
Advances in Viroporin Function and Structure: A Comparative Analysis of Alphavirus 6K with Well-Characterized Viroporins




Abstract
1. Introduction
2. Alphavirus 6K
2.1. Relative Abundance of 6K and TF
2.2. Membrane Topology and Orientation of 6K and TF
2.3. Pore-Formation and Ion Channel Activity of 6K and TF
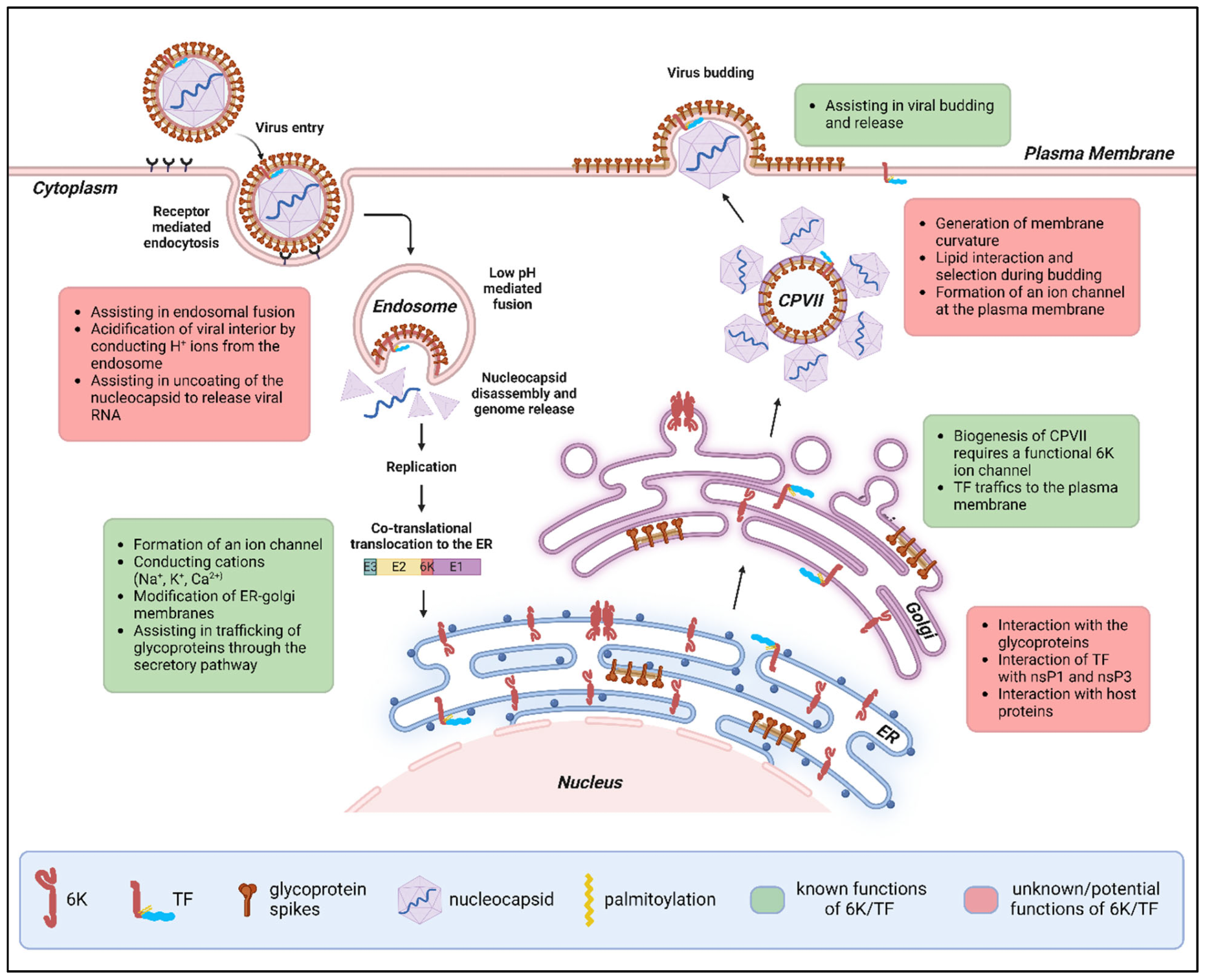
2.4. Intracellular Localization and Function of 6K and TF
2.4.1. Endoplasmic Reticulum (ER)
6K Is Needed for Efficient E1 Translocation and Polyprotein Processing
The 6K Ion Channel Permeabilizes Membranes to Conduct Cations
6K/TF May Interact with Other Viral Proteins in the ER
2.4.2. Golgi Compartment
6K Is Involved in Glycoprotein Trafficking and the Biogenesis of Cytopathic Vacuoles Type II (CPV-IIs)
2.4.3. Plasma Membrane (PM)
6K/TF Modifies Membrane Curvature and Affects Particle Morphology
The 6K/TF Ion Channel Permeabilizes the PM During Budding
6K/TF Affects the Thermostability of the Virus and Assists in Budding
6K/TF May Interact with Lipids to Promote Viral Budding
6K/TF May Interact with E2 During Budding
2.4.4. In the Virus Particle
Does the Alphavirus Particle Contain an Ion Channel?
Does 6K or TF Affect E1 Trimerization and Fusion During Entry?
