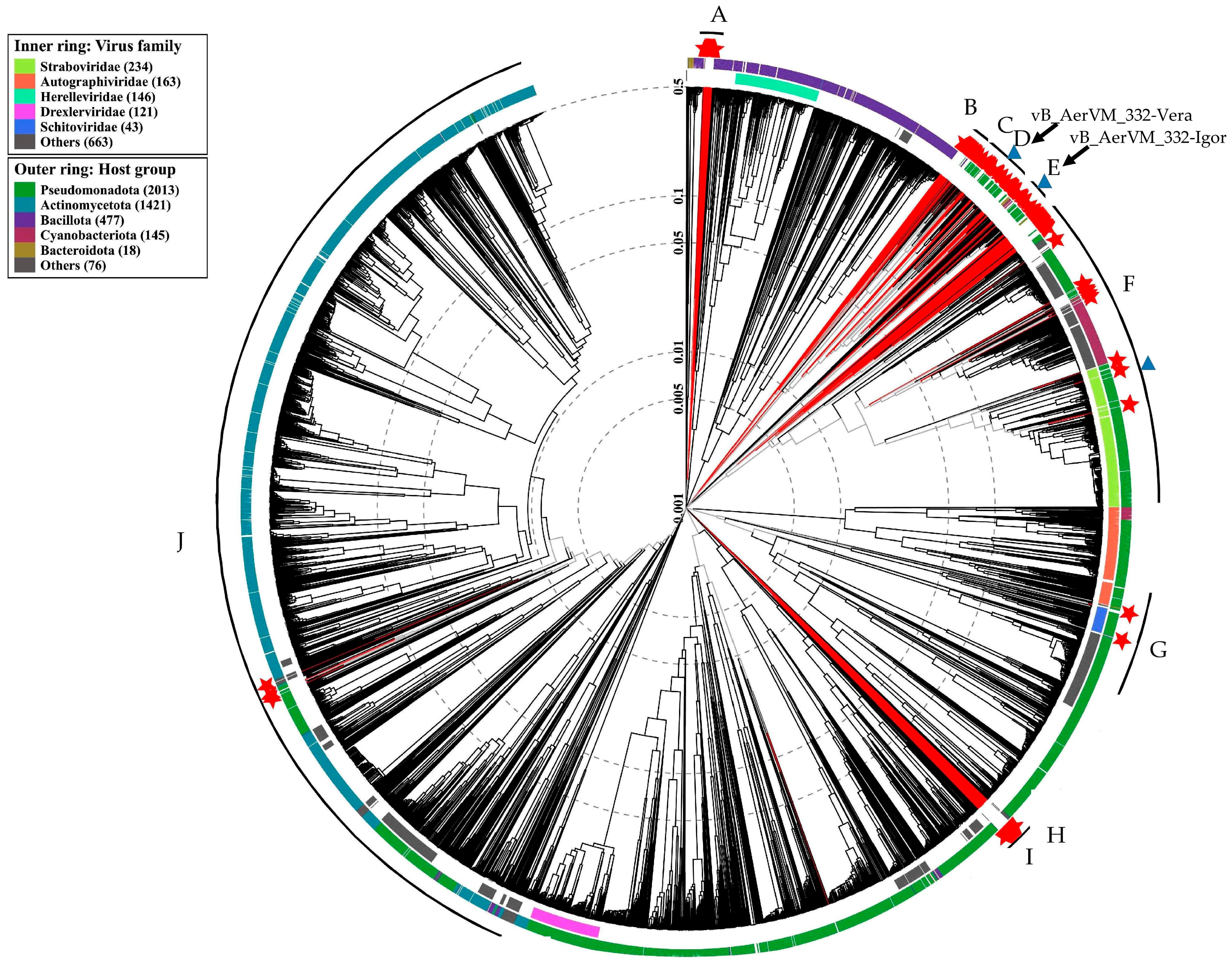
3.8. Clusters Containing Giant Phages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clokie, M.R.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef]
- Hook, A.E.; Beard, D. Isolation and characterization of the T2 bacteriophage of Escherichia coli. J. Biol. Chem. 1946, 165, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Donelli, G. Isolamento di un batteriofago di eccezionali dimensioni attivo su B. Megaterium. Cl. Sci. Fis. Mat. Nat. 1969, 44, 95–97. [Google Scholar]
- Krylov, V.N.; Zhazykov, I. Pseudomonas bacteriophage phiKZ-possible model for studying the genetic control of morphogenesis. Genetika 1978, 14, 678–685. [Google Scholar] [PubMed]
- Mesyanzhinov, V.V.; Robben, J.; Grymonprez, B.; Kostyuchenko, V.A.; Bourkaltseva, M.V.; Sykilinda, N.N.; Krylov, V.N.; Volckaert, G. The genome of bacteriophage φKZ of Pseudomonas aeruginosa. J. Mol. Biol. 2002, 317, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, R.W. Jumbo bacteriophages. In Lesser Known Large dsDNA Viruses; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2009; Volume 328, pp. 229–240. [Google Scholar] [CrossRef]
- Korn, A.M.; Hillhouse, A.E.; Sun, L.; Gill, J.J. Comparative Genomics of Three Novel Jumbo Bacteriophages Infecting Staphylococcus aureus. J. Virol. 2021, 95, e0239120. [Google Scholar] [CrossRef]
- Harding, K.R.; Kyte, N.; Fineran, P.C. Jumbo phages. Curr. Biol. 2023, 33, R750–R751. [Google Scholar] [CrossRef]
- Michniewski, S.; Rihtman, B.; Cook, R.; Jones, M.A.; Wilson, W.H.; Scanlan, D.J.; Millard, A. A new family of “megaphages” abundant in the marine environment. ISME Commun. 2021, 1, 58. [Google Scholar] [CrossRef]
- Al-Shayeb, B.; Sachdeva, R.; Chen, L.-X.; Ward, F.; Munk, P.; Devoto, A.; Castelle, C.J.; Olm, M.R.; Bouma-Gregson, K.; Amano, Y.; et al. Clades of Huge Phages from across Earth’s Ecosystems. Nature 2020, 578, 425–431. [Google Scholar] [CrossRef]
- Nazir, A.; Ali, A.; Qing, H.; Tong, Y. Emerging aspects of jumbo bacteriophages. Infect. Drug Resist. 2021, 14, 5041–5055. [Google Scholar] [CrossRef]
- M. Iyer, L.; Anantharaman, V.; Krishnan, A.; Burroughs, A.M.; Aravind, L. Jumbo phages: A comparative genomic overview of core functions and adaptions for biological conflicts. Viruses 2021, 13, 63. [Google Scholar] [CrossRef] [PubMed]
- Hardies, S.C.; Thomas, J.A.; Serwer, P. Comparative genomics of Bacillus thuringiensis phage 0305phi8-36: Defining patterns of descent in a novel ancient phage lineage. Virol. J. 2007, 4, 97. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shao, Q.; Guo, M.; Han, L.; Zhao, X.; Wang, A.; Li, X.; Wang, B.; Pan, J.A.; Chen, Z.; et al. Capsid structure of bacteriophage ΦKZ provides insights into assembly and stabilization of jumbo phages. Nat. Commun. 2024, 15, 6551. [Google Scholar] [CrossRef] [PubMed]
