
Genomic Characterization and Molecular Epidemiology of Tusaviruses and Related Novel Protoparvoviruses (Family Parvoviridae) from Ruminant Species (Bovine, Ovine and Caprine) in Hungary
 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
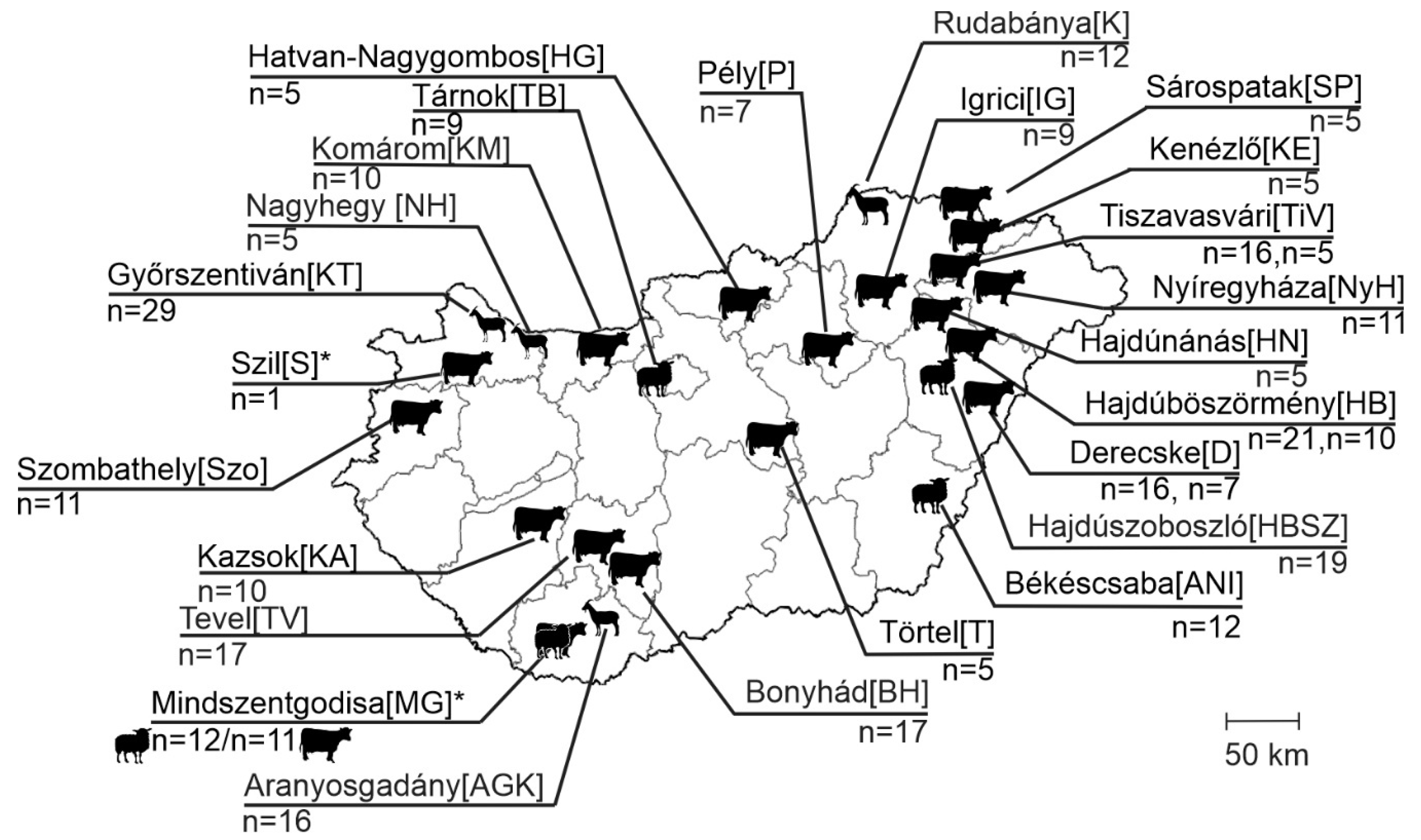
2.1. Background Information on Samples, Animals and Farms
2.2. Viral Metagenomics and Next Generation Sequencing
2.3. Genome Determination and Screening Reactions Using PCR and Sanger Sequencing Methods
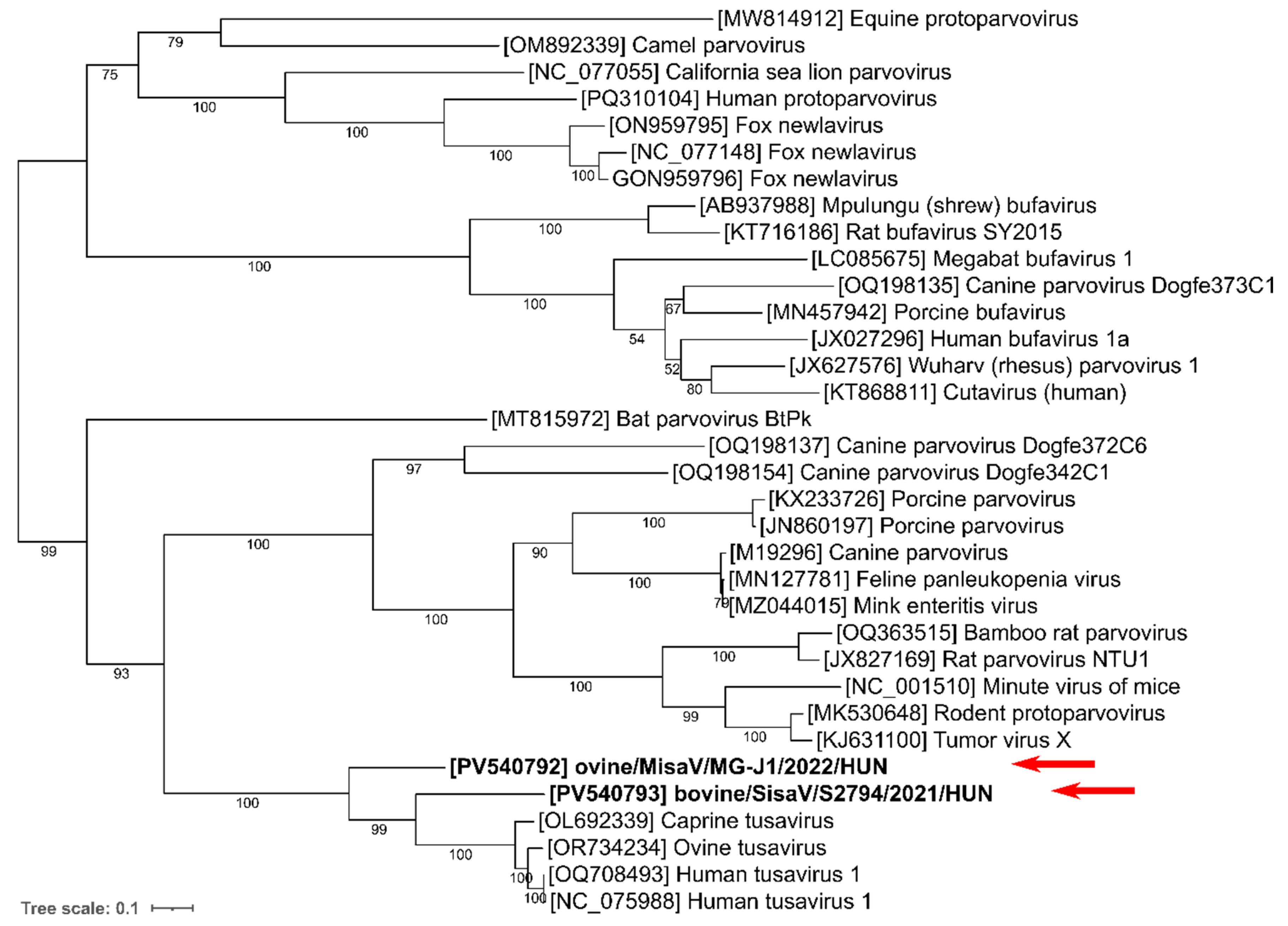
2.4. In Silico Sequence, Phylogenetic and Recombination Analyses
3. Results
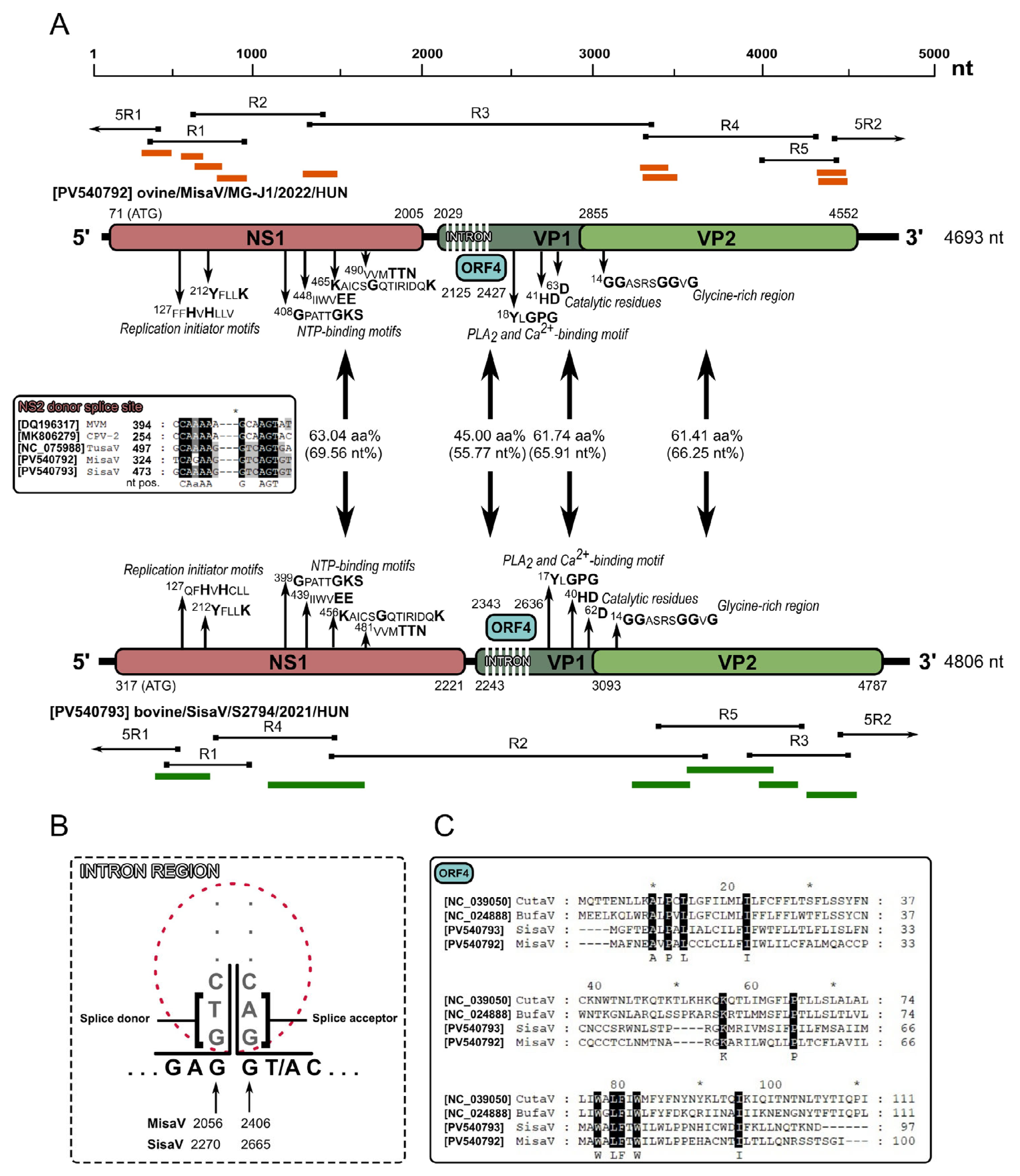
3.1. Genome Analysis of Ovine and Bovine Protoparvoviruses
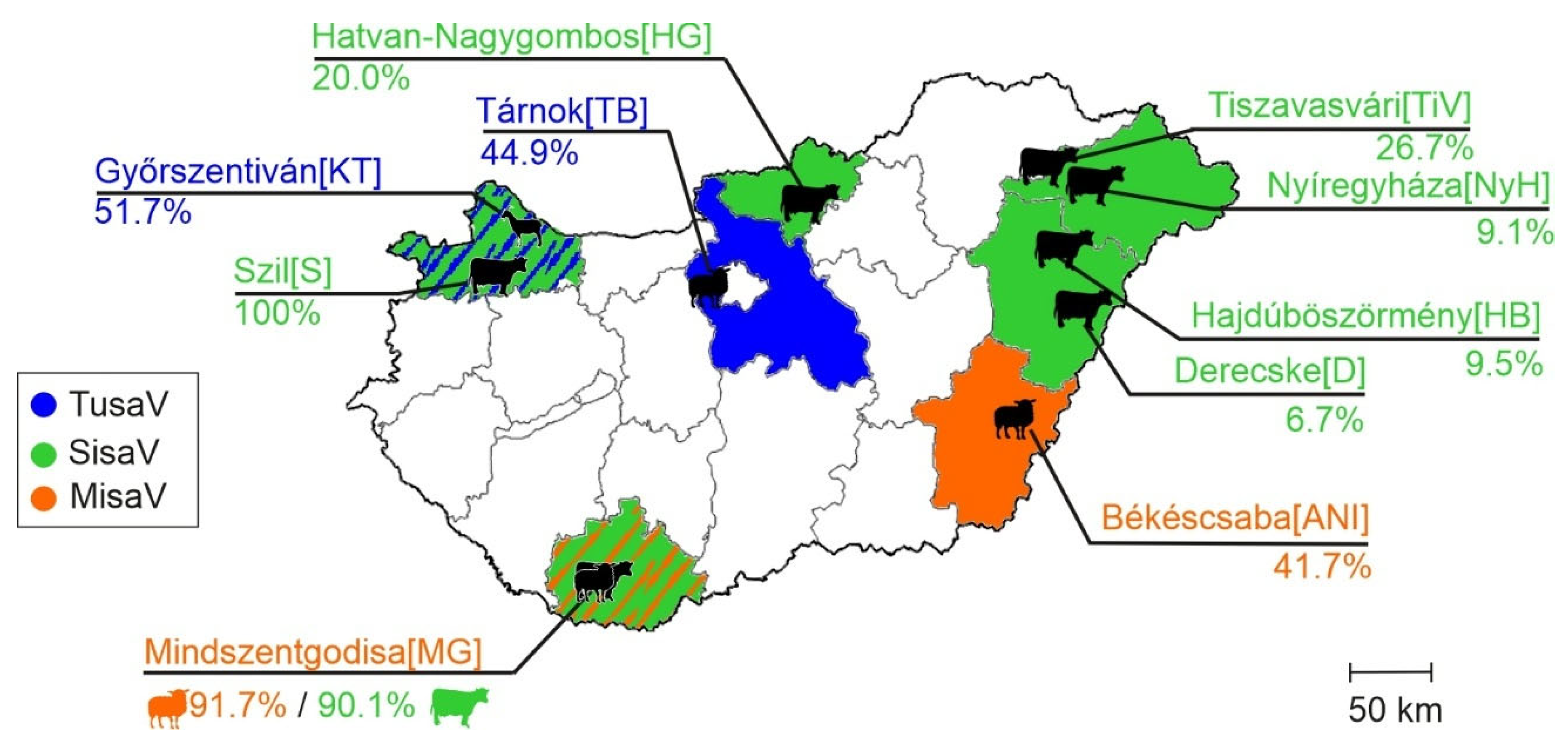
3.2. Epidemiological Investigations of Ruminant Protoparvoviruses
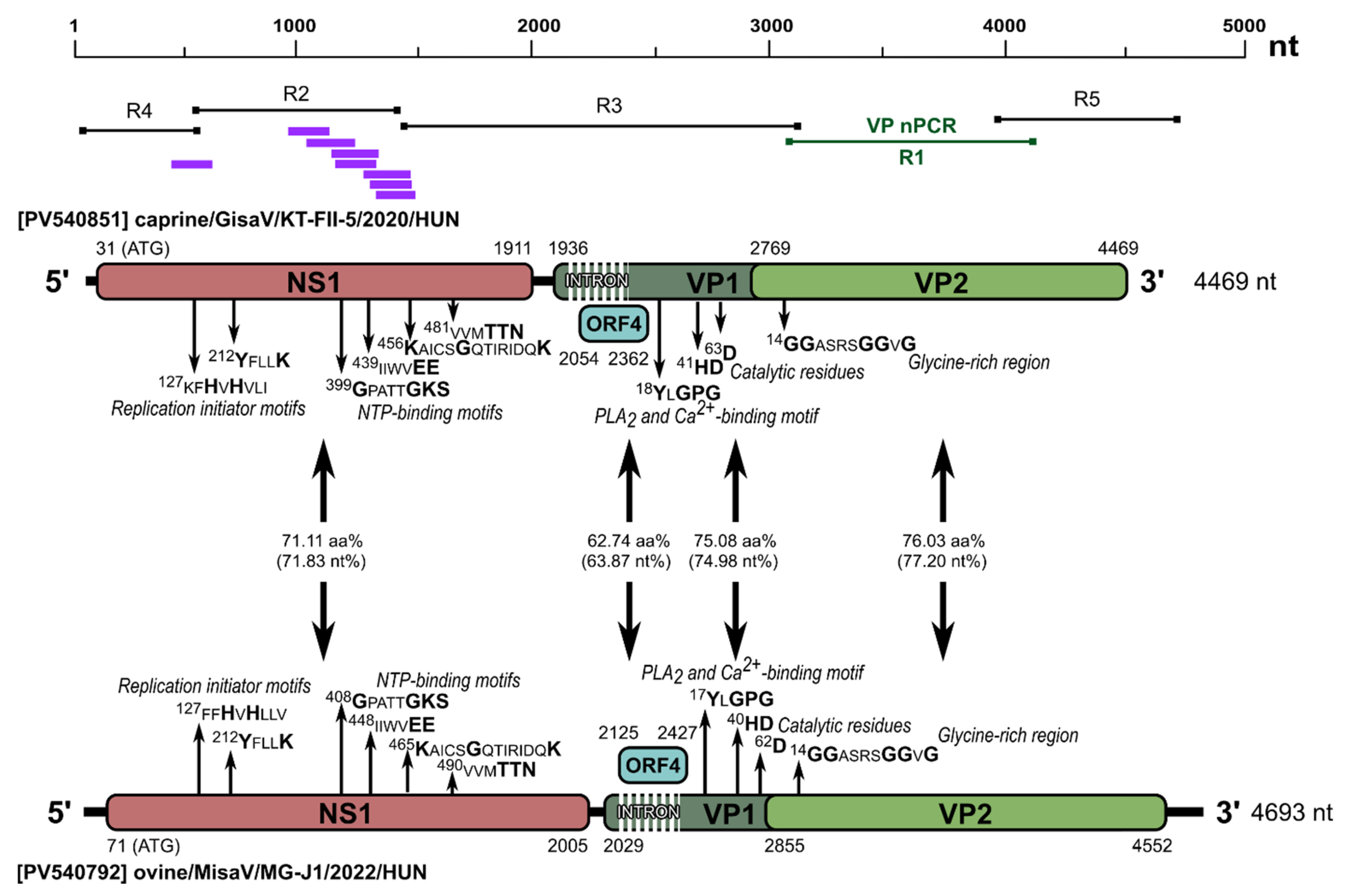
3.3. Determination and Analyses of a Misavirus-Related Goat Protoparvoviruses (Gisaviruses)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.-M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Pénzes, J.J.; et al. ICTV Virus Taxonomy Profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368. [Google Scholar] [CrossRef] [PubMed]
- Cotmore; Tattersall, P. Parvoviruses: Small Does Not Mean Simple. Annu. Rev. Virol. 2014, 1, 517–537. [Google Scholar] [CrossRef] [PubMed]
- Pénzes, J.J.; Söderlund-Venermo, M.; Canuti, M.; Eis-Hübinger, A.M.; Hughes, J.; Cotmore, S.F.; Harrach, B. Reorganizing the Family Parvoviridae: A Revised Taxonomy Independent of the Canonical Approach Based on Host Association. Arch. Virol. 2020, 165, 2133–2146. [Google Scholar] [CrossRef]
- Mietzsch, M.; Pénzes, J.J.; Agbandje-McKenna, M. Twenty-Five Years of Structural Parvovirology. Viruses 2019, 11, 362. [Google Scholar] [CrossRef]
- Cotmore; Tattersall, P. Genome Packaging Sense Is Controlled by the Efficiency of the Nick Site in the Right-End Replication Origin of Parvoviruses Minute Virus of Mice and LuIII. J. Virol. 2005, 79, 2287–2300. [Google Scholar] [CrossRef]
- Tijssen, P. CRC Handbook of Parvoviruses, 1st ed.; CRC Press: Boca Raton, FL, USA, 1989; Volume I, ISBN 978-0-8493-3217-3. [Google Scholar]