3. Comparison to Other Viroporins
3.1. Functional Complementation Studies with 6K, M2, Vpu, p7, and E
3.2. Subcellular Localization of 6K, M2, Vpu, p7, and E
3.3. Ion Selectivity of 6K, M2, Vpu, p7, and E
3.4. Ion Channel Activity of 6K M2, Vpu, p7, and E Involved in Glycoprotein Trafficking, Particle Assembly, and Budding
3.5. Interaction of 6K M2, Vpu, p7, and E with Viral and Host Proteins to Assist in Infection and Immune Evasion

3.6. Comparison of Structural Features of 6K, M2, Vpu, p7, and E
4. Viroporin Inhibition and Therapeutic Potential for Pandemic Preparedness
4.1. Amantadine
4.2. HMA (Amiloride Derivatives)
4.3. BIT225
5. Conclusions and Future Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.E.; Carrasco, L. Viroporins. FEBS Lett. 2003, 552, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Hyser, J.M.; Estes, M.K. Pathophysiological Consequences of Calcium-Conducting Viroporins. Annu. Rev. Virol. 2015, 2, 473–496. [Google Scholar] [CrossRef] [PubMed]
- Martín, C.S.-S.; Liu, C.Y.; Kielian, M. Dealing with low pH: Entry and exit of alphaviruses and flaviviruses. Trends Microbiol. 2009, 17, 514–521. [Google Scholar] [CrossRef]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef]
- de Armas-Rillo, L.; Valera, M.-S.; Marrero-Hernández, S.; Valenzuela-Fernández, A. Membrane dynamics associated with viral infection. Rev. Med. Virol. 2016, 26, 146–160. [Google Scholar] [CrossRef]
- Carrasco, L. Modification of Membrane Permeability by Animal Viruses. Adv. Virus Res. 1995, 45, 61–112. [Google Scholar] [CrossRef]
- Rossman, J.S.; Jing, X.; Leser, G.P.; Lamb, R.A. Influenza Virus M2 Protein Mediates ESCRT-Independent Membrane Scission. Cell 2010, 142, 902–913. [Google Scholar] [CrossRef]
- Schubert, U.; Ferrer-Montiel, A.V.; Oblatt-Montal, M.; Henklein, P.; Strebel, K.; Montal, M. Identification of an ion channel activity of the Vpu transmembrane domain and its involvement in the regulation of virus release from HIV-1-infected cells. FEBS Lett. 1996, 398, 12–18. [Google Scholar] [CrossRef]
- Steinmann, E.; Penin, F.; Kallis, S.; Patel, A.H.; Bartenschlager, R.; Pietschmann, T. Hepatitis C Virus p7 Protein Is Crucial for Assembly and Release of Infectious Virions. PLoS Pathog. 2007, 3, e103. [Google Scholar] [CrossRef]
- Boson, B.; Legros, V.; Zhou, B.; Siret, E.; Mathieu, C.; Cosset, F.-L.; Lavillette, D.; Denolly, S. The SARS-CoV-2 envelope and membrane proteins modulate maturation and retention of the spike protein, allowing assembly of virus-like particles. J. Biol. Chem. 2021, 296, 100111. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-A.; Carreras-Sureda, A.; Demaurex, N. SARS-CoV-2 infection alkalinizes the ERGIC and lysosomes through the viroporin activity of the viral envelope protein. J. Cell Sci. 2023, 136, jcs260685. [Google Scholar] [CrossRef] [PubMed]
- Elmasri, Z.; Negi, V.; Kuhn, R.J.; Jose, J. Requirement of a functional ion channel for Sindbis virus glycoprotein transport, CPV-II formation, and efficient virus budding. PLoS Pathog. 2022, 18, e1010892. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, T.; Leser, G.P.; Lamb, R.A. The ion channel activity of the influenza virus M2 protein affects transport through the Golgi apparatus. J. Cell Biol. 1996, 133, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, A.L.; Griffin, S.; Rowlands, D.; Harris, M.; Yi, M.; Lemon, S.M.; Weinman, S.A. Intracellular Proton Conductance of the Hepatitis C Virus p7 Protein and Its Contribution to Infectious Virus Production. PLoS Pathog. 2010, 6, e1001087. [Google Scholar] [CrossRef]
- Stauffer, S.; Feng, Y.; Nebioglu, F.; Heilig, R.; Picotti, P.; Helenius, A. Stepwise priming by acidic pH and a high K+ concentration is required for efficient uncoating of influenza A virus cores after penetration. J. Virol. 2014, 88, 13029–13046. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Verdiá-Báguena, C.; Castaño-Rodriguez, C.; Aguilella, V.M.; Enjuanes, L. Relevance of Viroporin Ion Channel Activity on Viral Replication and Pathogenesis. Viruses 2015, 7, 3552–3573. [Google Scholar] [CrossRef]
- To, J.; Torres, J. Beyond Channel Activity: Protein-Protein Interactions Involving Viroporins. In Virus Protein and Nucleoprotein Complexes; Harris, J.R., Bhella, D., Eds.; Springer: Singapore, 2018; pp. 329–377. [Google Scholar] [CrossRef]
- Farag, N.S.; Breitinger, U.; Breitinger, H.G.; El Azizi, M.A. Viroporins and inflammasomes: A key to understand virus-induced inflammation. Int. J. Biochem. Cell Biol. 2020, 122, 105738. [Google Scholar] [CrossRef]
- Gannagé, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Rämer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix Protein 2 of Influenza A Virus Blocks Autophagosome Fusion with Lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef]
