
Potential Resistance Mechanisms Exhibited by Cystic Fibrosis Patients Against SARS-CoV-2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
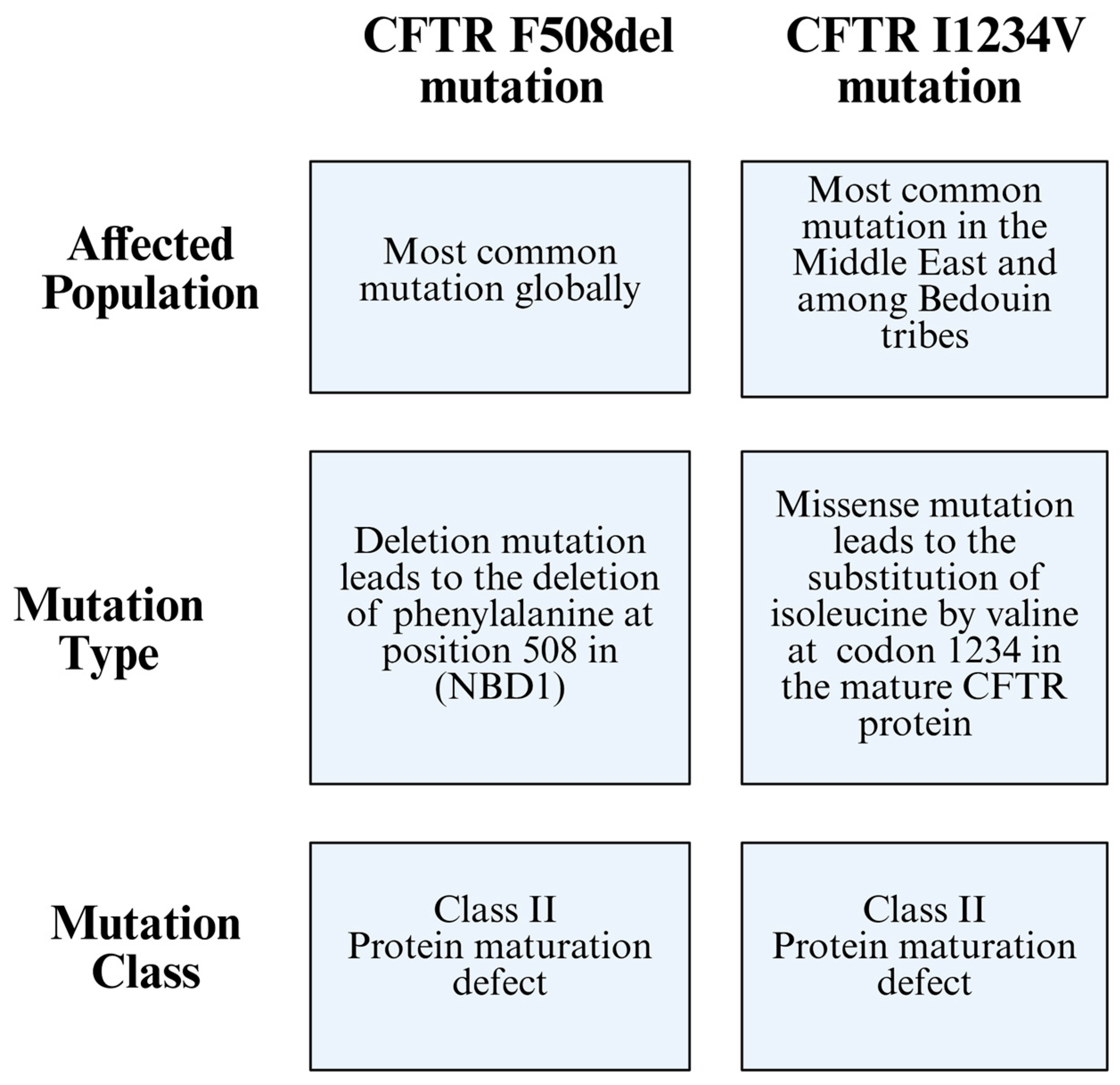
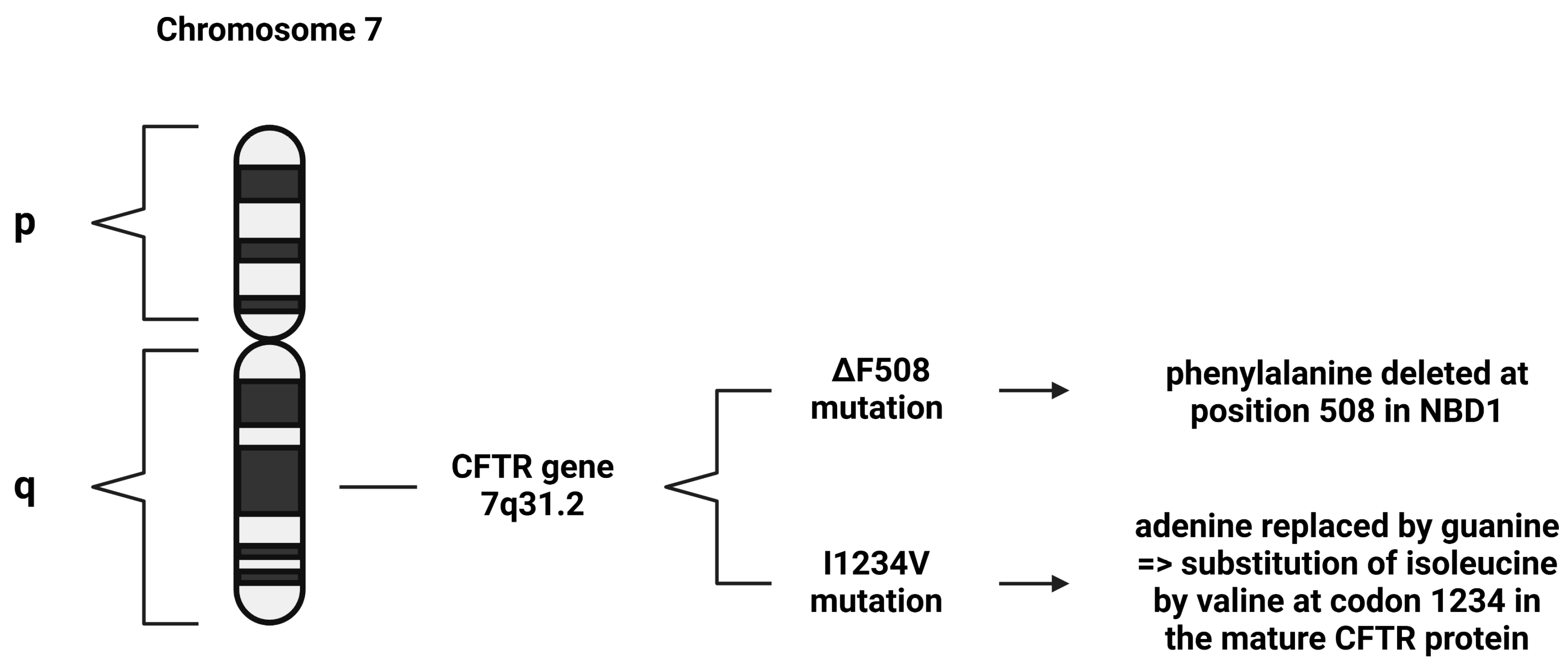
2. Cystic Fibrosis Genetics
3. Respiratory Viral Infections Associated with Cystic Fibrosis
3.1. Human Rhinovirus and Other Predominant Respiratory Viruses
3.2. Respiratory Syncytial Virus
3.3. Influenza Viruses
3.4. Human Parainfluenza Viruses
3.5. Severe Acute Respiratory Syndrome Coronavirus 2
4. SARS-CoV-2 Invasion Mechanisms and Pathogenicity
5. Potential Resistance Factors Against SARS-CoV-2 in CF Patients
5.1. Adenosine Triphosphate (ATP)
5.2. Deleted/Dysfunctional CFTR Gene
5.3. ACE and ACE2 Regulation and Expression
6. SARS-CoV-2 Resistance Mechanisms in CF Patients
6.1. ACE and ACE2 Polymorphism Effects
6.2. Host Proteins and SARS-CoV-2 Interactions
6.3. SMN1 and ACE/ACE2 Interactions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| COVID-19 | Coronavirus disease 2019 |
| ARDS | Acute respiratory distress syndrome |
| CF | Cystic fibrosis |
| CFTR gene | Cystic fibrosis transmembrane conductance regulator gene |
| ACE | Angiotensin-converting enzyme |
| TMPRSS2 | Transmembrane serine protease 2 |
| DNA | Deoxyribonucleic acid |
| RNA | Ribonucleic acid |
| mRNA | Messenger RNA |
| PFT | Pulmonary function test |
| CT | Computed tomography |
| IgM/G | Immunoglobulin M/G |
| cAMP | Cyclic adenosine monophosphate |
| TGFB1 | Transforming growth factor beta 1 |
| COPD | Chronic obstructive pulmonary disease |
| NBD1 | Nucleotide-binding domain 1 |
| WHO | World Health Organization |
| RSV | Respiratory syncytial virus |
| BAL | Bronchoalveolar lavage |
| IL-1/-6 | Interleukin-1/-6 |
| NE | Neutrophil elastase |
| HRV | Human rhinovirus |
| HMPV | Human metapneumovirus |
| FEV1 | Forced expiratory volume in 1 s |
| AE | Asthma exacerbation |
| RNA-Seq | RNA sequencing |
| AECs | Airway epithelial cells |
| IAV | Influenza A virus |
| HA | Hemagglutinin |
| NA | Neuraminidase |
| HPIVs | Human parainfluenza viruses |
| LRTIs | Lower respiratory tract infections |
| CD4 | Helper T cells |
| CD8 | Cytotoxic T cells |
| ssRNA | Single-stranded RNA |
| Orfs | Open reading frames |
| CDC | Center for Disease Control and Prevention |
| RdRp | RNA-dependent RNA polymerase |