- Bates, R.C.; Snyder, C.E.; Banerjee, P.T.; Mitra, S. Autonomous Parvovirus LuIII Encapsidates Equal Amounts of plus and Minus DNA Strands. J. Virol. 1984, 49, 319–324. [Google Scholar] [CrossRef]
- Shackelton, L.A.; Parrish, C.R.; Truyen, U.; Holmes, E.C. High Rate of Viral Evolution Associated with the Emergence of Carnivore Parvovirus. Proc. Natl. Acad. Sci. USA 2005, 102, 379–384. [Google Scholar] [CrossRef]
- Decaro, N.; Desario, C.; Miccolupo, A.; Campolo, M.; Parisi, A.; Martella, V.; Amorisco, F.; Lucente, M.S.; Lavazza, A.; Buonavoglia, C. Genetic Analysis of Feline Panleukopenia Viruses from Cats with Gastroenteritis. J. Gen. Virol. 2008, 89, 2290–2298. [Google Scholar] [CrossRef]
- Shackelton, L.A.; Hoelzer, K.; Parrish, C.R.; Holmes, E.C. Comparative Analysis Reveals Frequent Recombination in the Parvoviruses. J. Gen. Virol. 2007, 88, 3294–3301. [Google Scholar] [CrossRef]
- Ohshima, T.; Mochizuki, M. Evidence for Recombination Between Feline Panleukopenia Virus and Canine Parvovirus Type 2. J. Vet. Med. Sci. 2009, 71, 403–408. [Google Scholar] [CrossRef]
- Mochizuki, M.; Ohshima, T.; Une, Y.; Yachi, A. Recombination between Vaccine and Field Strains of Canine Parvovirus Is Revealed by Isolation of Virus in Canine and Feline Cell Cultures. J. Vet. Med. Sci. 2008, 70, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cheng, S.; Yi, L.; Cheng, Y.; Yang, S.; Xu, H.; Zhao, H.; Yan, X.; Wu, H. Evidence for Natural Recombination between Mink Enteritis Virus and Canine Parvovirus. Virol. J. 2012, 9, 252. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Vo, N.P.; Bonkoungou, I.J.O.; Kapoor, A.; Barro, N.; O’Ryan, M.; Kapusinszky, B.; Wang, C.; Delwart, E. Acute Diarrhea in West African Children: Diverse Enteric Viruses and a Novel Parvovirus Genus. J. Virol. 2012, 86, 11024–11030. [Google Scholar] [CrossRef]
- Sasaki, M.; Orba, Y.; Anindita, P.D.; Ishii, A.; Ueno, K.; Hang’ombe, B.M.; Mweene, A.S.; Ito, K.; Sawa, H. Distinct Lineages of Bufavirus in Wild Shrews and Nonhuman Primates. Emerg. Infect. Dis. J. 2015, 21, 1230–1233. [Google Scholar] [CrossRef] [PubMed]