- Ren, Y.; Li, C.; Feng, L.; Pan, W.; Li, L.; Wang, Q.; Li, J.; Li, N.; Han, L.; Zheng, X.; et al. Proton Channel Activity of Influenza A Virus Matrix Protein 2 Contributes to Autophagy Arrest. J. Virol. 2015, 90, 591–598. [Google Scholar] [CrossRef]
- Verma, S.; Ali, A.; Arora, S.; Banerjea, A.C. Inhibition of β-TrcP–dependent ubiquitination of p53 by HIV-1 Vpu promotes p53–mediated apoptosis in human T cells. Blood 2011, 117, 6600–6607. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Yin, L.; Mei, S.; Li, J.; Xu, F.; Sun, H.; Liu, X.; Cen, S.; Liang, C.; Li, A.; et al. BST-2 restricts IAV release and is countered by the viral M2 protein. Biochem. J. 2017, 474, 715–730. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- McNatt, M.W.; Zang, T.; Bieniasz, P.D. Vpu Binds Directly to Tetherin and Displaces It from Nascent Virions. PLoS Pathog. 2013, 9, e1003299. [Google Scholar] [CrossRef]
- Comardelle, A.M.; Norris, C.H.; Plymale, D.R.; Gatti, P.J.; Choi, B.; Fermin, C.D.; Haislip, A.M.; Tencza, S.B.; Mietzner, T.A.; Montelaro, R.C.; et al. A Synthetic Peptide Corresponding to the Carboxy Terminus of Human Immunodeficiency Virus Type 1 Transmembrane Glycoprotein Induces Alterations in the Ionic Permeability of Xenopus laevis Oocytes. AIDS Res. Hum. Retroviruses 1997, 13, 1525–1532. [Google Scholar] [CrossRef]
- Arroyo, J.; Boceta, M.; González, M.E.; Michel, M.; Carrasco, L. Membrane permeabilization by different regions of the human immunodeficiency virus type 1 transmembrane glycoprotein gp41. J. Virol. 1995, 69, 4095–4102. [Google Scholar] [CrossRef]
- Ciccaglione, A.R.; Marcantonio, C.; Costantino, A.; Equestre, M.; Geraci, A.; Rapicetta, M. Hepatitis C virus E1 protein induces modification of membrane permeability in E. coli cells. Virology 1998, 250, 1–8. [Google Scholar] [CrossRef]
- Denisova, E.; Dowling, W.; LaMonica, R.; Shaw, R.; Scarlata, S.; Ruggeri, F.; Mackow, E.R. Rotavirus Capsid Protein VP5* Permeabilizes Membranes. J. Virol. 1999, 73, 3147–3153. [Google Scholar] [CrossRef]
- Chang, Y.-S.; Liao, C.-L.; Tsao, C.-H.; Chen, M.-C.; Liu, C.-I.; Chen, L.-K.; Lin, Y.-L. Membrane Permeabilization by Small Hydrophobic Nonstructural Proteins of Japanese Encephalitis Virus. J. Virol. 1999, 73, 6257–6264. [Google Scholar] [CrossRef]
- Blanco, R.; Carrasco, L.; Ventoso, I. Cell Killing by HIV-1 Protease. J. Biol. Chem. 2003, 278, 1086–1093. [Google Scholar] [CrossRef]
- Liljeström, P.; Lusa, S.; Huylebroeck, D.; Garoff, H. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: The small 6,000-molecular-weight membrane protein modulates virus release. J. Virol. 1991, 65, 4107–4113. [Google Scholar] [CrossRef]
- Klimkait, T.; Strebel, K.; Hoggan, M.D.; Martin, M.A.; Orenstein, J.M. The human immunodeficiency virus type 1-specific protein vpu is required for efficient virus maturation and release. J. Virol. 1990, 64, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; DeDiego, M.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Alcaraz, A.; Torres, J.; Aguilella, V.M.; et al. Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Ion Channel Activity Promotes Virus Fitness and Pathogenesis. PLoS Pathog. 2014, 10, e1004077. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.E.; Kulcsar, K.A.; Schultz, K.L.W.; Riley, C.P.; Neary, J.T.; Marr, S.; Jose, J.; Griffin, D.E.; Kuhn, R.J. Functional Characterization of the Alphavirus TF Protein. J. Virol. 2013, 87, 8511–8523. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Shen, X.; He, Y.; Pan, X.; Liu, F.-L.; Wang, Y.; Yang, F.; Fang, S.; Wu, Y.; Duan, Z.; et al. SARS-CoV-2 envelope protein causes acute respiratory distress syndrome (ARDS)-like pathological damages and constitutes an antiviral target. Cell Res. 2021, 31, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Melton, J.V.; Herrero, L.J.; Thaa, B.; Karo-Astover, L.; Gage, P.W.; Nelson, M.A.; Sheng, K.-C.; Lidbury, B.A.; Ewart, G.D.; et al. Effects of an In-Frame Deletion of the 6k Gene Locus from the Genome of Ross River Virus. J. Virol. 2016, 90, 4150–4159. [Google Scholar] [CrossRef]
- Park, S.H.; Siddiqi, H.; Castro, D.V.; Angelis, A.A.D.; Oom, A.L.; Stoneham, C.A.; Lewinski, M.K.; Clark, A.E.; Croker, B.A.; Carlin, A.F.; et al. Interactions of SARS-CoV-2 envelope protein with amilorides correlate with antiviral activity. PLoS Pathog. 2021, 17, e1009519. [Google Scholar] [CrossRef]
- Jing, X.; Ma, C.; Ohigashi, Y.; Oliveira, F.A.; Jardetzky, T.S.; Pinto, L.H.; Lamb, R.A. Functional studies indicate amantadine binds to the pore of the influenza A virus M2 proton-selective ion channel. Proc. Natl. Acad. Sci. USA 2008, 105, 10967–10972. [Google Scholar] [CrossRef]