- Effantin, G.; Hamasaki, R.; Kawasaki, T.; Bacia, M.; Moriscot, C.; Weissenhorn, W.; Yamada, T.; Schoehn, G. Cryo-electron microscopy three-dimensional structure of the jumbo phage PhiRSL1 infecting the phytopathogen Ralstonia solanacearum. Structure 2013, 21, 298–305. [Google Scholar] [CrossRef]
- Chaikeeratisak, V.; Nguyen, K.; Khanna, K.; Brilot, A.F.; Erb, M.L.; Coker, J.K.; Vavilina, A.; Newton, G.L.; Buschauer, R.; Pogliano, K.; et al. Assembly of a nucleus-like structure during viral replication in bacteria. Science 2017, 355, 194–197. [Google Scholar] [CrossRef]
- Malone, L.M.; Warring, S.L.; Jackson, S.A.; Warnecke, C.; Gardner, P.P.; Gumy, L.F.; Fineran, P.C. A jumbo phage that forms a nucleus-like structure evades CRISPR-Cas DNA targeting but is vulnerable to type III RNA-based immunity. Nat. Microbiol. 2020, 5, 48–55. [Google Scholar] [CrossRef]
- Laughlin, T.G.; Deep, A.; Prichard, A.M.; Seitz, C.; Gu, Y.; Enustun, E.; Suslov, S.; Khanna, K.; Birkholz, E.A.; Armbruster, E.; et al. Architecture and self-assembly of the jumbo bacteriophage nuclear shell. Nature 2022, m608, 429–435. [Google Scholar] [CrossRef]
- Magar, S.; Kolte, V.; Sharma, G.; Govindarajan, S. Exploring pangenomic diversity and CRISPR-Cas evasion potential in jumbo phages: A comparative genomics study. Microbiol. Spectr. 2024, 12, e04200-23. [Google Scholar] [CrossRef]
- Petrov, V.M.; Ratnayaka, S.; Nolan, J.M.; Miller, E.S.; Karam, J.D. Genomes of the T4-related bacteriophages as windows on microbial genome evolution. Virol. J. 2010, 7, 292. [Google Scholar] [CrossRef]
- Hou, Y.; Wu, Z.; Ren, L.; Chen, Y.; Zhang, Y.A.; Zhou, Y. Characterization and application of a lytic jumbo phage ZPAH34 against multidrug-resistant Aeromonas hydrophila. Front. Microbiol. 2023, 14, 1178876. [Google Scholar] [CrossRef]
- Devoto, A.E.; Santini, J.M.; Olm, M.R.; Anantharaman, K.; Munk, P.; Tung, J.; Archie, E.A.; Turnbaugh, P.J.; Seed, K.D.; Blekhman, R.; et al. Megaphages Infect Prevotella and Variants Are Widespread in Gut Microbiomes. Nat. Microbiol. 2019, 4, 693–700. [Google Scholar] [CrossRef]
- Kallies, R.; Hu, D.; Abdulkadir, N.U.; Schloter, M.; Rocha, U. Identification of Huge Phages from Wastewater Metagenomes. Viruses 2023, 15, 2330. [Google Scholar] [CrossRef]
- Yuan, Y.; Gao, M. Jumbo Bacteriophages: An Overview. Front. Microbiol. 2017, 8, 403. [Google Scholar] [CrossRef]
