
A Type I IFN-Inducing Oncolytic Virus Improves NK Cell-Mediated Killing of Tumor Cells In Vitro Through Multiple Mechanisms



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells Lines, Viruses, and Infections
2.2. PM21-NK Cell Preparation and Cryopreservation
2.3. Flow Cytometry
2.4. Western Blotting
2.5. Reverse Transcription and Real-Time PCR
2.6. ELISA
2.7. UV Media Inactivation and Cytokine Treatment
2.8. Cell Viability and Cytotoxicity Assays
2.9. Figures, Statistics, and Images
3. Results
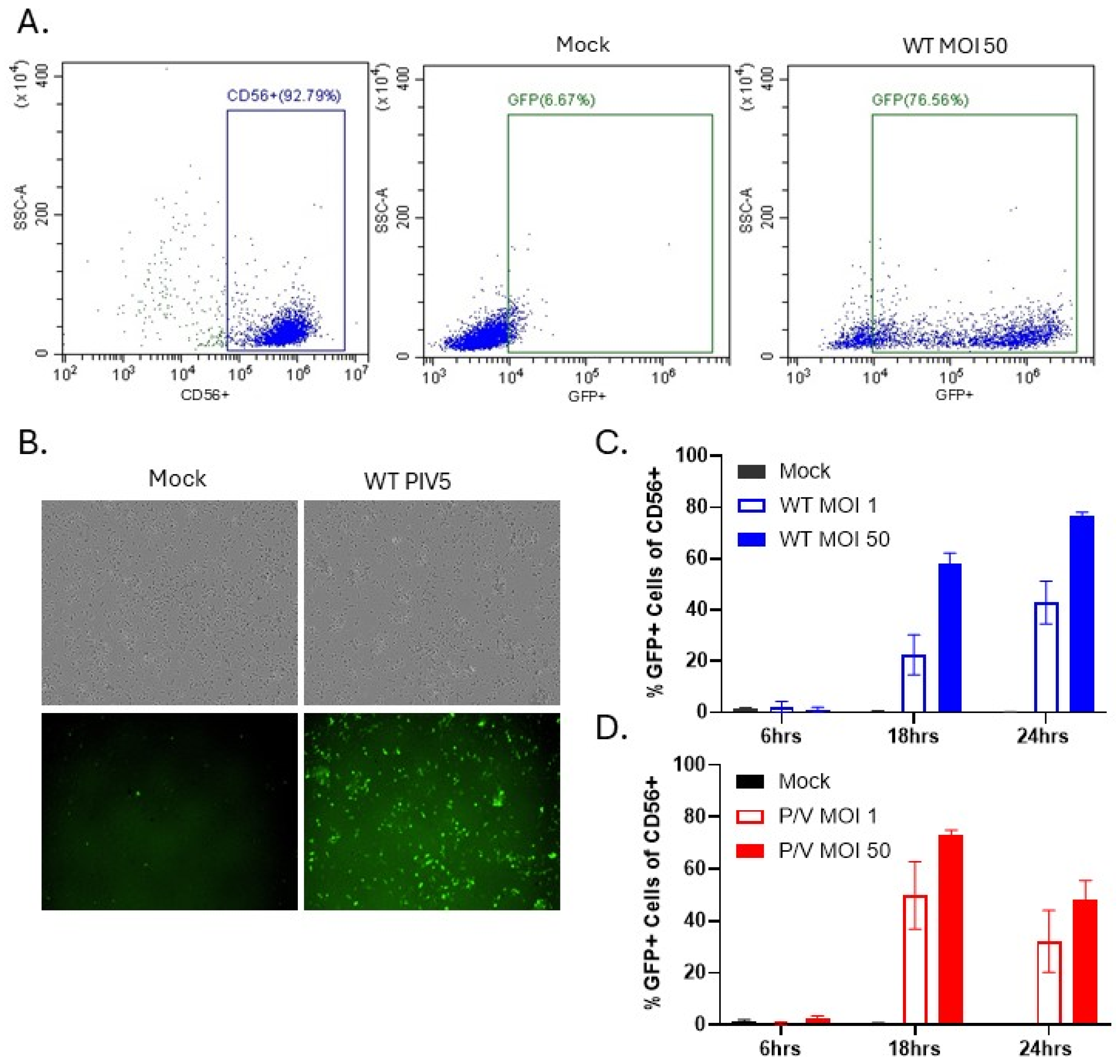
3.1. PM21-NK Cells Are Susceptible to PIV5 Infection
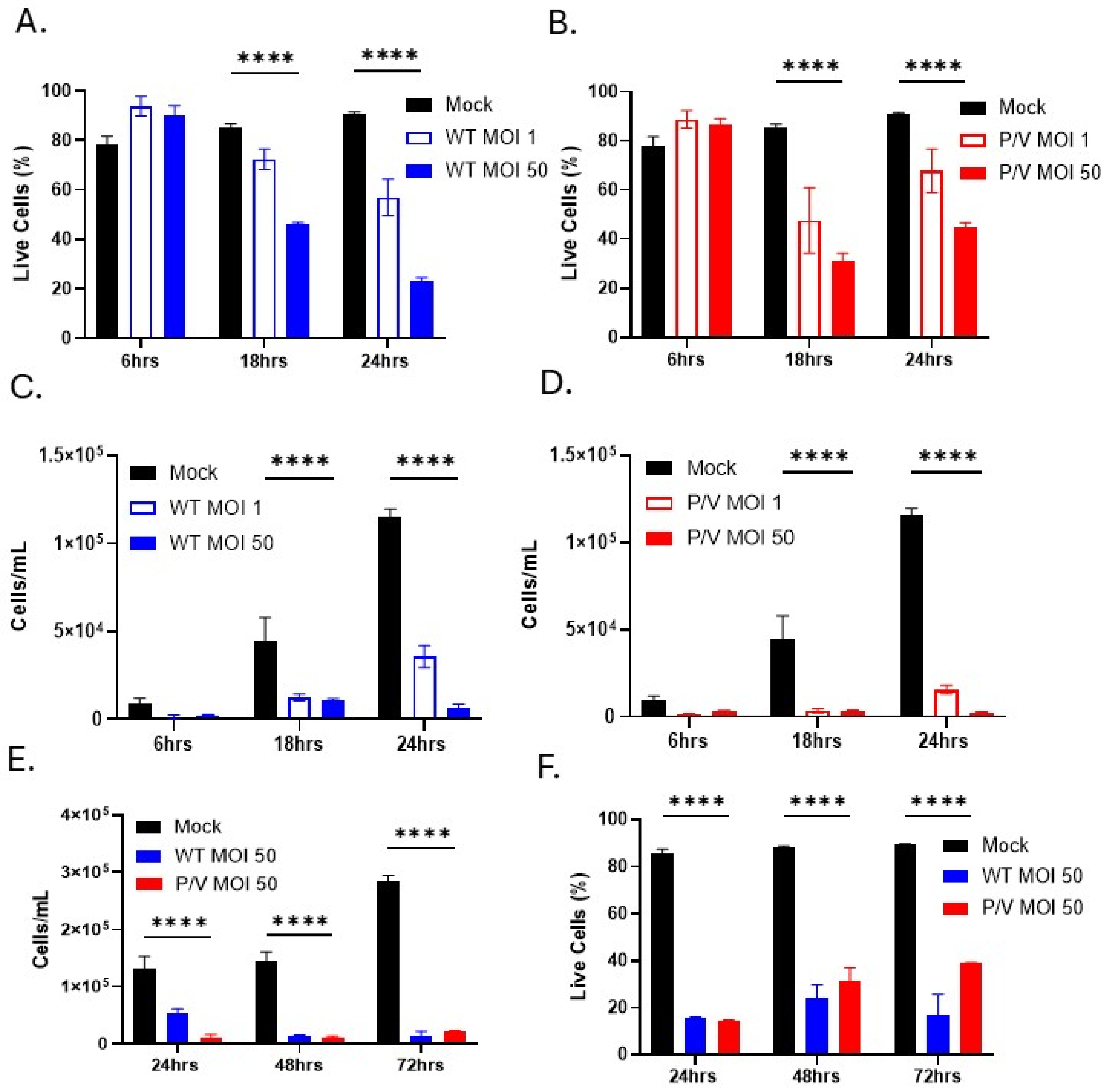
3.2. WT PIV5 and P/V Mutant Are Cytopathic to PM21-NK Cells
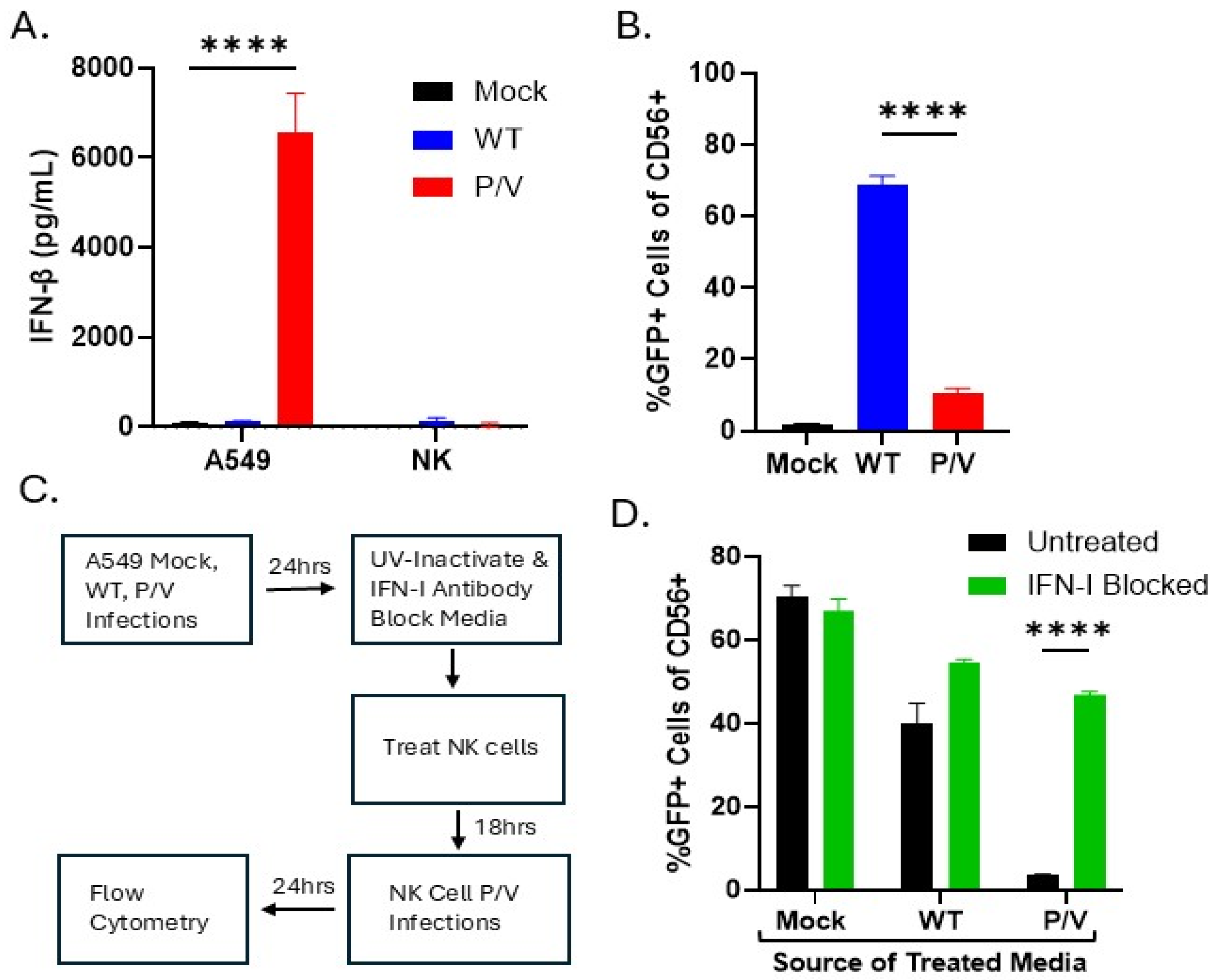
3.3. IFN-I Derived from P/V Mutant Infection of Tumor Cells Can Induce an Antiviral State in PM21-NK Cells and Prevents Tumor-Derived Virus from Spreading to NK Cells
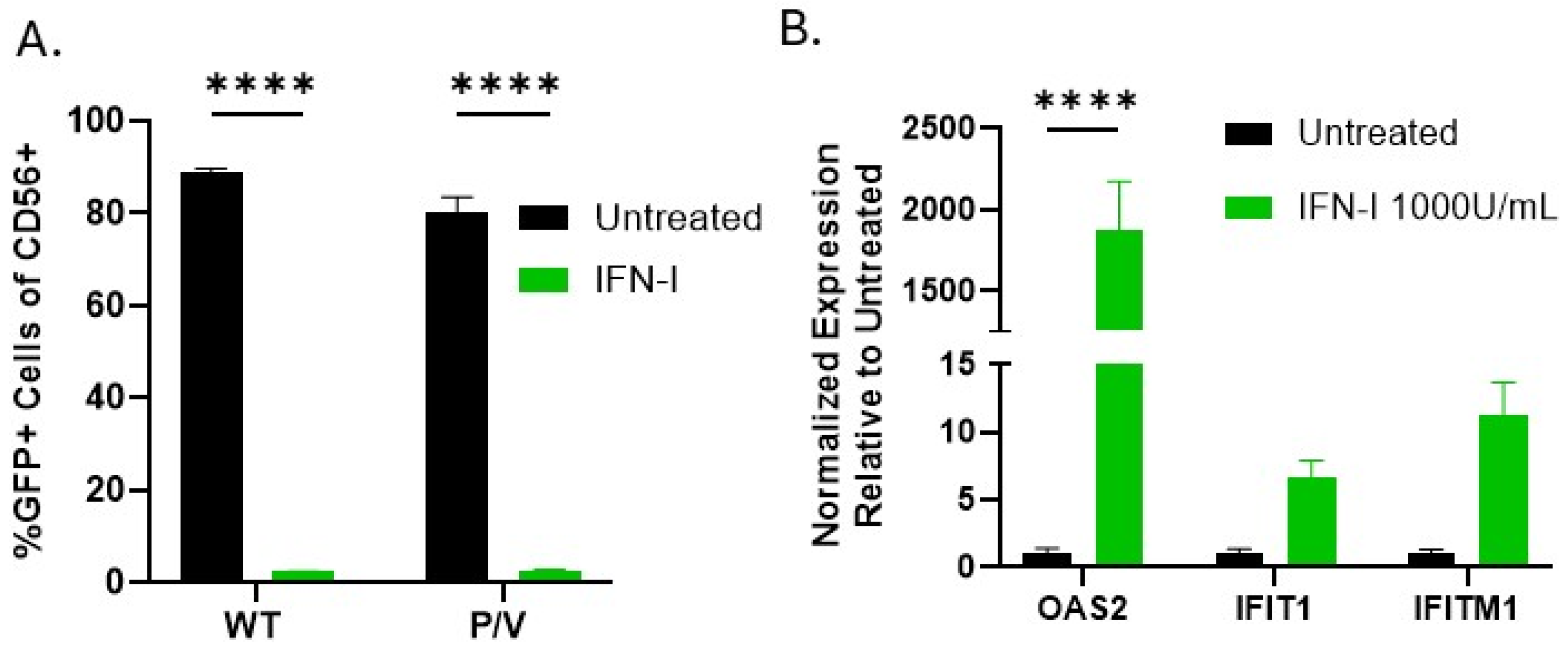
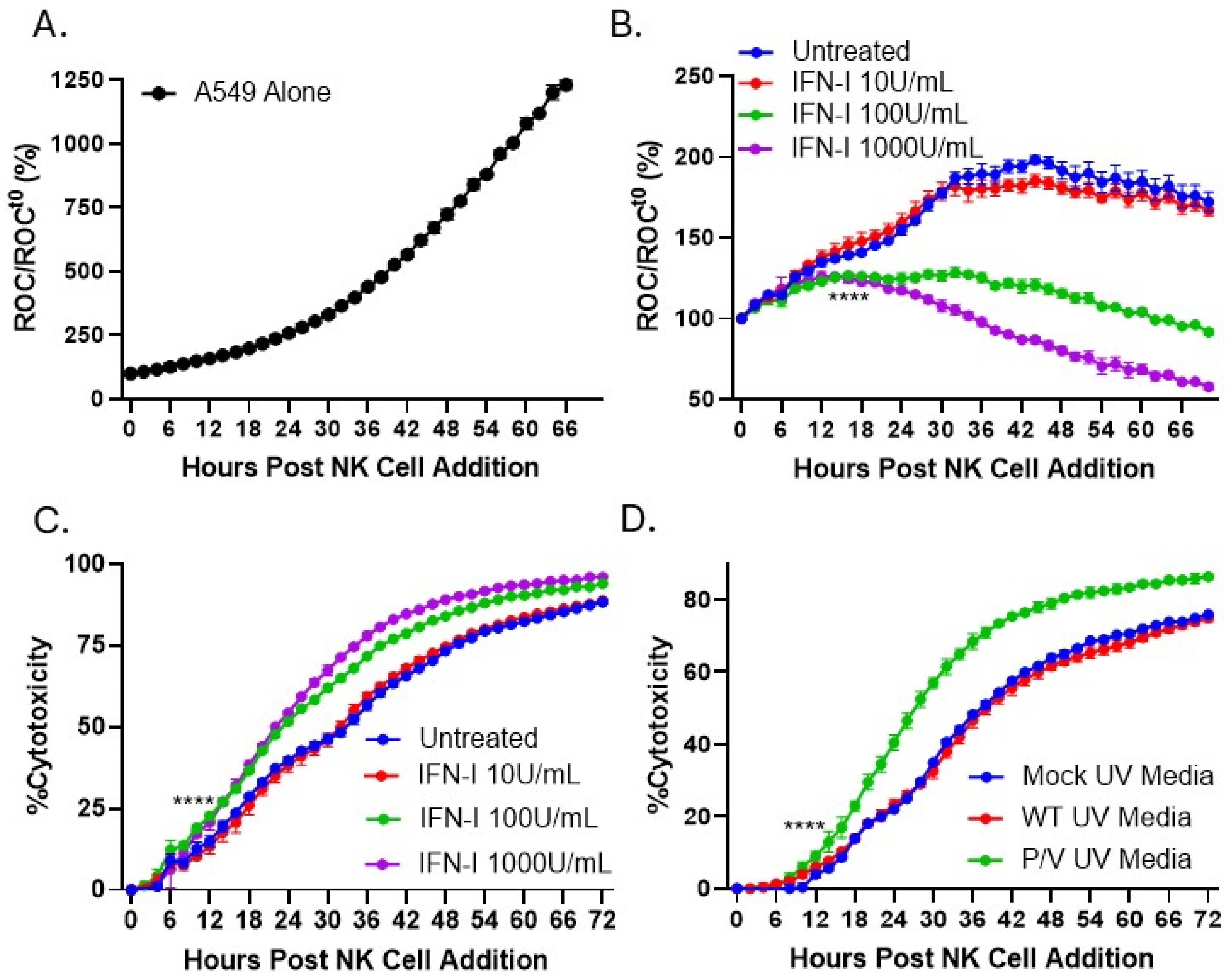
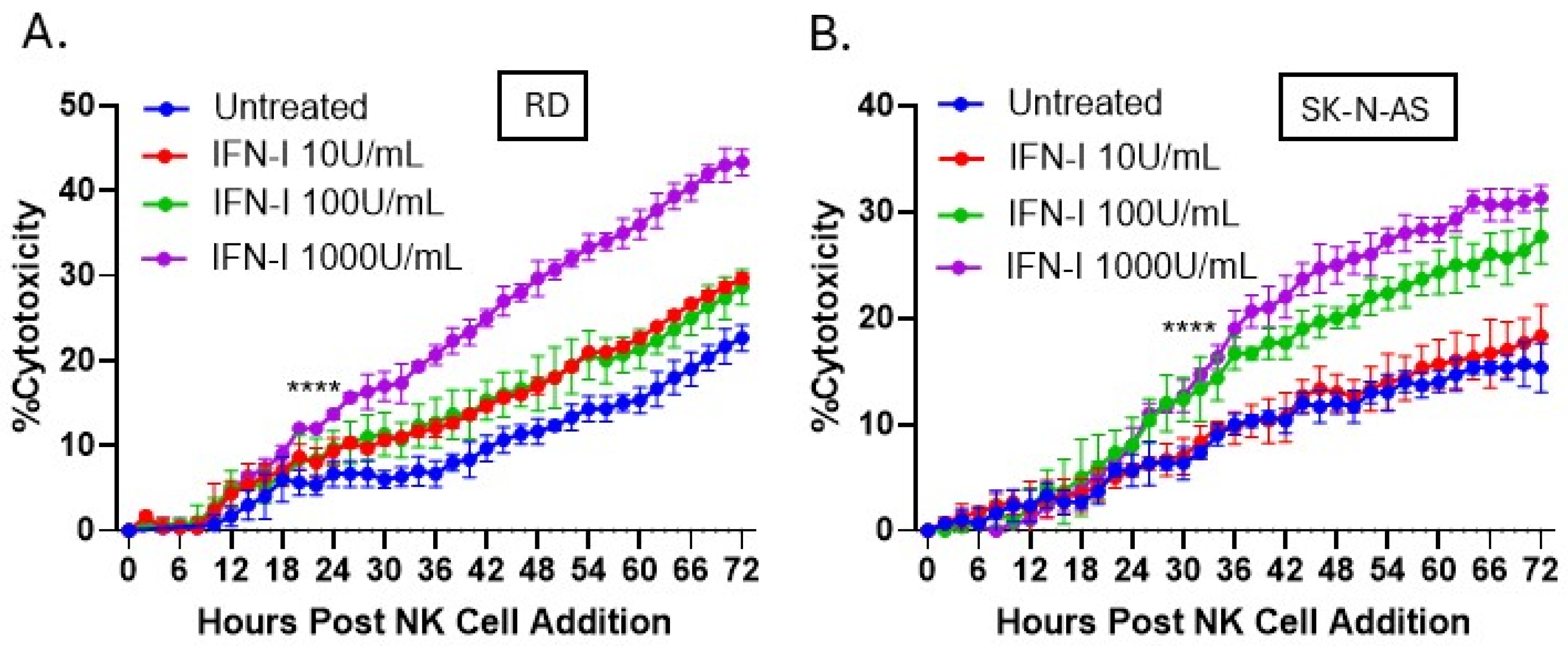
3.4. IFN-I Treatment of PM21-NK Cells Enhances Their Ability to Kill Lung, Neuroblastoma, and Rhabdomyosarcoma Tumor Cells In Vitro
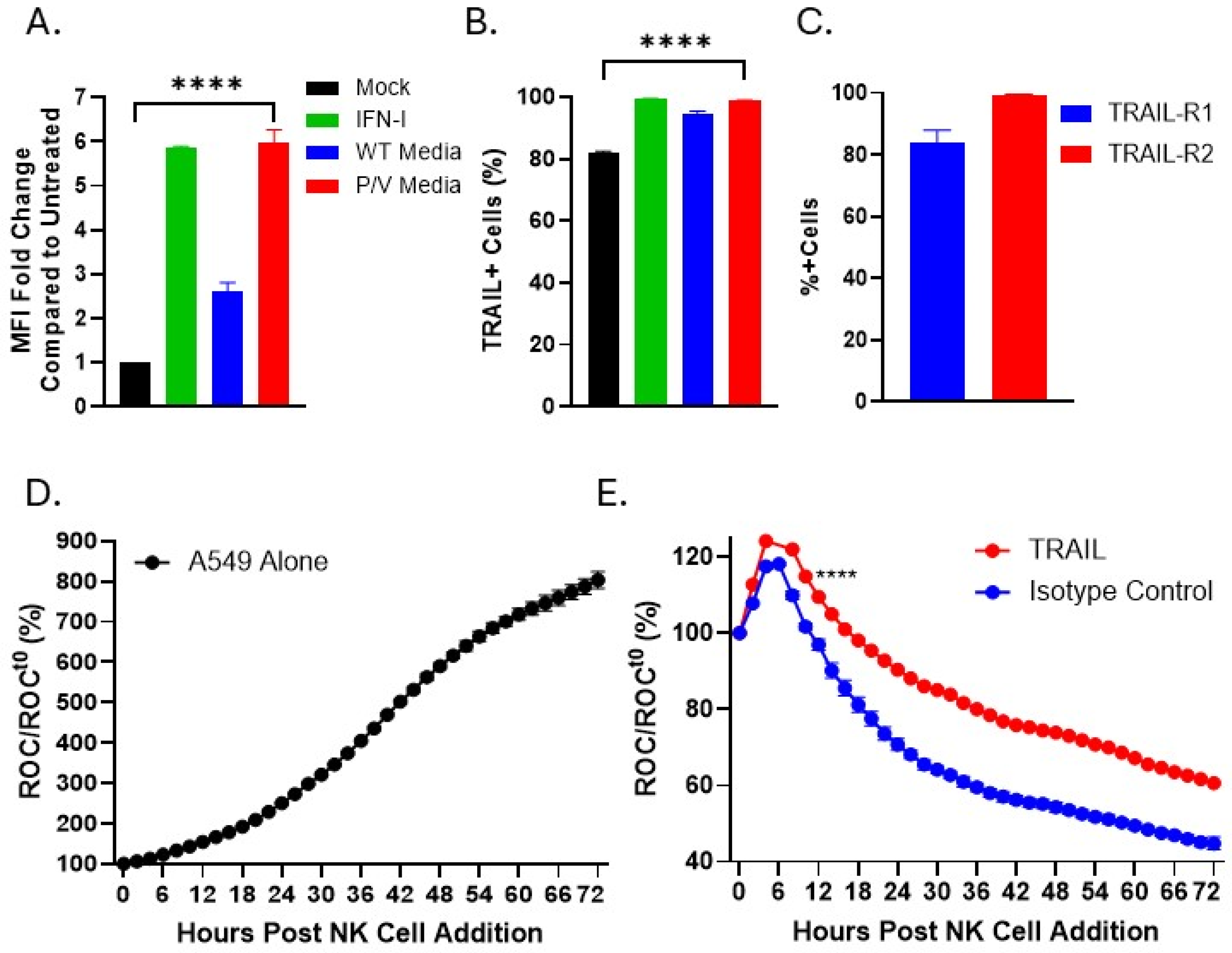
3.5. IFN-I or Media from P/V-Mutant-Infected Tumor Cells Upregulates Expression of TRAIL on PM21-NK Cells
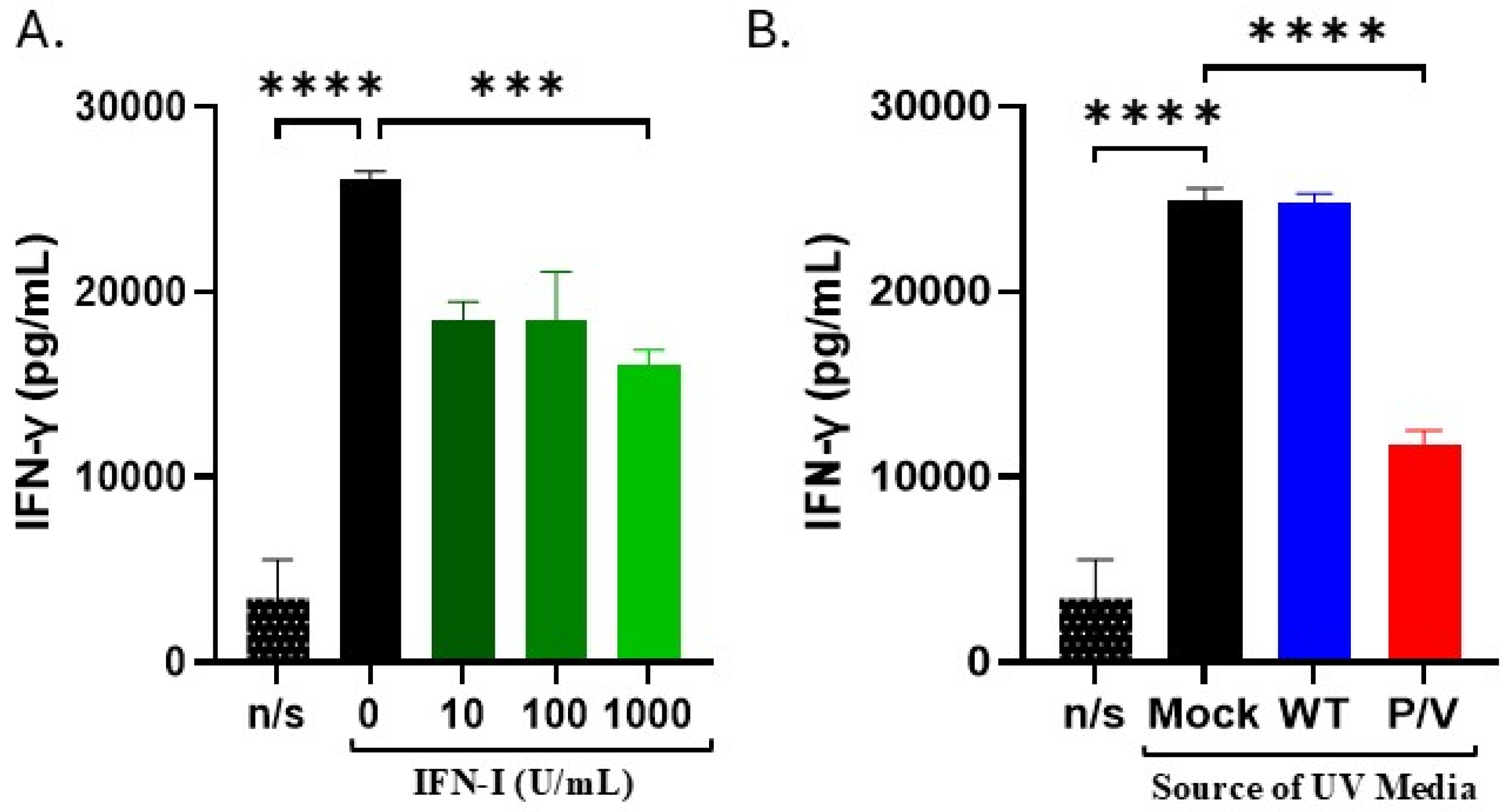
3.6. IFN-I and Media from P/V-Mutant-Infected Tumor Cells Decreases IFN-γ Release by PM21-NK Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wolf, N.K.; Kissiov, D.U.; Raulet, D.H. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat. Rev. Immunol. 2023, 23, 90–105. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Labrada, A.; Pesini, C.; Santiago, L.; Hidalgo, S.; Calvo-Perez, A.; Onate, C.; Andres-Tovar, A.; Garzon-Tituana, M.; Uranga-Murillo, I.; Arias, M.A.; et al. All About (NK Cell-Mediated) Death in Two Acts and an Unexpected Encore: Initiation, Execution and Activation of Adaptive Immunity. Front Immunol 2022, 13, 896228. [Google Scholar] [CrossRef] [PubMed]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Lupo, K.B.; Matosevic, S. Natural Killer Cells as Allogeneic Effectors in Adoptive Cancer Immunotherapy. Cancers 2019, 11, 769. [Google Scholar] [CrossRef] [PubMed]