| S glycoprotein | Spike glycoprotein |
| HE | Hemagglutinin-esterase |
| CoVs | Coronaviruses |
| MERS-CoV | Middle east respiratory syndrome coronavirus |
| CME | Clathrin-mediated endocytosis |
| CNS | Central nervous system |
| PNS | Peripheral nervous system |
| BBB | Blood–brain barrier |
| ATP | Adenosine triphosphate |
| PaO2 | Arterial oxygen pressure |
| Ang-I/-II | Angiotensin I/-II |
| Ang-1-7 | Angiotensin-1-7 |
| siRNAs | Small interfering RNAs |
| miRNAs | Micro RNAs |
| BioID | Proximity-dependent biotinylation |
| TNF | Tumor necrosis factor |
| SMN1 | Survival motor neuron 1 |
References
- Zaim, S.; Chong, J.H.; Sankaranarayanan, V.; Harky, A. COVID-19 and Multiorgan Response. Curr. Probl. Cardiol. 2020, 45, 100618. [Google Scholar] [CrossRef]
- Felsenstein, S.; Herbert, J.A.; McNamara, P.S.; Hedrich, C.M. COVID-19: Immunology and treatment options. Clin. Immunol. 2020, 215, 108448. [Google Scholar] [CrossRef] [PubMed]
- Salian, V.S.; Wright, J.A.; Vedell, P.T.; Nair, S.; Li, C.; Kandimalla, M.; Tang, X.; Porquera, E.M.C.; Kalari, K.R.; Kandimalla, K.K. COVID-19 Transmission, Current Treatment, and Future Therapeutic Strategies. Mol. Pharm. 2021, 18, 754–771. [Google Scholar] [CrossRef] [PubMed]
- Thanh Le, T.; Andreadakis, Z.; Kumar, A.; Gómez Román, R.; Tollefsen, S.; Saville, M.; Mayhew, S. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 19, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181, S4–S15.e1. [Google Scholar] [CrossRef]
- Hammoudeh, S.; Gadelhak, W.; Abdulwahab, A.; Al-Langawi, M.; Janahi, I.A. Approaching two decades of cystic fibrosis research in Qatar: A historical perspective and future directions. Multidiscip. Respir. Med. 2019, 14, 29. [Google Scholar] [CrossRef]
- Thomas, M.; Raja, M.; Albakri, M.; Najim, M.; Chandra, P.; Allangawi, M. CT score and correlation with lung function and microbiology of adult patients with cystic fibrosis with predominant I1234V genotype in Qatar. Qatar. Med. J. 2020, 2020, 4. [Google Scholar] [CrossRef]
- Quinton, P.M. Physiological Basis of Cystic Fibrosis: A Historical Perspective. Physiol. Rev. 1999, 79, S3–S22. [Google Scholar] [CrossRef] [PubMed]
- Colombo, C.; Burgel, P.R.; Gartner, S.; van Koningsbruggen-Rietschel, S.; Naehrlich, L.; Sermet-Gaudelus, I.; Southern, K.W. Impact of COVID-19 on people with cystic fibrosis. Lancet Respir. Med. 2020, 8, e35–e36. [Google Scholar] [CrossRef]