- Pallavi, B.; Puneeth, T.G.; Shekar, M.; Girisha, S.K. Isolation, characterization and genomic analysis of vB-AhyM-AP1, a lytic bacteriophage infecting Aeromonas hydrophila. J. Appl. Microbiol. 2021, 131, 695–705. [Google Scholar] [CrossRef]
- Vincent, A.T.; Paquet, V.E.; Bernatchez, A.; Tremblay, D.M.; Moineau, S.; Charette, S.J. Characterization and diversity of phages infecting Aeromonas salmonicida subsp. salmonicida. Sci. Rep. 2017, 7, 7054. [Google Scholar] [CrossRef]
- Morozova, V.; Babkin, I.; Kozlova, Y.; Tikunov, A.; Ushakova, T.; Bardasheva, A.; Fedorets, V.; Zhirakovskaya, E.; Tikunova, N. Isolation, Characterization and Genomic Analysis of a Novel Jumbo Phage, AerS_266, That Infects Aeromonas salmonicida. Microorganisms 2023, 11, 2649. [Google Scholar] [CrossRef]
- Wang, Y.; Qian, P.Y. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS ONE 2009, 4, e7401. [Google Scholar] [CrossRef]
- Bacteriophage λ and its vectors. In Molecular Cloning, 3rd ed.; Sambrook, J., Russell, D., Eds.; Cold Spring Harbour Laboratory Press: New York, NY, USA, 2001; Volume 1, pp. 2.25–2.106. [Google Scholar]
- Kutter, E. Phage host range and efficiency of plating. In Bacteriophages: Methods and Protocols; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: New York, NY, USA, 2009; pp. 141–149. [Google Scholar] [CrossRef]
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.J.; Luo, H.; Gao, F. Ori-Finder 2022: A Comprehensive Web server for prediction and analysis of bacterial replication origins. Genom. Proteom. Bioinform. 2022, 20, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. In Gene Prediction; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1962, pp. 1–14. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The Viral Proteomic Tree Server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 8, 3022–3027. [Google Scholar] [CrossRef]
- Chen, F.; Sun, J.; Han, Z.; Yang, X.; Xian, J.A.; Lv, A.; Hu, X.; Shi, H. Isolation, identification and characteristics of Aeromonas veronii from diseased crucian carp (Carassius auratus gibelio). Front. Microbiol. 2019, 10, 2742. [Google Scholar] [CrossRef]
- Cuervo, A.; Fàbrega-Ferrer, M.; Machón, C.; Conesa, J.J.; Fernández, F.J.; Pérez-Luque, R.; Pérez-Ruiz, M.; Pous, J.; Vega, M.C.; Carrascosa, J.L.; et al. Structures of T7 bacteriophage portal and tail suggest a viral DNA retention and ejection mechanism. Nat. Commun. 2019, 10, 3746. [Google Scholar] [CrossRef]
- Bhattacharyya, S.P.; Rao, V.B. A novel terminase activity associated with the DNA packaging protein gp17 of bacteriophage T4. Virology 1993, 196, 34–44. [Google Scholar] [CrossRef]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for genome-based phage taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Sokolova, M.L.; Misovetc, I.V.; Severinov, K. Multisubunit RNA polymerases of Jumbo bacteriophages. Viruses 2020, 12, 1064. [Google Scholar] [CrossRef] [PubMed]