- Geller, M.A.; Miller, J.S. Use of allogeneic NK cells for cancer immunotherapy. Immunotherapy 2011, 3, 1445–1459. [Google Scholar] [CrossRef]
- Laskowski, T.J.; Biederstadt, A.; Rezvani, K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer 2022, 22, 557–575. [Google Scholar] [CrossRef]
- Oyer, J.L.; Igarashi, R.Y.; Kulikowski, A.R.; Colosimo, D.A.; Solh, M.M.; Zakari, A.; Khaled, Y.A.; Altomare, D.A.; Copik, A.J. Generation of highly cytotoxic natural killer cells for treatment of acute myelogenous leukemia using a feeder-free, particle-based approach. Biol. Blood Marrow Transplant. 2015, 21, 632–639. [Google Scholar] [CrossRef]
- Oyer, J.L.; Pandey, V.; Igarashi, R.Y.; Somanchi, S.S.; Zakari, A.; Solh, M.; Lee, D.A.; Altomare, D.A.; Copik, A.J. Natural killer cells stimulated with PM21 particles expand and biodistribute in vivo: Clinical implications for cancer treatment. Cytotherapy 2016, 18, 653–663. [Google Scholar] [CrossRef]
- Oyer, J.L.; Croom-Perez, T.J.; Dieffenthaller, T.A.; Robles-Carillo, L.D.; Gitto, S.B.; Altomare, D.A.; Copik, A.J. Cryopreserved PM21-Particle-Expanded Natural Killer Cells Maintain Cytotoxicity and Effector Functions In Vitro and In Vivo. Front. Immunol. 2022, 13, 861681. [Google Scholar] [CrossRef]
- Oyer, J.L.; Gitto, S.B.; Altomare, D.A.; Copik, A.J. PD-L1 blockade enhances anti-tumor efficacy of NK cells. Oncoimmunology 2018, 7, e1509819. [Google Scholar] [CrossRef]
- Jhawar, S.R.; Thandoni, A.; Bommareddy, P.K.; Hassan, S.; Kohlhapp, F.J.; Goyal, S.; Schenkel, J.M.; Silk, A.W.; Zloza, A. Oncolytic Viruses-Natural and Genetically Engineered Cancer Immunotherapies. Front. Oncol. 2017, 7, 202. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.; Ho, L.; Wan, Y. Chemotherapy and Oncolytic Virotherapy: Advanced Tactics in the War against Cancer. Front. Oncol. 2014, 4, 145. [Google Scholar] [CrossRef]
- Singh, P.K.; Doley, J.; Kumar, G.R.; Sahoo, A.P.; Tiwari, A.K. Oncolytic viruses & their specific targeting to tumour cells. Indian. J. Med. Res. 2012, 136, 571–584. [Google Scholar]
- Enow, J.A.; Sheikh, H.I.; Rahman, M.M. Tumor Tropism of DNA Viruses for Oncolytic Virotherapy. Viruses 2023, 15, 2262. [Google Scholar] [CrossRef] [PubMed]