- Mason, K.; Hasan, S.; Darukhanavala, A.; Kutney, K. COVID-19: Pathophysiology and implications for cystic fibrosis, diabetes and cystic fibrosis-related diabetes. J. Clin. Transl. Endocrinol. 2021, 26, 100268. [Google Scholar] [CrossRef]
- Hamad, S.G.; Kammouh, H.; Alamri, M.; Zahraldin, K. The clinical features and impact of SARS-CoV-2/COVID-19 infection in children with Cystic Fibrosis (CF): A Qatari experience. Qatar. Med. J. 2023, 2023, 19. [Google Scholar] [CrossRef] [PubMed]
- Viviani, L.; Assael, B.M.; Kerem, E. Impact of the A (H1N1) pandemic influenza (season 2009–2010) on patients with cystic fibrosis. J. Cyst. Fibros. 2011, 10, 370–376. [Google Scholar] [CrossRef]
- Kiedrowski, M.R.; Bomberger, J.M. Viral-Bacterial Co-infections in the Cystic Fibrosis Respiratory Tract. Front. Immunol. 2018, 9, 3067. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.A.; Hartl, D. CFTR: Cystic fibrosis and beyond. Eur. Respir. J. 2014, 44, 1042–1054. [Google Scholar] [CrossRef]
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef]
- Sharma, N.; Cutting, G.R. The genetics and genomics of cystic fibrosis. J. Cyst. Fibros. 2020, 19, S5–S9. [Google Scholar] [CrossRef] [PubMed]
- Hammoudeh, S.; Gadelhak, W.; AbdulWahab, A.; Al-Langawi, M.; Janahi, I.A. An Overview of the Homozygous Cystic Fibrosis Transmembrane Conductance Regulator Mutation c.3700 A>G (p.Ile1234Val) in Qatar. Curr. Genet. Med. Rep. 2019, 7, 187–190. [Google Scholar] [CrossRef]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell. 2016, 27, 424–433. [Google Scholar] [CrossRef]
- Anwar, S.; Peng, J.L.; Zahid, K.R.; Zhou, Y.M.; Ali, Q.; Qiu, C.R. Cystic Fibrosis: Understanding Cystic Fibrosis Transmembrane Regulator Mutation Classification and Modulator Therapies. Adv. Respir. Med. 2024, 92, 263–277. [Google Scholar] [CrossRef]
- Hodson, M.; Andrew, B.; Duncan, G. Cystic Fibrosis; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- van Ewijk, B.E.; van der Zalm, M.M.; Wolfs, T.F.W.; van der Ent, C.K. Viral respiratory infections in cystic fibrosis. J. Cyst. Fibros. 2005, 4 (Suppl. 2), 31–36. [Google Scholar] [CrossRef]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Lung infections associated with cystic fibrosis. Clin. Microbiol. Rev. 2002, 15, 194–222. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.; Grimwood, K.; Carlin, J.B.; Carzino, R.; Hull, J.; Olinsky, A.; Phelan, P.D. Severe Viral Respiratory Infections in Infants with Cystic Fibrosis. Pediatr. Pulmonol. 1998, 26, 371–379. [Google Scholar] [CrossRef]