- Cady, S.D.; Schmidt-Rohr, K.; Wang, J.; Soto, C.S.; DeGrado, W.F.; Hong, M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature 2010, 463, 689–692. [Google Scholar] [CrossRef]
- Griffin, S.D.C. Plugging the holes in hepatitis C virus antiviral therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 12567–12568. [Google Scholar] [CrossRef]
- Griffin, S.D.C.; Beales, L.P.; Clarke, D.S.; Worsfold, O.; Evans, S.D.; Jaeger, J.; Harris, M.P.G.; Rowlands, D.J. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett. 2003, 535, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Pavlović, D.; Neville, D.C.A.; Argaud, O.; Blumberg, B.; Dwek, R.A.; Fischer, W.B.; Zitzmann, N. The hepatitis C virus p7 protein forms an ion channel that is inhibited by long-alkyl-chain iminosugar derivatives. Proc. Natl. Acad. Sci. USA 2003, 100, 6104–6108. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Watanabe, T.; Kawaoka, Y. Influenza A Virus Lacking M2 Protein as a Live Attenuated Vaccine. J. Virol. 2009, 83, 5947–5950. [Google Scholar] [CrossRef] [PubMed]
- Braz Gomes, K.; Zhang, Y.-N.; Lee, Y.-Z.; Eldad, M.; Lim, A.; Ward, G.; Auclair, S.; He, L.; Zhu, J. Single-Component Multilayered Self-Assembling Protein Nanoparticles Displaying Extracellular Domains of Matrix Protein 2 as a Pan-influenza A Vaccine. ACS Nano 2023, 17, 23545–23567. [Google Scholar] [CrossRef] [PubMed]
- Zaid, A.; Burt, F.J.; Liu, X.; Poo, Y.S.; Zandi, K.; Suhrbier, A.; Weaver, S.C.; Texeira, M.M.; Mahalingam, S. Arthritogenic alphaviruses: Epidemiological and clinical perspective on emerging arboviruses. Lancet Infect. Dis. 2021, 21, e123–e133. [Google Scholar] [CrossRef]
- Salimi, H.; Cain, M.D.; Klein, R.S. Encephalitic Arboviruses: Emergence, Clinical Presentation, and Neuropathogenesis. Neurotherapeutics 2016, 13, 514–534. [Google Scholar] [CrossRef]
- Eastern Equine Encephalitis|Eastern Equine Encephalitis|CDC. 2023. Available online: https://www.cdc.gov/easternequineencephalitis/index.html (accessed on 8 April 2025).
- McMahon, R.; Fuchs, U.; Schneider, M.; Hadl, S.; Hochreiter, R.; Bitzer, A.; Kosulin, K.; Koren, M.; Mader, R.; Zoihsl, O.; et al. A randomized, double-blinded Phase 3 study to demonstrate lot-to-lot consistency and to confirm immunogenicity and safety of the live-attenuated chikungunya virus vaccine candidate VLA1553 in healthy adults. J. Travel Med. 2024, 31, taad156. [Google Scholar] [CrossRef]
- Commissioner, O. FDA Approves First Vaccine to Prevent Disease Caused by Chikungunya Virus. 2023. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-vaccine-prevent-disease-caused-chikungunya-virus (accessed on 17 June 2025).
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [CrossRef]
- Jose, J.; Snyder, J.E.; Kuhn, R.J. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 2009, 4, 837–856. [Google Scholar] [CrossRef]
- Suhrbier, A.; Jaffar-Bandjee, M.-C.; Gasque, P. Arthritogenic alphaviruses—An overview. Nat. Rev. Rheumatol. 2012, 8, 420–429. [Google Scholar] [CrossRef]
- Skidmore, A.M.; Bradfute, S.B. The life cycle of the alphaviruses: From an antiviral perspective. Antivir. Res. 2023, 209, 105476. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, M.; Zhu, D.; Sun, Z.; Ma, J.; Wang, J.; Kong, L.; Wang, S.; Liu, Z.; Wei, L.; et al. Implication for alphavirus host-cell entry and assembly indicated by a 3.5 Å resolution cryo-EM structure. Nat. Commun. 2018, 9, 5326. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.H.; Holsinger, L.J.; Lamb, R.A. Influenza virus M2 protein has ion channel activity. Cell 1992, 69, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Lama, J.; Carrasco, L. Expression of poliovirus nonstructural proteins in Escherichia coli cells. Modification of membrane permeability induced by 2B and 3A. J. Biol. Chem. 1992, 267, 15932–15937. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Pérez, L.; Carrasco, L. Semliki Forest virus 6K protein modifies membrane permeability after inducible expression in Escherichia coli cells. J. Biol. Chem. 1994, 269, 12106–12110. [Google Scholar] [CrossRef]
- Melancon, P.; Garoff, H. Reinitiation of translocation in the Semliki Forest virus structural polyprotein: Identification of the signal for the E1 glycoprotein. EMBO J. 1986, 5, 1551–1560. [Google Scholar] [CrossRef]
- Liljeström, P.; Garoff, H. Internally located cleavable signal sequences direct the formation of Semliki Forest virus membrane proteins from a polyprotein precursor. J. Virol. 1991, 65, 147–154. [Google Scholar] [CrossRef]