- Matveeva, O.V.; Chumakov, P.M. Defects in interferon pathways as potential biomarkers of sensitivity to oncolytic viruses. Rev. Med. Virol. 2018, 28, e2008. [Google Scholar] [CrossRef] [PubMed]
- Shobana, R.; Samal, S.K.; Elankumaran, S. Prostate-specific antigen-retargeted recombinant newcastle disease virus for prostate cancer virotherapy. J. Virol. 2013, 87, 3792–3800. [Google Scholar] [CrossRef]
- Keshavarz, M.; Solaymani-Mohammadi, F.; Miri, S.M.; Ghaemi, A. Oncolytic paramyxoviruses-induced autophagy; a prudent weapon for cancer therapy. J. Biomed. Sci. 2019, 26, 48. [Google Scholar]
- Ammayappan, A.; Russell, S.J.; Federspiel, M.J. Recombinant mumps virus as a cancer therapeutic agent. Mol. Ther. Oncolytics 2016, 3, 16019. [Google Scholar] [CrossRef]
- Andrejeva, J.; Childs, K.S.; Young, D.F.; Carlos, T.S.; Stock, N.; Goodbourn, S.; Randall, R.E. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. USA 2004, 101, 17264–17269. [Google Scholar] [CrossRef]
- Didcock, L.; Young, D.F.; Goodbourn, S.; Randall, R.E. The V protein of simian virus 5 inhibits interferon signalling by targeting STAT1 for proteasome-mediated degradation. J. Virol. 1999, 73, 9928–9933. [Google Scholar] [CrossRef]
- Sun, M.; Rothermel, T.A.; Shuman, L.; Aligo, J.A.; Xu, S.; Lin, Y.; Lamb, R.A.; He, B. Conserved cysteine-rich domain of paramyxovirus simian virus 5 V protein plays an important role in blocking apoptosis. J. Virol. 2004, 78, 5068–5078. [Google Scholar] [CrossRef] [PubMed]
- Dillon, P.J.; Wansley, E.K.; Young, V.A.; Alexander-Miller, M.A.; Parks, G.D. Exchange of P/V genes between two non-cytopathic simian virus 5 variants results in a recombinant virus that kills cells through death pathways that are sensitive to caspase inhibitors. J. Gen. Virol. 2006, 87 Pt 12, 3643–3648. [Google Scholar] [CrossRef]
- Wansley, E.K.; Parks, G.D. Naturally occurring substitutions in the P/V gene convert the noncytopathic paramyxovirus simian virus 5 into a virus that induces alpha/beta interferon synthesis and cell death. J. Virol. 2002, 76, 10109–10121. [Google Scholar] [CrossRef] [PubMed]