- Deschamp, A.R.; Hatch, J.E.; Slaven, J.E.; Gebregziabher, N.; Storch, G.; Hall, G.L.; Stick, S.; Ranganathan, S.; Ferkol, T.W.; Davis, S.D. Early respiratory viral infections in infants with cystic fibrosis. J. Cyst. Fibros. 2019, 18, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Daccò, V.; Daleno, C.; Gambazza, S.; Montinaro, V.; Bisogno, A.; Principi, N.; Colombo, C. Human Rhinovirus Infection in Children with Cystic Fibrosis [Internet]. Jpn. J. Infect. Dis. 2014, 67, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Flight, W.G.; Bright-Thomas, R.J.; Tilston, P.; Mutton, K.J.; Guiver, M.; Morris, J.; Webb, A.K.; Jones, A.M. Incidence and clinical impact of respiratory viruses in adults with cystic fibrosis. Thorax 2014, 69, 247–253. [Google Scholar] [CrossRef]
- Ling, K.-M.; Garratt, L.W.; Gill, E.E.; Lee, A.H.Y.; Agudelo-Romero, P.; Sutanto, E.N.; Iosifidis, T.; Rosenow, T.; Turvey, S.E.; Lassmann, T.; et al. Rhinovirus Infection Drives Complex Host Airway Molecular Responses in Children with Cystic Fibrosis. Front. Immunol. 2020, 11, 1327. [Google Scholar] [CrossRef]
- Falsey, A.R.; Walsh, E.E. Respiratory Syncytial Virus Infection in Adults. Clin. Microbiol. Rev. 2000, 13, 371–384. [Google Scholar] [CrossRef]
- Giebels, K.; Marcotte, J.-E.; Podoba, J.; Rousseau, C.; Denis, M.-H.; Fauvel, V.; Laberge, S. Prophylaxis against respiratory syncytial virus in young children with cystic fibrosis. Pediatr. Pulmonol. 2008, 43, 169–174. [Google Scholar] [CrossRef]
- Peteranderl, C.; Herold, S.; Schmoldt, C. Human Influenza Virus Infections. Semin. Respir. Crit. Care Med. 2016, 37, 487–500. [Google Scholar] [CrossRef]
- Renk, H.; Regamey, N.; Hartl, D. Influenza a(H1N1)pdm09 and cystic fibrosis lung disease: A systematic meta-analysis. PLoS ONE 2014, 9, e78583. [Google Scholar] [CrossRef]
- Somayaji, R.; Goss, C.H.; Khan, U.; Neradilek, M.; Neuzil, K.M.; Ortiz, J.R. Cystic fibrosis pulmonary exacerbations attributable to respiratory syncytial virus and influenza: A population-based study. Clin. Infect. Dis. 2017, 64, 1760–1767. [Google Scholar] [CrossRef]