- Yahiro, T.; Wangchuk, S.; Tshering, K.; Bandhari, P.; Zangmo, S.; Dorji, T.; Tshering, K.; Matsumoto, T.; Nishizono, A.; Söderlund-Venermo, M.; et al. Novel Human Bufavirus Genotype 3 in Children with Severe Diarrhea, Bhutan. Emerg. Infect. Dis. 2014, 20, 1037–1039. [Google Scholar] [CrossRef]
- Di Martino, B.; Sarchese, V.; Di Profio, F.; Palombieri, A.; Melegari, I.; Fruci, P.; Aste, G.; Bányai, K.; Fulvio, M.; Martella, V. Genetic Heterogeneity of Canine Bufaviruses. Transbounding Emerg. Dis. 2021, 68, 802–812. [Google Scholar] [CrossRef]
- Janus, L.M.; Bleich, A. Coping with Parvovirus Infections in Mice: Health Surveillance and Control. Lab. Anim. 2012, 46, 14–23. [Google Scholar] [CrossRef]
- Mészáros, I.; Olasz, F.; Cságola, A.; Tijssen, P.; Zádori, Z. Biology of Porcine Parvovirus (Ungulate parvovirus 1). Viruses 2017, 9, 393. [Google Scholar] [CrossRef]
- Tuteja, D.; Banu, K.; Mondal, B. Canine Parvovirology—A Brief Updated Review on Structural Biology, Occurrence, Pathogenesis, Clinical Diagnosis, Treatment and Prevention. Comp. Immunol. Microbiol. Infect. Dis. 2022, 82, 101765. [Google Scholar] [CrossRef]
- Barrs, V.R. Feline Panleukopenia. Vet. Clin. N. Am. Small Anim. Pract. 2019, 49, 651–670. [Google Scholar] [CrossRef]
- Phan, T.G.; Sdiri-Loulizi, K.; Aouni, M.; Ambert-Balay, K.; Pothier, P.; Deng, X.; Delwart, E. New Parvovirus in Child with Unexplained Diarrhea, Tunisia. Emerg. Infect. Dis. 2014, 20, 1911–1913. [Google Scholar] [CrossRef] [PubMed]
- Mohanraj, U.; Jokinen, M.; Thapa, R.R.; Paloniemi, M.; Vesikari, T.; Lappalainen, M.; Tarkka, E.; Nora-Krūkle, Z.; Vilmane, A.; Vettenranta, K. Human Protoparvovirus DNA and IgG in Children and Adults with and without Respiratory or Gastrointestinal Infections. Viruses 2021, 13, 483. [Google Scholar] [CrossRef]
- He, H.; Li, Y.; Chen, J.; Xian, J.; Zheng, L.; Sun, H.; Fan, S.; Fu, J.; Li, Q.; Chen, C. Identification and Genetic Characteristics of Tusavirus in Fecal Samples of Patients with Chronic Diseases in Guangzhou, China. Front. Microbiol. 2023, 14, 1205134. [Google Scholar] [CrossRef] [PubMed]
- Väisänen, E.; Paloniemi, M.; Kuisma, I.; Lithovius, V.; Kumar, A.; Franssila, R.; Ahmed, K.; Delwart, E.; Vesikari, T.; Hedman, K. Epidemiology of Two Human Protoparvoviruses, Bufavirus and Tusavirus. Sci. Rep. 2016, 6, 39267. [Google Scholar] [CrossRef] [PubMed]