- Lavysh, D.; Sokolova, M.; Minakhin, L.; Yakunina, M.; Artamonova, T.; Kozyavkin, S.; Makarova, K.S.; Koonin, E.V.; Severinov, K. The genome of AR9, a giant transducing Bacillus phage encoding two multisubunit RNA polymerases. Virology 2016, 495, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Krylov, V.N.; Cruz, D.; Hertveldt, K.; Ackermann, H.W. “φKZ-like viruses”, a proposed new genus of myovirus bacteriophages. Arch. Virol. 2007, 152, 1955–1959. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, S.D.; Nieweglowska, E.S.; Govindarajan, S.; Leon, L.M.; Berry, J.D.; Tiwari, A.; Chaikeeratisak, V.; Pogliano, J.; Agard, D.A.; Bondy-Denomy, J. A bacteriophage nucleus-like compartment shields DNA from CRISPR nucleases. Nature 2020, 577, 244–248. [Google Scholar] [CrossRef]
- Thomas, J.A.; Weintraub, S.T.; Wu, W.; Winkler, D.C.; Cheng, N.; Steven, A.C.; Black, L.W. Exten-sive proteolysis of head and inner body proteins by a morphogenetic protease in the giant Pseudo-monas aeruginosa phage φKZ. Mol. Microbiol. 2012, 84, 324–339. [Google Scholar] [CrossRef]
- Timms, A.R.; Cambray-Young, J.; Scott, A.E.; Petty, N.K.; Connerton, P.L.; Clarke, L.; Seeger, K.; Quail, M.; Cummings, N.; Maskell, D.J.; et al. Evidence for a lineage of virulent bacteriophages that target Campylobacter. BMC Genom. 2010, 11, 214. [Google Scholar] [CrossRef]
- Day, A.; Ahn, J.; Salmond, G.P. Jumbo bacteriophages are represented within an increasing diversity of environmental viruses infecting the emerging phytopathogen, Dickeya solani. Front. Microbiol. 2018, 9, 2169. [Google Scholar] [CrossRef]
- Sykilinda, N.N.; Bondar, A.A.; Gorshkova, A.S.; Kurochkina, L.P.; Kulikov, E.E.; Shneider, M.M.; Kadykov, V.A.; Solovjeva, N.V.; Kabilov, M.R.; Mesyanzhinov, V.V.; et al. Complete Genome Sequence of the Novel Giant Pseudomonas Phage PaBG. Genome Announc. 2014, 2, e00929-13. [Google Scholar] [CrossRef]
- Fernández-Bravo, A.; Figueras, M.J. An Update on the Genus Aeromonas: Taxonomy, Epidemiology, and Pathogenicity. Microorganisms 2020, 8, 129. [Google Scholar] [CrossRef]
- Ghenghesh, K.S.; Ahmed, S.F.; El-Khalek, R.A.; Al-Gendy, A.; Klena, J. Aeromonas-associated infections in developing countries. J. Infect. Dev. Ctries. 2008, 2, 81–98. [Google Scholar] [CrossRef]
- Saad, A.M.; Soliman, A.M.; Kawasaki, T.; Fujie, M.; Nariya, H.; Shimamoto, T.; Yamada, T. Systemic method to isolate large bacteriophages for use in biocontrol of a wide-range of pathogenic bacteria. J. Biosci. Bioeng. 2019, 127, 73–78. [Google Scholar] [CrossRef]