- Gaedigk-Nitschko, K.; Schlesinger, M.J. Site-directed mutations in sindbis virus E2 glycoprotein’s cytoplasmic domain and the 6K protein lead to similar defects in virus assembly and budding. Virology 1991, 183, 206–214. [Google Scholar] [CrossRef]
- Rice, C.M.; Strauss, J.H. Nucleotide sequence of the 26S mRNA of Sindbis virus and deduced sequence of the encoded virus structural proteins. Proc. Natl. Acad. Sci. USA 1981, 78, 2062–2066. [Google Scholar] [CrossRef]
- Sanz, M.A.; Madan, V.; Carrasco, L.; Nieva, J.L. Interfacial Domains in Sindbis Virus 6K Protein: DETECTION AND FUNCTIONAL CHARACTERIZATION. J. Biol. Chem. 2003, 278, 2051–2057. [Google Scholar] [CrossRef]
- Melton, J.V.; Ewart, G.D.; Weir, R.C.; Board, P.G.; Lee, E.; Gage, P.W. Alphavirus 6K Proteins Form Ion Channels. J. Biol. Chem. 2002, 277, 46923–46931. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, J.; Mukhopadhyay, S. Disentangling the Frames, the State of Research on the Alphavirus 6K and TF Proteins. Viruses 2017, 9, 228. [Google Scholar] [CrossRef] [PubMed]
- Harrington, H.R.; Zimmer, M.H.; Chamness, L.M.; Nash, V.; Penn, W.D.; Miller, T.F.; Mukhopadhyay, S.; Schlebach, J.P. Cotranslational folding stimulates programmed ribosomal frameshifting in the alphavirus structural polyprotein. J. Biol. Chem. 2020, 295, 6798–6808. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.E.; Chung, B.Y.; Fleeton, M.N.; Atkins, J.F. Discovery of frameshifting in Alphavirus 6K resolves a 20-year enigma. Virol. J. 2008, 5, 108. [Google Scholar] [CrossRef]
- Gaedigk-Nitschko, K.; Schlesinger, M.J. The sindbis virus 6K protein can be detected in virions and is acylated with fatty acids. Virology 1990, 175, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Lusa, S.; Garoff, H.; Liueström, P. Fate of the 6K membrane protein of semliki forest virus during virus assembly. Virology 1991, 185, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, L.; Le, L.; Schlesinger, M.J. Characterization of revertants of a Sindbis virus 6K gene mutant that affects proteolytic processing and virus assembly. Virus Res. 1995, 39, 165–179. [Google Scholar] [CrossRef]
- Ramsey, J.; Renzi, E.C.; Arnold, R.J.; Trinidad, J.C.; Mukhopadhyay, S. Palmitoylation of Sindbis Virus TF Protein Regulates Its Plasma Membrane Localization and Subsequent Incorporation into Virions. J. Virol. 2017, 91, e02000-16. [Google Scholar] [CrossRef]
- Schlesinger, M.J.; London, S.D.; Ryan, C. An in-frame insertion into the Sindbis virus 6K gene leads to defective proteolytic processing of the virus glycoproteins, a trans-dominant negative inhibition of normal virus formation, and interference in virus shut off of host-cell protein synthesis. Virology 1993, 193, 424–432. [Google Scholar] [CrossRef]
- Guo, T.-C.; Johansson, D.X.; Haugland, Ø.; Liljeström, P.; Evensen, Ø. A 6K-Deletion Variant of Salmonid Alphavirus Is Non-Viable but Can Be Rescued through RNA Recombination. PLoS ONE 2014, 9, e100184. [Google Scholar] [CrossRef]
- Liu, N.; Brown, D.T. Transient translocation of the cytoplasmic (endo) domain of a type I membrane glycoprotein into cellular membranes. J. Cell Biol. 1993, 120, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Owen, K.E.; Choi, H.K.; Lee, H.; Lu, G.; Wengler, G.; Brown, D.T.; Rossmann, M.G.; Kuhn, R.J. Identification of a protein binding site on the surface of the alphavirus nucleocapsid and its implication in virus assembly. Structure 1996, 4, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Owen, K.E.; Kuhn, R.J. Alphavirus Budding Is Dependent on the Interaction between the Nucleocapsid and Hydrophobic Amino Acids on the Cytoplasmic Domain of the E2 Envelope Glycoprotein. Virology 1997, 230, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.Y.-W.; Firth, A.E.; Atkins, J.F. Frameshifting in Alphaviruses: A Diversity of 3′ Stimulatory Structures. J. Mol. Biol. 2010, 397, 448–456. [Google Scholar] [CrossRef]
- Antoine, A.-F.; Montpellier, C.; Cailliau, K.; Browaeys-Poly, E.; Vilain, J.-P.; Dubuisson, J. The Alphavirus 6K Protein Activates Endogenous Ionic Conductances when Expressed in Xenopus Oocytes. J. Membr. Biol. 2007, 215, 37–48. [Google Scholar] [CrossRef]
- Gaedigk-Nitschko, K.; Ding, M.; Levy, M.A.; Schlesinger, M.J. Site-directed mutations in the sindbis virus 6K protein reveal sites for fatty acylation and the underacylated protein affects virus release and virion structure. Virology 1990, 175, 282–291. [Google Scholar] [CrossRef]
- Sanz, M.A.; Carrasco, L. Sindbis Virus Variant with a Deletion in the 6K Gene Shows Defects in Glycoprotein Processing and Trafficking: Lack of Complementation by a Wild-Type 6K Gene intrans. J. Virol. 2001, 75, 7778–7784. [Google Scholar] [CrossRef]