- Branche, A.R.; Falsey, A.R. Parainfluenza Virus Infection. Semin. Respir. Crit. Care Med. 2016, 37, 538–554. [Google Scholar] [CrossRef] [PubMed]
- Pawełczyk, M.; Kowalski, M.L. The Role of Human Parainfluenza Virus Infections in the Immunopathology of the Respiratory Tract. Curr. Allergy Asthma Rep. 2017, 17, 16. [Google Scholar] [CrossRef] [PubMed]
- Terlizzi, V.; Motisi, M.A.; Pellegrino, R.; Padoan, R.; Chiappini, E. Risk factors for severe COVID-19 in people with cystic fibrosis: A systematic review. Front. Pediatr. 2022, 10, 958658. [Google Scholar] [CrossRef]
- Wu, Y.C.; Chen, C.S.; Chan, Y.J. The outbreak of COVID-19: An overview. J. Chin. Med. Assoc. 2020, 83, 217–220. [Google Scholar] [CrossRef]
- Wiegand, R.E.; Deng, Y.; Deng, X.; Lee, A.; Meyer, W.A.; Letovsky, S.; Charles, M.D.; Gundlapalli, A.V.; MacNeil, A.; Hall, A.J.; et al. Estimated SARS-CoV-2 antibody seroprevalence trends and relationship to reported case prevalence from a repeated, cross-sectional study in the 50 states and the District of Columbia, United States—25 October 2020–26 February 2022. Lancet Reg. Health–Am. 2023, 18, 100403. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Williams, C.M.; Decker, J.; Fletcher, E.; Sze, S.; Assadi, S.; Haigh, R.; Saleem, B.; Nazareth, J.; Garton, N.J.; et al. Exhaled SARS-CoV-2 RNA viral load kinetics measured by facemask sampling associates with household transmission. Clin. Microbiol. Infect. 2023, 29, 254.e1–254.e6. [Google Scholar] [CrossRef]
- Sette, A.; Crotty, S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 2021, 184, 861–880. [Google Scholar] [CrossRef]
- Monteiro, M.E.S.; Lechuga, G.C.; Napoleão-Pêgo, P.; Carvalho, J.P.R.S.; Gomes, L.R.; Morel, C.M.; Provance, D.W.; De-Simone, S.G. Humoral Immune Response to SARS-CoV-2 Spike Protein Receptor-Binding Motif Linear Epitopes. Vaccines 2024, 12, 342. [Google Scholar] [CrossRef]
- Das, A.; Ahmed, R.; Akhtar, S.; Begum, K.; Banu, S. An overview of basic molecular biology of SARS-CoV-2 and current COVID-19 prevention strategies. Gene Rep. 2021, 23, 101122. [Google Scholar] [CrossRef]
- World Health Organization (WHO) [Internet]. Tracking SARS-CoV-2 Variants. 2025. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants/2020-DON305 (accessed on 16 June 2025).