- Väisänen, E.; Mohanraj, U.; Kinnunen, P.M.; Jokelainen, P.; Al-Hello, H.; Barakat, A.M.; Sadeghi, M.; Jalilian, F.A.; Majlesi, A.; Masika, M. Global Distribution of Human Protoparvoviruses. Emerg. Infect. Dis. 2018, 24, 1292. [Google Scholar] [CrossRef]
- Väisänen, E.; Fu, Y.; Koskenmies, S.; Fyhrquist, N.; Wang, Y.; Keinonen, A.; Mäkisalo, H.; Väkevä, L.; Pitkänen, S.; Ranki, A. Cutavirus DNA in Malignant and Nonmalignant Skin of Cutaneous T-Cell Lymphoma and Organ Transplant Patients but Not of Healthy Adults. Clin. Infect. Dis. 2019, 68, 1904–1910. [Google Scholar] [CrossRef]
- Chesnut, S.K.; Mohanraj, U.; Rayamajhi Thapa, R.; Jalilian, F.A.; Amini, R.; Sedighi, I.; Sedighi, P.; Al-Hello, H.; Barakat, A.M.; Masika, M.; et al. In Search of Human Protoparvovirus Acute Infections. Virology 2025, 608, 110529. [Google Scholar] [CrossRef]
- Reuter, G.; Pankovics, P.; László, Z.; Gáspár, G.; Hui, A.; Delwart, E.; Boros, Á. Human-Stool-Associated Tusavirus (Parvoviridae) in Domestic Goats and Sheep. Arch. Virol. 2022, 167, 1307–1310. [Google Scholar] [CrossRef]
- Davies, H.; Dastjerdi, A.; Everest, D.; Floyd, T.; Collins, R.; McFadzean, H.; Reuter, G.; Reichel, R. Incidental Finding of a Human-like Tusavirus in a Lamb with Lip Lesions and Fatal Pneumonia. J. Gen. Virol. 2024, 105, 001968. [Google Scholar] [CrossRef]
- Boros, Á.; Albert, M.; Urbán, P.; Herczeg, R.; Gáspár, G.; Balázs, B.; Cságola, A.; Pankovics, P.; Gyenesei, A.; Reuter, G. Unusual “Asian-Origin” 2c to 2b Point Mutant Canine Parvovirus (Parvoviridae) and Canine Astrovirus (Astroviridae) Co-Infection Detected in Vaccinated Dogs with an Outbreak of Severe Haemorrhagic Gastroenteritis with High Mortality Rate in Hungary. Vet. Res. Commun. 2022, 46, 1355–1361. [Google Scholar] [CrossRef]