- Loewy, A.; Smyth, J.; von Bonsdorff, C.H.; Liljeström, P.; Schlesinger, M.J. The 6-kilodalton membrane protein of Semliki Forest virus is involved in the budding process. J. Virol. 1995, 69, 469–475. [Google Scholar] [CrossRef]
- McInerney, G.M.; Smit, J.M.; Liljeström, P.; Wilschut, J. Semliki Forest virus produced in the absence of the 6K protein has an altered spike structure as revealed by decreased membrane fusion capacity. Virology 2004, 325, 200–206. [Google Scholar] [CrossRef]
- Yao, J.S.; Strauss, E.G.; Strauss, J.H. Interactions between PE2, E1, and 6K required for assembly of alphaviruses studied with chimeric viruses. J. Virol. 1996, 70, 7910–7920. [Google Scholar] [CrossRef]
- Strauss, E.G.; Lenches, E.M.; Strauss, J.H. Molecular Genetic Evidence that the Hydrophobic Anchors of Glycoproteins E2 and E1 Interact during Assembly of Alphaviruses. J. Virol. 2002, 76, 10188–10194. [Google Scholar] [CrossRef] [PubMed]
- González, M.E.; Carrasco, L. Human immunodeficiency virus type 1 VPU protein affects Sindbis virus glycoprotein processing and enhances membrane permeabilization. Virology 2001, 279, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Hallengärd, D.; Kakoulidou, M.; Lulla, A.; Kümmerer, B.M.; Johansson, D.X.; Mutso, M.; Lulla, V.; Fazakerley, J.K.; Roques, P.; Le Grand, R.; et al. Novel Attenuated Chikungunya Vaccine Candidates Elicit Protective Immunity in C57BL/6 mice. J. Virol. 2014, 88, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Mou, C.; Zhang, L.; Zhou, J.; Lu, T.; Chen, Z. The roles of 6K protein on Getah virus replication and pathogenicity. J. Med. Virol. 2023, 95, e29302. [Google Scholar] [CrossRef]
- Dey, D.; Siddiqui, S.I.; Mamidi, P.; Ghosh, S.; Kumar, C.S.; Chattopadhyay, S.; Ghosh, S.; Banerjee, M. The effect of amantadine on an ion channel protein from Chikungunya virus. PLoS Negl. Trop. Dis. 2019, 13, e0007548. [Google Scholar] [CrossRef]
- Suhag, K.; Borkotoky, S.; Siddiqui, S.I.; Kumar, J.; Kumar, C.S.; Tatiya, P.; Ghosh, S.; Banerjee, M. Mechanistic Insights into the Divergent Membrane Activities of a Viroporin from Chikungunya Virus and Its Transframe Variant. ACS Infect. Dis. 2025, 11, 430–441. [Google Scholar] [CrossRef]
- Bretscher, M.S.; Munro, S. Cholesterol and the Golgi Apparatus. Science 1993, 261, 1280–1281. [Google Scholar] [CrossRef]
- Zentgraf, H.; Deumling, B.; Jarasch, E.-D.; Franke, W.W. Nuclear Membranes and Plasma Membranes from Hen Erythrocytes: I. ISOLATION, CHARACTERIZATION, AND COMPARISON. J. Biol. Chem. 1971, 246, 2986–2995. [Google Scholar] [CrossRef]
- Lanzrein, M.; Weingart, R.; Kempf, C. pH-dependent pore formation in Semliki forest virus-infected Aedes albopictus cells. Virology 1993, 193, 296–302. [Google Scholar] [CrossRef]
- Garry, R.F.; Bishop, J.M.; Parker, S.; Westbrook, K.; Lewis, G.; Waite, M.R.F. Na+ and K+ concentrations and the regulation of protein synthesis in Sindbis virus-infected chick cells. Virology 1979, 96, 108–120. [Google Scholar] [CrossRef]
- Ulug, E.T.; Garry, R.F.; Bose, H.R. The role of monovalent cation transport in Sindbis virus maturation and release. Virology 1989, 172, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Yin, P.; Zhao, H.; Zhang, N.; Jian, X.; Song, S.; Gao, S.; Zhang, L. Intraviral interactome of Chikungunya virus reveals the homo-oligomerization and palmitoylation of structural protein TF. Biochem. Biophys. Res. Commun. 2019, 513, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Elmasri, Z.; Nasal, B.L.; Jose, J. Alphavirus-Induced Membrane Rearrangements during Replication, Assembly, and Budding. Pathogens 2021, 10, 984. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, D.; Schmid, M.F.; Simmons, G.; Jin, J.; Chiu, W. Chikungunya virus assembly and budding visualized in situ using cryogenic electron tomography. Nat. Microbiol. 2022, 7, 1270–1279. [Google Scholar] [CrossRef]
- Ivanova, L.; Lustig, S.; Schlesinger, M.J. A pseudo-revertant of a Sindbis virus 6K protein mutant, which corrects for aberrant particle formation, contains two new mutations that map to the ectodomain of the E2 glycoprotein. Virology 1995, 206, 1027–1034. [Google Scholar] [CrossRef]
- Ramsey, J.; Chavez, M.; Mukhopadhyay, S. Domains of the TF protein important in regulating its own palmitoylation. Virology 2019, 531, 31–39. [Google Scholar] [CrossRef]
- Kumar, R.; Tatiya, P.; Dey, D.; Ratra, Y.; Mian, S.Y.; Chaudhary, S.; Suhag, K.; Basak, S.; Banerjee, M. Downregulation of a cell polarity protein potentiates Chikungunya Virus infection in host cells. bioRxiv 2023, 2023.07.24.550336. [Google Scholar] [CrossRef]
- Rogers, K.J.; Jones-Burrage, S.; Maury, W.; Mukhopadhyay, S. TF protein of Sindbis virus antagonizes host type I interferon responses in a palmitoylation-dependent manner. Virology 2020, 542, 63–70. [Google Scholar] [CrossRef]