- Chen, L.; He, Y.; Liu, H.; Shang, Y.; Guo, G. Potential immune evasion of the severe acute respiratory syndrome coronavirus 2 Omicron variants. Front. Immunol. 2024, 15, 1339660. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Bhattacharya, M.; Nag, S.; Dhama, K.; Chakraborty, C. A Detailed Overview of SARS-CoV-2 Omicron: Its Sub-Variants, Mutations and Pathophysiology, Clinical Characteristics, Immunological Landscape, Immune Escape, and Therapies. Viruses 2023, 15, 167. [Google Scholar] [CrossRef]
- Perveen, S.; Negi, A.; Gopalakrishnan, V.; Panda, S.; Sharma, V.; Sharma, R. COVID-19 diagnostics: Molecular biology to nanomaterials. Clin. Chim. Acta 2023, 538, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Pagani, I.; Venturini, A.; Capurro, V.; Nonis, A.; Ghezzi, S.; Lena, M.; Alcalá-Franco, B.; Gianferro, F.; Guidone, D.; Colombo, C.; et al. Distinct Responses of Cystic Fibrosis Epithelial Cells to SARS-CoV-2 and Influenza A Virus. Am. J. Respir. Cell. Mol. Biol. 2025, 72, 308–319. [Google Scholar] [CrossRef]
- Agrahari, R.; Mohanty, S.; Vishwakarma, K.; Nayak, S.K.; Samantaray, D.; Mohapatra, S. Update vision on COVID-19: Structure, immune pathogenesis, treatment and safety assessment. Sens. Int. 2021, 2, 100073. [Google Scholar] [CrossRef] [PubMed]
- Nile, S.H.; Nile, A.; Qiu, J.; Li, L.; Jia, X.; Kai, G. COVID-19: Pathogenesis, cytokine storm and therapeutic potential of interferons. Cytokine Growth Factor Rev. 2020, 53, 66–70. [Google Scholar] [CrossRef]
- Swain, O.; Romano, S.K.; Miryala, R.; Tsai, J.; Parikh, V.; Umanah, G.K.E. SARS-CoV-2 Neuronal Invasion and Complications: Potential Mechanisms and Therapeutic Approaches. J. Neurosci. 2021, 41, 5338–5349. [Google Scholar] [CrossRef]
- Yamauchi, Y.; Helenius, A. Virus entry at a glance. J. Cell Sci. 2013, 16, 1289–1295. [Google Scholar] [CrossRef]
- Marsh, M.; Heleniust, A. Virus Entry into Animal Cells. Adv. Virus Res. 1989, 36, 107–151. [Google Scholar]
- Bezzerri, V.; Gentili, V.; Api, M.; Finotti, A.; Papi, C.; Tamanini, A.; Boni, C.; Baldisseri, E.; Olioso, D.; Duca, M.; et al. SARS-CoV-2 viral entry and replication is impaired in Cystic Fibrosis airways due to ACE2 downregulation. Nat. Commun. 2023, 14, 132. [Google Scholar] [CrossRef]
- Falcone, C.; Caracciolo, M.; Correale, P.; Macheda, S.; Vadalà, E.G.; La Scala, S.; Tescione, M.; Danieli, R.; Ferrarelli, A.; Tarsitano, M.G.; et al. Can adenosine fight COVID-19 acute respiratory distress syndrome? J. Clin. Med. 2020, 9, 3045. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; He, H.; Wang, L.; Zhang, N.; Huang, H.; Xiong, Q.; Yan, Y.; Wu, N.; Ren, H.; Han, H.; et al. Virus-Triggered ATP Release Limits Viral Replication through Facilitating IFN-β Production in a P2X7-Dependent Manner. J. Immunol. 2017, 199, 1372–1381. [Google Scholar] [CrossRef]