- László, Z.; Pankovics, P.; Reuter, G.; Cságola, A.; Bálint, Á.; Albert, M.; Boros, Á. Multiple Types of Novel Enteric Bopiviruses (Picornaviridae) with the Possibility of Interspecies Transmission Identified from Cloven-Hoofed Domestic Livestock (Ovine, Caprine and Bovine) in Hungary. Viruses 2021, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.S.; Kapoor, A.; Lukashov, V.V.; Simmonds, P.; Hecht, F.; Delwart, E. New DNA Viruses Identified in Patients with Acute Viral Infection Syndrome. J. Virol. 2005, 79, 8230–8236. [Google Scholar] [CrossRef] [PubMed]
- Wakuda, M.; Pongsuwanna, Y.; Taniguchi, K. Complete Nucleotide Sequences of Two RNA Segments of Human Picobirnavirus. J. Virol. Methods 2005, 126, 165–169. [Google Scholar] [CrossRef]
- Reuter, G.; Boros, Á.; Delwart, E.; Pankovics, P. Novel Seadornavirus (Family Reoviridae) Related to Banna Virus in Europe. Arch. Virol. 2013, 158, 2163–2167. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Nicholas, K.B.; Nicholas, H.B., Jr.; Deerfield, D.W., II. GeneDoc: Analysis and Visualization of Genetic Variation. GeneDoc: Anal. Vis. Genet. Var. 1997, 4, 14. [Google Scholar]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v6: Recent Updates to the Phylogenetic Tree Display and Annotation Tool. Nucleic Acids Res. 2024, 52, W78–W82. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A Computer Program for Analyzing Recombination in, and Removing Signals of Recombination from, Nucleotide Sequence Datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Scalzitti, N.; Kress, A.; Orhand, R.; Weber, T.; Moulinier, L.; Jeannin-Girardon, A.; Collet, P.; Poch, O.; Thompson, J.D. Spliceator: Multi-Species Splice Site Prediction Using Convolutional Neural Networks. BMC Bioinform. 2021, 22, 561. [Google Scholar] [CrossRef] [PubMed]
- Vashisht, K.; Faaberg, K.S.; Aber, A.L.; Brown, K.E.; O’Sullivan, M.G. Splice Junction Map of Simian Parvovirus Transcripts. J. Virol. 2004, 78, 10911–10919. [Google Scholar] [CrossRef]
- Takáts, K.; Pankovics, P.; Balázs, B.; Boros, Á.; Mátics, R.; Reuter, G. Novel Relatives of Mecsek Mountains Mammarenavirus (Family Arenaviridae) in Hedgehogs Living in Different Sampling Areas in Hungary. Sci. Rep. 2025, 15, 2907. [Google Scholar] [CrossRef] [PubMed]
- Borg, I.; Groenen, P.J.F. (Eds.) Methods Related to MDS. In Modern Multidimensional Scaling: Theory and Applications; Springer: New York, NY, USA, 2005; pp. 519–540. ISBN 978-0-387-28981-6. [Google Scholar]