- Sanz, M.A.; Madan, V.; Nieva, J.L.; Carrasco, L. The Alphavirus 6K Protein. In Viral Membrane Proteins: Structure, Function, and Drug Design; Fischer, W.B., Ed.; Springer: Boston, MA, USA, 2005; pp. 233–244. [Google Scholar] [CrossRef]
- Waite, M.R.F.; Pfefferkorn, E.R. Inhibition of Sindbis Virus Production by Media of Low Ionic Strength: Intracellular Events and Requirements for Reversal. J. Virol. 1970, 5, 60–71. [Google Scholar] [CrossRef]
- Li, M.-L.; Stollar, V. A Mutant of Sindbis Virus Which Is Released Efficiently from Cells Maintained in Low Ionic Strength Medium. Virology 1995, 210, 237–243. [Google Scholar] [CrossRef]
- Miller, M.L.; Brown, D.T. Morphogenesis of Sindbis virus in three subclones of Aedes albopictus (mosquito) cells. J. Virol. 1992, 66, 4180–4190. [Google Scholar] [CrossRef] [PubMed]
- Yondola, M.; Carter, C. Un-“ESCRT”-ed Budding. Viruses 2011, 3, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, M.T.; Phalen, T.; Kielian, M. Cholesterol is required in the exit pathway of Semliki Forest virus. J. Cell Biol. 1993, 123, 57–65. [Google Scholar] [CrossRef] [PubMed]
- David, A.E. Lipid composition of Sindbis virus. Virology 1971, 46, 711–720. [Google Scholar] [CrossRef]
- Scheiffele, P.; Rietveld, A.; Wilk, T.; Simons, K. Influenza Viruses Select Ordered Lipid Domains during Budding from the Plasma Membrane. J. Biol. Chem. 1999, 274, 2038–2044. [Google Scholar] [CrossRef]
- Elkins, M.R.; Williams, J.K.; Gelenter, M.D.; Dai, P.; Kwon, B.; Sergeyev, I.V.; Pentelute, B.L.; Hong, M. Cholesterol-binding site of the influenza M2 protein in lipid bilayers from solid-state NMR. Proc. Natl. Acad. Sci. USA 2017, 114, 12946–12951. [Google Scholar] [CrossRef]
- Luukkonen, A.; Kääriäinen, L.; Renkonen, O. Phospholipids of Semliki Forest virus grown in cultured mosquito cells. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1976, 450, 109–120. [Google Scholar] [CrossRef]
- Kalvodova, L.; Sampaio, J.L.; Cordo, S.; Ejsing, C.S.; Shevchenko, A.; Simons, K. The Lipidomes of Vesicular Stomatitis Virus, Semliki Forest Virus, and the Host Plasma Membrane Analyzed by Quantitative Shotgun Mass Spectrometry. J. Virol. 2009, 83, 7996–8003. [Google Scholar] [CrossRef]
- Hafer, A.; Whittlesey, R.; Brown, D.T.; Hernandez, R. Differential Incorporation of Cholesterol by Sindbis Virus Grown in Mammalian or Insect Cells. J. Virol. 2009, 83, 9113–9121. [Google Scholar] [CrossRef]
- Vial, T.; Marti, G.; Missé, D.; Pompon, J. Lipid Interactions Between Flaviviruses and Mosquito Vectors. Front. Physiol. 2021, 12, 763195. [Google Scholar] [CrossRef]
- Wharton, S.A.; Belshe, R.B.; Skehel, J.J.; Hay, A.J. Role of virion M2 protein in influenza virus uncoating: Specific reduction in the rate of membrane fusion between virus and liposomes by amantadine. J. Gen. Virol. 1994, 75 Pt 4, 945–948. [Google Scholar] [CrossRef] [PubMed]
- White, J.; Helenius, A. pH-dependent fusion between the Semliki Forest virus membrane and liposomes. Proc. Natl. Acad. Sci. USA 1980, 77, 3273–3277. [Google Scholar] [CrossRef] [PubMed]
- Helenius, A.; Kartenbeck, J.; Simons, K.; Fries, E. On the entry of semliki forest virus into BHK-21 cells. J. Cell Biol. 1980, 84, 404–420. [Google Scholar] [CrossRef] [PubMed]
- White, J.; Kartenbeck, J.; Helenius, A. Fusion of Semliki forest virus with the plasma membrane can be induced by low pH. J. Cell Biol. 1980, 87, 264–272. [Google Scholar] [CrossRef]
- Söderlund, H.; Kääriäinen, L.; von Bonsdorff, C.H. Properties of Semliki Forest virus nucleocapsid. Med. Biol. 1975, 53, 412–417. [Google Scholar] [CrossRef]
- Stubbs, M.J.; Miller, A.; Sizer, P.J.H.; Stephenson, J.R.; Crooks, A.J. X-ray solution scattering of Sindbis virus: Changes in conformation induced at low pH. J. Mol. Biol. 1991, 221, 39–42. [Google Scholar] [CrossRef]
- Wahlberg, J.M.; Bron, R.; Wilschut, J.; Garoff, H. Membrane fusion of Semliki Forest virus involves homotrimers of the fusion protein. J. Virol. 1992, 66, 7309–7318. [Google Scholar] [CrossRef]
- Bron, R.; Wahlberg, J.M.; Garoff, H.; Wilschut, J. Membrane fusion of Semliki Forest virus in a model system: Correlation between fusion kinetics and structural changes in the envelope glycoprotein. EMBO J. 1993, 12, 693–701. [Google Scholar] [CrossRef]
- Klimjack, M.R.; Jeffrey, S.; Kielian, M. Membrane and protein interactions of a soluble form of the Semliki Forest virus fusion protein. J. Virol. 1994, 68, 6940–6946. [Google Scholar] [CrossRef]
- Ahn, A.; Gibbons, D.L.; Kielian, M. The Fusion Peptide of Semliki Forest Virus Associates with Sterol-Rich Membrane Domains. J. Virol. 2002, 76, 3267–3275. [Google Scholar] [CrossRef]