- Gainey, M.D.; Manuse, M.J.; Parks, G.D. A hyperfusogenic F protein enhances the oncolytic potency of a paramyxovirus simian virus 5 P/V mutant without compromising sensitivity to type I interferon. J. Virol. 2008, 82, 9369–9380. [Google Scholar] [CrossRef] [PubMed]
- Wansley, E.K.; Dillon, P.J.; Gainey, M.D.; Tam, J.; Cramer, S.D.; Parks, G.D. Growth sensitivity of a recombinant simian virus 5 P/V mutant to type I interferon differs between tumor cell lines and normal primary cells. Virology 2005, 335, 131–144. [Google Scholar] [CrossRef]
- Mandelboim, O.; Lieberman, N.; Lev, M.; Paul, L.; Arnon, T.I.; Bushkin, Y.; Davis, D.M.; Strominger, J.L.; Yewdell, J.W.; Porgador, A. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 2001, 409, 1055–1060. [Google Scholar] [CrossRef]
- Varudkar, N.; Oyer, J.L.; Copik, A.; Parks, G.D. Oncolytic parainfluenza virus combines with NK cells to mediate killing of infected and non-infected lung cancer cells within 3D spheroids: Role of type I and type III interferon signaling. J. Immunother. Cancer 2021, 9, 16019. [Google Scholar] [CrossRef]
- Ogbomo, H.; Michaelis, M.; Geiler, J.; van Rikxoort, M.; Muster, T.; Egorov, A.; Doerr, H.W.; Cinatl, J., Jr. Tumor cells infected with oncolytic influenza A virus prime natural killer cells for lysis of resistant tumor cells. Med. Microbiol. Immunol. 2010, 199, 93–101. [Google Scholar] [CrossRef]
- Ogbomo, H.; Zemp, F.J.; Lun, X.; Zhang, J.; Stack, D.; Rahman, M.M.; McFadden, G.; Mody, C.H.; Forsyth, P.A. Myxoma virus infection promotes NK lysis of malignant gliomas in vitro and in vivo. PLoS ONE 2013, 8, e66825. [Google Scholar] [CrossRef]
- Paolini, R.; Bernardini, G.; Molfetta, R.; Santoni, A. NK cells and interferons. Cytokine Growth Factor. Rev. 2015, 26, 113–120. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Shiffer, E.M.; Oyer, J.L.; Copik, A.J.; Parks, G.D. Parainfluenza Virus 5 V Protein Blocks Interferon Gamma-Mediated Upregulation of NK Cell Inhibitory Ligands and Improves NK Cell Killing of Neuroblastoma Cells. Viruses 2024, 16, 1270. [Google Scholar] [CrossRef] [PubMed]
- Capraro, G.A.; Johnson, J.B.; Kock, N.D.; Parks, G.D. Virus growth and antibody responses following respiratory tract infection of ferrets and mice with WT and P/V mutants of the paramyxovirus Simian Virus 5. Virology 2008, 376, 416–428. [Google Scholar] [CrossRef]