- Abraham, E.H.; Guidotti, G.; Rapaport, E.; Bower, D.; Brown, J.; Griffin, R.J.; Donnelly, A.; Waitzkin, E.D.; Qamar, K.; Thompson, M.A.; et al. Cystic fibrosis improves COVID-19 survival and provides clues for treatment of SARS-CoV-2. Purinergic Signal 2021, 17, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Lotti, V.; Merigo, F.; Lagni, A.; Di Clemente, A.; Ligozzi, M.; Bernardi, P.; Rossini, G.; Concia, E.; Plebani, R.; Romano, M.; et al. CFTR Modulation Reduces SARS-CoV-2 Infection in Human Bronchial Epithelial Cells. Cells 2022, 11, 1347. [Google Scholar] [CrossRef]
- Colombo, C.; Alicandro, G.; Daccó, V.; Gagliano, V.; Morlacchi, L.C.; Casciaro, R.; Pisi, G.; Francalanci, M.; Badolato, R.; Bignamini, E.; et al. SARS-CoV-2 infection in cystic fibrosis: A multicentre prospective study with a control group, Italy, February-July 2020. PLoS ONE 2021, 16, e0251527. [Google Scholar] [CrossRef] [PubMed]
- Mathew, H.R.; Choi, M.Y.; Parkins, M.D.; Fritzler, M.J. Systematic review: Cystic fibrosis in the SARS-CoV-2/COVID-19 pandemic. BMC Pulm. Med. 2021, 21, 173. [Google Scholar] [CrossRef]
- Leowattana, W.; Leowattana, T.; Leowattana, P. Circulating angiotensin converting enzyme 2 and COVID-19. World J. Clin. Cases 2022, 10, 12462–12803. [Google Scholar] [CrossRef]
- Hammoud, S.H.; Wehbe, Z.; Abdelhady, S.; Kobeissy, F.; Eid, A.H.; El-Yazbi, A.F. Dysregulation of angiotensin converting enzyme 2 expression and function in comorbid disease conditions possibly contributes to coronavirus infectious disease 2019 complication severity. Mol. Pharmacol. 2021, 99, 17–28. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, S.; Gou, J.; Wen, Y.; Fan, L.; Zhou, J.; Zhou, G.; Xu, G.; Zhang, Z. Spike-mediated ACE2 down-regulation was involved in the pathogenesis of SARS-CoV-2 infection. J. Infect. 2022, 85, 418–427. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef]
- Stanton, B.A.; Thomas, X.; Hampton, H.; Ashare, A. SARS-CoV-2 (COVID-19) and cystic fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, 408–415. Available online: www.ajplung.org (accessed on 21 August 2024). [CrossRef]
- Bakhshandeh, B.; Sorboni, S.G.; Javanmard, A.-R.; Mottaghi, S.S.; Mehrabi, M.-R.; Sorouri, F.; Abbasi, A.; Jahanafrooz, Z. Variants in ACE2; potential influences on virus infection and COVID-19 severity. Infect. Genet. Evol. 2021, 90, 104773. [Google Scholar] [CrossRef]
- Ali, F.; Elserafy, M.; Alkordi, M.H.; Amin, M. ACE2 coding variants in different populations and their potential impact on SARS-CoV-2 binding affinity. Biochem. Biophys. Rep. 2020, 24, 100798. [Google Scholar] [CrossRef] [PubMed]
- Calcagnile, M.; Forgez, P.; Iannelli, A.; Bucci, C.; Alifano, M.; Alifano, P. Molecular docking simulation reveals ACE2 polymorphisms that may increase the affinity of ACE2 with the SARS-CoV-2 Spike protein. Biochimie 2021, 180, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Gómez, J.; Albaiceta, G.M.; García-Clemente, M.; López-Larrea, C.; Amado-Rodríguez, L.; Lopez-Alonso, I.; Hermida, T.; Enriquez, A.I.; Herrero, P.; Melón, S.; et al. Angiotensin-converting enzymes (ACE, ACE2) gene variants and COVID-19 outcome. Gene 2020, 762, 145102. [Google Scholar] [CrossRef]