- Serwer, P.; Wright, E.T. In-Gel Isolation and Characterization of Large (and Other) Phages. Viruses 2020, 12, 410. [Google Scholar] [CrossRef]
- Betts, A.; Gray, C.; Zelek, M.; MacLean, R.C.; King, K.C. High parasite diversity accelerates host adaptation and diversification. Science 2018, 360, 907–911. [Google Scholar] [CrossRef]
- Bujak, K.; Decewicz, P.; Kitowicz, M.; Radlinska, M. Characterization of Three Novel Virulent Aeromonas Phages Provides Insights into the Diversity of the Autographiviridae Family. Viruses 2022, 14, 1016. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage | Name | Sequence (5’–3’) | Reaction | Product Size (bp) |
|---|---|---|---|---|
| vB_AerVM_332-Yuliya | 43_58U22 | CGCTGCTGCTCCTGCTGTCTGG | PCR | 646 |
| 43_59_L24 | TGCTGAAGCTGGCCTTGAATGGTG | |||
| 43_56U | TCCAGAGCGTATCGACTTCAACGG | Real time PCR | 124 | |
| 43_56L | ACCGAACTCGTCCACGTCAAAGG | |||
| 43_63P | FAM-ATGCCTACCGAGTCTACAACCGGCAGA-TAMRA | |||
| vB_AerVM_332-Vera | 294_58U24 | TCTTTGGCTGAGCGTTGGAACACC GGTCCTGACCCATCTTGTCGGACG | PCR | 439 |
| 294_59_L24 | ||||
| 294_55U | ATTGGCTAAGGCTCGCTCTATTGC | Real time PCR | 131 | |
| 294_56L | CTTCTTTCCAAGTCCAGACCATCCC | |||
| 294_64P | FAM-CCCGCGTTGCCGTGGCTTGTTC-TAMRA | |||
| vB_AerVM_332-Igor | 237_59U22 | CGACGGGCTGCGGTAAGACGAC | PCR | 598 |
| 237_58_L22 | ACGGCTTGCAGAATGGACAGCG | |||
| 237_56U | GCTGGTCTGAACACCCATCGTCAC | Real time PCR | 134 | |
| 237_55L | AGTGGTTGACCGTCTTTGGTCTCG | |||
| 237_63P | FAM-ACGCTGGGAACTCACGTCCGCAGA-TAMRA |
| 332-Yuliya | 332-Vera | 332-Igor | |
|---|---|---|---|
| Sequenced genome size (bp) | 43,457 | 294,685 | 237,907 |
| Physical genome size (bp) | 43,584 | 294,685 | 237,907 |
| Terminal repeats size (bp) | 127 | - | - |
| GC content (%) | 47.86 | 51.46 | 54.18 |
| Number of predicted genes | 68 | 317 | 272 |
| Number of predicted tRNA genes | 0 | 0 | 0 |
| Number of predicted gene products | 35 | 110 | 99 |
| Number of hypothetical gene products | 33 | 207 | 173 |
| Positions of the origin of replication (bp) | 42,181–42,892 | 126,966–127,056 | 210,883–211,002 |
| Temperature, °C | vB_AerVM_332-Vera | vB_AerVM_332-Igor | vB_AerVS_332-Yuliya | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Cq Mean | Cq Std. Dev | E, % * | Cq Mean | Cq Std. Dev | E, % * | Cq Mean | Cq Std. Dev | E, % * | |
| 37 | - | - | - | - | - | - | 30.41 | 0.025 | 98.0 |
| 25 | - | - | - | 15.96 | 0.144 | 92.9 | 13.52 | 0.101 | 96.5 |
| 18 | - | - | - | 20.40 | 0.168 | 94.2 | 13.48 | 0.016 | 98.0 |
| 10 | 30.17 | 0.334 | 98.9 | 28.15 | 0.093 | 94.7 | 13.34 | 0.266 | 99.9 |