- Jolliffe, I.T.; Cadima, J. Principal Component Analysis: A Review and Recent Developments. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef]
- Kozak, M. Point Mutations Define a Sequence Flanking the AUG Initiator Codon That Modulates Translation by Eukaryotic Ribosomes. Cell 1986, 44, 283–292. [Google Scholar] [CrossRef]
- Kozak, M. An Analysis of 5′-Noncoding Sequences from 699 Vertebrate Messenger RNAs. Nucleic Acids Res. 1987, 15, 8125–8148. [Google Scholar] [CrossRef]
- Ilyina, T.V.; Koonin, E.V. Conserved Sequence Motifs in the Initiator Proteins for Rolling Circle DNA Replication Encoded by Diverse Replicons from Eubacteria, Eucaryotes and Archaebacteria. Nucleic Acids Res. 1992, 20, 3279–3285. [Google Scholar] [CrossRef]
- Walker, J.E.; Saraste, M.; Runswick, M.J.; Gay, N.J. Distantly Related Sequences in the Alpha- and Beta-Subunits of ATP Synthase, Myosin, Kinases and Other ATP-Requiring Enzymes and a Common Nucleotide Binding Fold. EMBO J. 1982, 1, 945–951. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Koonin, E.V.; Wolf, Y.I. A New Superfamily of Putative NTP-Binding Domains Encoded by Genomes of Small DNA and RNA Viruses. FEBS Lett. 1990, 262, 145–148. [Google Scholar] [CrossRef]
- Zádori, Z.; Szelei, J.; Lacoste, M.-C.; Li, Y.; Gariépy, S.; Raymond, P.; Allaire, M.; Nabi, I.R.; Tijssen, P. A Viral Phospholipase A2 Is Required for Parvovirus Infectivity. Dev. Cell 2001, 1, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Dreno, B.; da Costa, A.C.; Li, L.; Orlandi, P.; Deng, X.; Kapusinszky, B.; Siqueira, J.; Knol, A.-C.; Halary, F.; et al. A New Protoparvovirus in Human Fecal Samples and Cutaneous T Cell Lymphomas (Mycosis Fungoides). Virology 2016, 496, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P.; Kerr, J.; Bloom, M.E.; Parrish, R.M.; Linden, C.R. Structure and Organization of the Viral Genome. In Parvoviruses; Hodder Arnold: London, UK, 2005; pp. 73–94. [Google Scholar]
- Polz, M.F.; Cavanaugh, C.M. Bias in template-to-product ratios in multitemplate PCR. Appl. Environ. Microbiol. 1998, 64, 3724–3730. [Google Scholar] [CrossRef]