- Parks, G.D.; Ward, K.R.; Rassa, J.C. Increased readthrough transcription across the simian virus 5 M-F gene junction leads to growth defects and a global inhibition of viral mRNA synthesis. J. Virol. 2001, 75, 2213–2223. [Google Scholar] [CrossRef]
- Kedarinath, K.; Fox, C.R.; Crowgey, E.; Mazar, J.; Phelan, P.; Westmoreland, T.J.; Alexander, K.A.; Parks, G.D. CD24 Expression Dampens the Basal Antiviral State in Human Neuroblastoma Cells and Enhances Permissivity to Zika Virus Infection. Viruses 2022, 14, 1735. [Google Scholar] [CrossRef]
- Aquino, J.R.; Fox, C.R.; Parks, G.D. Role of Defective Interfering Particles in Complement-Mediated Lysis of Parainfluenza Virus-Infected Cells. Viruses 2025, 17, 488. [Google Scholar] [CrossRef]
- Didcock, L.; Young, D.F.; Goodbourn, S.; Randall, R.E. Sendai virus and simian virus 5 block activation of interferon-responsive genes: Importance for virus pathogenesis. J. Virol. 1999, 73, 3125–3133. [Google Scholar] [CrossRef]
- Varudkar, N.; Shiffer, E.M.; Oyer, J.L.; Copik, A.; Parks, G.D. Delivery of a novel membrane-anchored Fc chimera enhances NK cell-mediated killing of tumor cells and persistently virus-infected cells. PLoS ONE 2023, 18, e0285532. [Google Scholar] [CrossRef]
- Coenon, L.; Geindreau, M.; Ghiringhelli, F.; Villalba, M.; Bruchard, M. Natural Killer cells at the frontline in the fight against cancer. Cell Death Dis. 2024, 15, 614. [Google Scholar] [CrossRef]
- Hofle, J.; Trenkner, T.; Kleist, N.; Schwane, V.; Vollmers, S.; Barcelona, B.; Niehrs, A.; Fittje, P.; Huynh-Tran, V.H.; Sauter, J.; et al. Engagement of TRAIL triggers degranulation and IFNgamma production in human natural killer cells. EMBO Rep. 2022, 23, e54133. [Google Scholar] [CrossRef] [PubMed]
- Muller, L.; Aigner, P.; Stoiber, D. Type I Interferons and Natural Killer Cell Regulation in Cancer. Front. Immunol. 2017, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Hida, S.; Takayanagi, H.; Yokochi, T.; Kayagaki, N.; Takeda, K.; Yagita, H.; Okumura, K.; Tanaka, N.; Taniguchi, T.; et al. Antiviral response by natural killer cells through TRAIL gene induction by IFN-alpha/beta. Eur. J. Immunol. 2001, 31, 3138–3146. [Google Scholar] [CrossRef]