| 5 | 30.22 | 0.010 | 95.0 | 29.76 | 0.023 | 92.4 | 20.35 | 0.061 | 93.0 |
| Cluster | Subcluster | Contain Only Giant Phages | The Size of Giant Phage Genomes, kb | Contain Aeromonas Giant Phages | Known Bacterial Hosts |
|---|---|---|---|---|---|
| A | 1 | + | 208–237 | - | Gram-positive |
| 2 | + | 386 | - | - | |
| 3 | + | 309 | - | - | |
| 4 | + | 227–286 | - | - | |
| 5 | + | 203–227 | - | - | |
| 6 | + | 205–213 | - | - | |
| B | 7 | + | 222 | - | - |
| 8 | + | 229–241 | - | Gram-positive and Gram-negative | |
| 9 | + | 201–288 | - | - | |
| C | 10 | + | 236–241 | - | Gram-negative |
| D | 11 | + | 201–321 | + | Gram-negative |
| E | 12 | + | 200–267 | + | Gram-negative |
| F | 13 | + | 447–595 | - | - |
| 14 | + | 214–235 | - | Gram-negative | |
| 15 | + | 257–735 | - | - | |
| 16 | + | 203–490 | - | Gram-negative | |
| 17 | - | 200–261 | + | Gram-negative | |
| G | 18 | - | 203–208 | - | Gram-negative |
| 19 | + | 235 | - | Gram-negative | |
| H | 20 | + | 258 | - | - |
| I | 21 | + | 209–501 | - | Gram-negative |
| 22 | + | 274 | - | Gram-negative | |
| 23 | + | 208–449 | - | Gram-positive and Gram-negative | |
| J | 24 | - | 200–308 | - | Gram-positive and Gram-negative |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babkin, I.V.; Morozova, V.V.; Kozlova, Y.N.; Fedorets, V.A.; Tikunov, A.Y.; Ushakova, T.A.; Bardasheva, A.V.; Zhirakovskaya, E.V.; Tikunova, N.V. Novel Giant Phages vB_AerVM_332-Vera and vB_AerVM_332-Igor and Siphophage vB_AerVS_332-Yulya Infecting the Same Aeromonas veronii Strain. Viruses 2025, 17, 1027. https://doi.org/10.3390/v17081027
Babkin IV, Morozova VV, Kozlova YN, Fedorets VA, Tikunov AY, Ushakova TA, Bardasheva AV, Zhirakovskaya EV, Tikunova NV. Novel Giant Phages vB_AerVM_332-Vera and vB_AerVM_332-Igor and Siphophage vB_AerVS_332-Yulya Infecting the Same Aeromonas veronii Strain. Viruses. 2025; 17(8):1027. https://doi.org/10.3390/v17081027
Chicago/Turabian StyleBabkin, Igor V., Vera V. Morozova, Yuliya N. Kozlova, Valeria A. Fedorets, Artem Y. Tikunov, Tatyana A. Ushakova, Alevtina V. Bardasheva, Elena V. Zhirakovskaya, and Nina V. Tikunova. 2025. "Novel Giant Phages vB_AerVM_332-Vera and vB_AerVM_332-Igor and Siphophage vB_AerVS_332-Yulya Infecting the Same Aeromonas veronii Strain" Viruses 17, no. 8: 1027. https://doi.org/10.3390/v17081027
APA StyleBabkin, I. V., Morozova, V. V., Kozlova, Y. N., Fedorets, V. A., Tikunov, A. Y., Ushakova, T. A., Bardasheva, A. V., Zhirakovskaya, E. V., & Tikunova, N. V. (2025). Novel Giant Phages vB_AerVM_332-Vera and vB_AerVM_332-Igor and Siphophage vB_AerVS_332-Yulya Infecting the Same Aeromonas veronii Strain. Viruses, 17(8), 1027. https://doi.org/10.3390/v17081027