- Hejenkowska, E.D.; Mitash, N.; Donovan, J.E.; Chandra, A.; Bertrand, C.; De Santi, C.; Greene, C.M.; Mu, F.; Swiatecka-Urban, A. TGF-β1 Inhibition of ACE2 Mediated by miRNA Uncovers Novel Mechanism of SARS-CoV-2 Pathogenesis. J. Innate Immun. 2023, 15, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 422. [Google Scholar] [CrossRef]
- Montaldo, C.; Messina, F.; Abbate, I.; Antonioli, M.; Bordoni, V.; Aiello, A.; Ciccosanti, F.; Colavita, F.; Farroni, C.; Fard, S.N.; et al. Multi-omics approach to COVID-19: A domain-based literature review. J. Transl. Med. 2021, 19, 501. [Google Scholar] [CrossRef]
- Messina, F.; Giombini, E.; Montaldo, C.; Sharma, A.A.; Zoccoli, A.; Sekaly, R.-P.; Locatelli, F.; Zumla, A.; Maeurer, M.; Capobianchi, M.R.; et al. Looking for pathways related to COVID-19: Confirmation of pathogenic mechanisms by SARS-CoV-2–host interactome. Cell Death Dis. 2021, 12, 788. [Google Scholar] [CrossRef]
- St-Germain, J.R.; Astori, A.; Samavarchi-Tehrani, P.; Abdouni, H.; Macwan, V.; Kim, D.-K.; Knapp, J.J.; Roth, F.P.; Gingras, A.-C.; Raught, B. A SARS-CoV-2 BioID-based virus-host membrane protein interactome and virus peptide compendium: New proteomics resources for COVID-19 research [Internet]. bioRxiv 2020. [Google Scholar] [CrossRef]
- Viinikainen, A.; Nyman, T.; Fyhrquist, F.; Saijonmaa, O. Downregulation of angiotensin converting enzyme by TNF-α in differentiating human macrophages. Cytokine 2002, 18, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Marson, F.A.L.; Bertuzzo, C.S.; Hortencio, T.D.R.; Ribeiro, J.D.; Bonadia, L.C.; Ribeiro, A.F. The ACE gene D/I polymorphism as a modulator of severity of cystic fibrosis. BMC Pulm. Med. 2012, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, X.; Shen, J.; Tan, H.; Rong, T.; Lin, Y.; Feng, E.; Chen, Z.; Jiao, Y.; Liu, G.; et al. Bioinformatic analysis of SMN1-ACE/ACE2 interactions hinted at a potential protective effect of spinal muscular atrophy against COVID-19-induced lung injury. Brief. Bioinform. 2021, 22, 1291–1296. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. SMN1 functions as a novel inhibitor for TRAF6-mediated NF-κB signaling. Biochim. Biophys. Acta Mol. Cell. Res. 2017, 1864, 760–770. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsharabassi, Y.K.; Swaidan, N.T.; Emara, M.M. Potential Resistance Mechanisms Exhibited by Cystic Fibrosis Patients Against SARS-CoV-2. Viruses 2025, 17, 919. https://doi.org/10.3390/v17070919
Elsharabassi YK, Swaidan NT, Emara MM. Potential Resistance Mechanisms Exhibited by Cystic Fibrosis Patients Against SARS-CoV-2. Viruses. 2025; 17(7):919. https://doi.org/10.3390/v17070919
Chicago/Turabian StyleElsharabassi, Yasmin K., Nuha T. Swaidan, and Mohamed M. Emara. 2025. "Potential Resistance Mechanisms Exhibited by Cystic Fibrosis Patients Against SARS-CoV-2" Viruses 17, no. 7: 919. https://doi.org/10.3390/v17070919
APA StyleElsharabassi, Y. K., Swaidan, N. T., & Emara, M. M. (2025). Potential Resistance Mechanisms Exhibited by Cystic Fibrosis Patients Against SARS-CoV-2. Viruses, 17(7), 919. https://doi.org/10.3390/v17070919