- Lee, A.J.; Mian, F.; Poznanski, S.M.; Stackaruk, M.; Chan, T.; Chew, M.V.; Ashkar, A.A. Type I Interferon Receptor on NK Cells Negatively Regulates Interferon-gamma Production. Front. Immunol. 2019, 10, 1261. [Google Scholar]
- Lamers-Kok, N.; Panella, D.; Georgoudaki, A.M.; Liu, H.; Ozkazanc, D.; Kucerova, L.; Duru, A.D.; Spanholtz, J.; Raimo, M. Natural killer cells in clinical development as non-engineered, engineered, and combination therapies. J. Hematol. Oncol. 2022, 15, 164. [Google Scholar] [CrossRef]
- Page, A.; Chuvin, N.; Valladeau-Guilemond, J.; Depil, S. Development of NK cell-based cancer immunotherapies through receptor engineering. Cell. Mol. Immunol. 2024, 21, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Teng, W.; Tian, Y.; Li, S.; Xia, N.; Huang, C. Immune landscape and response to oncolytic virus-based immunotherapy. Front. Med. 2024, 18, 411–429. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, M.; Chi, H.; Liu, Y.; Yu, G. The combination therapy of oncolytic virotherapy. Front. Pharmacol. 2024, 15, 1380313. [Google Scholar] [CrossRef]
- Mamola, J.A.; Chen, C.Y.; Currier, M.A.; Cassady, K.; Lee, D.A.; Cripe, T.P. Opportunities and challenges of combining adoptive cellular therapy with oncolytic virotherapy. Mol. Ther. Oncolytics 2023, 29, 118–124. [Google Scholar] [CrossRef]
- Marotel, M.; Hasim, M.S.; Hagerman, A.; Ardolino, M. The two-faces of NK cells in oncolytic virotherapy. Cytokine Growth Factor. Rev. 2020, 56, 59–68. [Google Scholar] [CrossRef]
- Geoffroy, K.; Bourgeois-Daigneault, M.C. The pros and cons of interferons for oncolytic virotherapy. Cytokine Growth Factor. Rev. 2020, 56, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Kulaeva, O.I.; Draghici, S.; Tang, L.; Kraniak, J.M.; Land, S.J.; Tainsky, M.A. Epigenetic silencing of multiple interferon pathway genes after cellular immortalization. Oncogene 2003, 22, 4118–4127. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Tan, F.; Wang, Y.; Liu, X.; Kong, X.; Meng, J.; Yang, L.; Cen, S. The gamble between oncolytic virus therapy and IFN. Front. Immunol. 2022, 13, 971674. [Google Scholar] [CrossRef] [PubMed]