- Razizadeh, M.H.; Khatami, A.; Zarei, M. Global Status of Bufavirus, Cosavirus, and Saffold Virus in Gastroenteritis: A Systematic Review and Meta-Analysis. Front. Med. 2021, 8, 775698. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.; Nagaro, K. Cutavirus: A Newly Discovered Parvovirus on the Rise. Infect. Genet. Evol. 2020, 80, 104175. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Region | Primer Name | 5′-3′ Sequence | Reaction Type | Product Length (bp) # |
|---|---|---|---|---|
| NS | MiSiTuV-NS-Screen-F | TATGTRCACATIATGACTCA | Screening nPCR 1st PCR round | 585 * |
| NS | MiSiTuV-NS-Screen-R | TCTGATYGGTTGAGTGTGTTC | Screening nPCR 1st PCR round | |
| NS | MiSiTuV-NS-Screen-F2 | ACCTAGTTAARGAAAGACACAC | Screening nPCR 2nd PCR round | 371 * |
| NS | MiSiTuV-NS-Screen-R2 | TTGRTCTATGCGGATAGTTTG | Screening nPCR 2nd PCR round | |
| VP | MiSiTuV-VP-Fgen | CTGGTGAYTTTGACAATACTAC | Typing nPCR 1st PCR round | 1285 |
| VP | MiSiTuV-VP-Rgen | TTGTCCCAGATTTGTCCATGTGG | Typing nPCR 1st PCR round | |
| VP | MiSiTuV-VP-F2gen | AGCCTTGTAGACTGTAATGCATGG | Typing nPCR 2nd PCR round | 1062 |
| VP | MiSiTuV-VP-R2gen | TGGATAGTATGGACCTTGATGGTC | Typing nPCR 2nd PCR round |
| Protein/Virus | Misavirus PV540792 | Sisavirus PV540793 | |||
|---|---|---|---|---|---|
| Tusavirus 1 NC_075988 | aa% | nt% | aa% | nt% | |
| NS1 | 63.04 | 67.02 | 62.14 | 67.61 | |
| VP1 | 62.15 | 59.90 | 62.79 | 58.57 | |
| VP2 | 62.12 | 67.66 | 63.89 | 67.07 | |
| No. of Positive Samples/Total by Age Groups | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| (I) <2 mo | (II) 2–12 mo | (III) >12 mo | ||||||||
| Host species | Farm location [ID] | MisaV | SisaV | TusaV | MisaV | SisaV | TusaV | MisaV | SisaV | TusaV |
| Ovine | Mindszentgodisa [MG] | n.a. | n.a. | n.a. | 11/12 | 0/12 | 0/12 | n.a. | n.a. | n.a. |
| Tárnok [TB] | 0/9 | 0/9 | 4/9 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | |
| Békéscsaba [ANI] | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | 5/12 | 0/12 | 0/12 | |
| Ʃ | 0/9 | 0/9 | 4/9 (44.4%) | 11/12 (91.7%) | 0/12 | 0/12 | 5/12 (41.7%) | 0/12 | 0/12 | |
| Bovine | Hajdúböszörmény [HB] | 0/19 | 0/19 | 0/19 | n.a. | n.a. | n.a. | 0/2 | 2/2 | 0/2 |