- Arimilli, S.; Alexander-Miller, M.A.; Parks, G.D. A simian virus 5 (SV5) P/V mutant is less cytopathic than wild-type SV5 in human dendritic cells and is a more effective activator of dendritic cell maturation and function. J. Virol. 2006, 80, 3416–3427. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Durbin, J.E. Contribution of type III interferons to antiviral immunity: Location, location, location. J. Biol. Chem. 2017, 292, 7295–7303. [Google Scholar] [CrossRef]
- Ruby, J.; Bluethmann, H.; Peschon, J.J. Antiviral activity of tumor necrosis factor (TNF) is mediated via p55 and p75 TNF receptors. J. Exp. Med. 1997, 186, 1591–1596. [Google Scholar] [CrossRef]
- Al-Qahtani, A.A.; Alhamlan, F.S.; Al-Qahtani, A.A. Pro-Inflammatory and Anti-Inflammatory Interleukins in Infectious Diseases: A Comprehensive Review. Trop. Med. Infect. Dis. 2024, 9, 13. [Google Scholar] [CrossRef]
- Valdes-Lopez, J.F.; Hernandez-Sarmiento, L.J.; Tamayo-Molina, Y.S.; Velilla-Hernandez, P.A.; Rodenhuis-Zybert, I.A.; Urcuqui-Inchima, S. Interleukin 27, like interferons, activates JAK-STAT signaling and promotes pro-inflammatory and antiviral states that interfere with dengue and chikungunya viruses replication in human macrophages. Front. Immunol. 2024, 15, 1385473. [Google Scholar] [CrossRef]
- Amsden, H.; Kourko, O.; Roth, M.; Gee, K. Antiviral Activities of Interleukin-27: A Partner for Interferons? Front. Immunol. 2022, 13, 902853. [Google Scholar] [CrossRef]
- Zannikou, M.; Fish, E.N.; Platanias, L.C. Signaling by Type I Interferons in Immune Cells: Disease Consequences. Cancers 2024, 16, 1600. [Google Scholar] [CrossRef]
- Cheon, H.; Borden, E.C.; Stark, G.R. Interferons and their stimulated genes in the tumor microenvironment. Semin. Oncol. 2014, 41, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Zamai, L.; Ahmad, M.; Bennett, I.M.; Azzoni, L.; Alnemri, E.S.; Perussia, B. Natural killer (NK) cell-mediated cytotoxicity: Differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J. Exp. Med. 1998, 188, 2375–2380. [Google Scholar] [CrossRef]
- Kwaa, A.K.R.; Talana, C.A.G.; Blankson, J.N. Interferon Alpha Enhances NK Cell Function and the Suppressive Capacity of HIV-Specific CD8+ T Cells. J. Virol. 2019, 93, e01541-18. [Google Scholar] [CrossRef]
- Aquino-Lopez, A.; Senyukov, V.V.; Vlasic, Z.; Kleinerman, E.S.; Lee, D.A. Interferon Gamma Induces Changes in Natural Killer (NK) Cell Ligand Expression and Alters NK Cell-Mediated Lysis of Pediatric Cancer Cell Lines. Front. Immunol. 2017, 8, 391. [Google Scholar] [CrossRef]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-gamma in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.R.; Merlino, G. The two faces of interferon-gamma in cancer. Clin. Cancer Res. 2011, 17, 6118–6124. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, T.; Gil, M.P.; Wang, X.; Louten, J.; Chu, W.M.; Biron, C.A. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J. Exp. Med. 2007, 204, 2383–2396. [Google Scholar] [CrossRef]
- Nguyen, K.B.; Cousens, L.P.; Doughty, L.A.; Pien, G.C.; Durbin, J.E.; Biron, C.A. Interferon alpha/beta-mediated inhibition and promotion of interferon gamma: STAT1 resolves a paradox. Nat. Immunol. 2000, 1, 70–76. [Google Scholar] [CrossRef]
- Nguyen, K.B.; Watford, W.T.; Salomon, R.; Hofmann, S.R.; Pien, G.C.; Morinobu, A.; Gadina, M.; O’Shea, J.J.; Biron, C.A. Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science 2002, 297, 2063–2066. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shiffer, E.M.; Oyer, J.L.; Copik, A.J.; Parks, G.D. A Type I IFN-Inducing Oncolytic Virus Improves NK Cell-Mediated Killing of Tumor Cells In Vitro Through Multiple Mechanisms. Viruses 2025, 17, 897. https://doi.org/10.3390/v17070897
Shiffer EM, Oyer JL, Copik AJ, Parks GD. A Type I IFN-Inducing Oncolytic Virus Improves NK Cell-Mediated Killing of Tumor Cells In Vitro Through Multiple Mechanisms. Viruses. 2025; 17(7):897. https://doi.org/10.3390/v17070897
Chicago/Turabian StyleShiffer, Elisabeth M., Jeremiah L. Oyer, Alicja J. Copik, and Griffith D. Parks. 2025. "A Type I IFN-Inducing Oncolytic Virus Improves NK Cell-Mediated Killing of Tumor Cells In Vitro Through Multiple Mechanisms" Viruses 17, no. 7: 897. https://doi.org/10.3390/v17070897
APA StyleShiffer, E. M., Oyer, J. L., Copik, A. J., & Parks, G. D. (2025). A Type I IFN-Inducing Oncolytic Virus Improves NK Cell-Mediated Killing of Tumor Cells In Vitro Through Multiple Mechanisms. Viruses, 17(7), 897. https://doi.org/10.3390/v17070897