| Nyíregyháza [NyH] | 0/5 | 0/5 | 0/5 | 0/4 | 0/4 | 0/4 | 0/2 | 1/2 | 0/2 | |
| Szil [S] | n.a. | n.a. | n.a. | 0/1 | 1/1 | 0/1 | n.a. | n.a. | n.a. | |
| Derecske [DR] | 0/3 | 0/3 | 0/3 | 0/1 | 0/1 | 0/1 | 0/11 | 1/11 | 0/11 | |
| Tiszavasvári [TiV] | 0/8 | 1/8 | 0/8 | 0/7 | 3/7 | 0/7 | 0/1 | 0/1 | 0/1 | |
| Mindszentgodisa [MG] | n.a. | n.a. | n.a. | 0/11 | 10/11 | 0/11 | n.a. | n.a. | n.a. | |
| Hatvan-Nagygombos [HG] | 0/5 | 1/5 | 0/5 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | |
| Ʃ | 0/40 | 2/40 (5.0%) | 0/40 | 0/23 | 14/23 (60.86%) | 0/23 | 0/16 | 4/16 (25.0%) | 0/16 | |
| Caprine | Győrszentiván [KT] | 0/9 | 0/9 | 2/9 | 0/10 | 0/10 | 10/10 | 0/10 | 0/10 | 3/10 |
| Ʃ | 0/9 | 0/9 | 2/9 (22.2%) | 0/10 | 0/10 | 10/10 (100%) | 0/10 | 0/10 | 3/10 (30.0%) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tóth, F.; Pankovics, P.; Urbán, P.; Herczeg, R.; Albert, E.; Reuter, G.; Boros, Á. Genomic Characterization and Molecular Epidemiology of Tusaviruses and Related Novel Protoparvoviruses (Family Parvoviridae) from Ruminant Species (Bovine, Ovine and Caprine) in Hungary. Viruses 2025, 17, 888. https://doi.org/10.3390/v17070888
Tóth F, Pankovics P, Urbán P, Herczeg R, Albert E, Reuter G, Boros Á. Genomic Characterization and Molecular Epidemiology of Tusaviruses and Related Novel Protoparvoviruses (Family Parvoviridae) from Ruminant Species (Bovine, Ovine and Caprine) in Hungary. Viruses. 2025; 17(7):888. https://doi.org/10.3390/v17070888
Chicago/Turabian StyleTóth, Fruzsina, Péter Pankovics, Péter Urbán, Róbert Herczeg, Ervin Albert, Gábor Reuter, and Ákos Boros. 2025. "Genomic Characterization and Molecular Epidemiology of Tusaviruses and Related Novel Protoparvoviruses (Family Parvoviridae) from Ruminant Species (Bovine, Ovine and Caprine) in Hungary" Viruses 17, no. 7: 888. https://doi.org/10.3390/v17070888
APA StyleTóth, F., Pankovics, P., Urbán, P., Herczeg, R., Albert, E., Reuter, G., & Boros, Á. (2025). Genomic Characterization and Molecular Epidemiology of Tusaviruses and Related Novel Protoparvoviruses (Family Parvoviridae) from Ruminant Species (Bovine, Ovine and Caprine) in Hungary. Viruses, 17(7), 888. https://doi.org/10.3390/v17070888