- Park, S.H.; De Angelis, A.A.; Nevzorov, A.A.; Wu, C.H.; Opella, S.J. Three-Dimensional Structure of the Transmembrane Domain of Vpu from HIV-1 in Aligned Phospholipid Bicelles. Biophys. J. 2006, 91, 3032–3042. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol. 2013, 11, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Duart, G.; García-Murria, M.J.; Grau, B.; Acosta-Cáceres, J.M.; Martínez-Gil, L.; Mingarro, I. SARS-CoV-2 envelope protein topology in eukaryotic membranes. Open Biol. 2020, 10, 200209. [Google Scholar] [CrossRef] [PubMed]
- Hout, D.R.; Gomez, M.L.; Pacyniak, E.; Gomez, L.M.; Fegley, B.; Mulcahy, E.R.; Hill, M.S.; Culley, N.; Pinson, D.M.; Nothnick, W.; et al. Substitution of the transmembrane domain of Vpu in simian–human immunodeficiency virus (SHIVKU1bMC33) with that of M2 of influenza A results in a virus that is sensitive to inhibitors of the M2 ion channel and is pathogenic for pig-tailed macaques. Virology 2006, 344, 541–559. [Google Scholar] [CrossRef] [PubMed]
- Griffin, S.D.C.; Harvey, R.; Clarke, D.S.; Barclay, W.S.; Harris, M.; Rowlands, D.J. A conserved basic loop in hepatitis C virus p7 protein is required for amantadine-sensitive ion channel activity in mammalian cells but is dispensable for localization to mitochondria. J. Gen. Virol. 2004, 85, 451–461. [Google Scholar] [CrossRef]
- Brohm, C.; Steinmann, E.; Friesland, M.; Lorenz, I.C.; Patel, A.; Penin, F.; Bartenschlager, R.; Pietschmann, T. Characterization of Determinants Important for Hepatitis C Virus p7 Function in Morphogenesis by Using trans-Complementation. J. Virol. 2009, 83, 11682–11693. [Google Scholar] [CrossRef]
- Negi, V.; Kuhn, R.J. “A BSL-2 chimeric system designed to screen SARS-CoV-2 E protein ion channel inhibitors. J. Virol. 2025, 99, e02252-24. [Google Scholar] [CrossRef]
- Pielak, R.M.; Chou, J.J. Influenza M2 proton channels. Biochim. Biophys. Acta 2011, 1808, 522–529. [Google Scholar] [CrossRef]
- Khan, N.; Geiger, J.D. Role of Viral Protein U (Vpu) in HIV-1 Infection and Pathogenesis. Viruses 2021, 13, 1466. [Google Scholar] [CrossRef]
- Steinmann, E.; Pietschmann, T. Hepatitis C Virus P7—A Viroporin Crucial for Virus Assembly and an Emerging Target for Antiviral Therapy. Viruses 2010, 2, 2078–2095. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; DeDiego, M.L.; Álvarez, E.; Jiménez-Guardeño, J.M.; Regla-Nava, J.A.; Llorente, M.; Kremer, L.; Shuo, S.; Enjuanes, L. Subcellular location and topology of severe acute respiratory syndrome coronavirus envelope protein. Virology 2011, 415, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Carrère-Kremer, S.; Montpellier-Pala, C.; Cocquerel, L.; Wychowski, C.; Penin, F.; Dubuisson, J. Subcellular Localization and Topology of the p7 Polypeptide of Hepatitis C Virus. J. Virol. 2002, 76, 3720–3730. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Garcia, D.; Bekdash, R.; Abbott, G.W.; Yazawa, M.; Harrison, N.L. The envelope protein of SARS-CoV-2 increases intra-Golgi pH and forms a cation channel that is regulated by pH. J. Physiol. 2021, 599, 2851–2868. [Google Scholar] [CrossRef] [PubMed]
- Mehregan, A.; Pérez-Conesa, S.; Zhuang, Y.; Elbahnsi, A.; Pasini, D.; Lindahl, E.; Howard, R.J.; Ulens, C.; Delemotte, L. Probing effects of the SARS-CoV-2 E protein on membrane curvature and intracellular calcium. Biochim. Biophys. Acta (BBA)-Biomembr. 2022, 1864, 183994. [Google Scholar] [CrossRef]
- Poggio, E.; Vallese, F.; Hartel, A.J.W.; Morgenstern, T.J.; Kanner, S.A.; Rauh, O.; Giamogante, F.; Barazzuol, L.; Shepard, K.L.; Colecraft, H.M.; et al. Perturbation of the host cell Ca2+ homeostasis and ER-mitochondria contact sites by the SARS-CoV-2 structural proteins E and M. Cell Death Dis. 2023, 14, 297. [Google Scholar] [CrossRef]
- Wang, C.; Lamb, R.A.; Pinto, L.H. Direct measurement of the influenza A virus M2 protein ion channel activity in mammalian cells. Virology 1994, 205, 133–140. [Google Scholar] [CrossRef]
- Lin, T.I.; Schroeder, C. Definitive assignment of proton selectivity and attoampere unitary current to the M2 ion channel protein of influenza A virus. J. Virol. 2001, 75, 3647–3656. [Google Scholar] [CrossRef]
- Vijayvergiya, V.; Wilson, R.; Chorak, A.; Gao, P.F.; Cross, T.A.; Busath, D.D. Proton conductance of influenza virus M2 protein in planar lipid bilayers. Biophys. J. 2004, 87, 1697–1704. [Google Scholar] [CrossRef]
- Ewart, G.D.; Sutherland, T.; Gage, P.W.; Cox, G.B. The Vpu protein of human immunodeficiency virus type 1 forms cation-selective ion channels. J. Virol. 1996, 70, 7108–7115. [Google Scholar] [CrossRef]
- Coady, M.J.; Daniel, N.G.; Tiganos, E.; Allain, B.; Friborg, J.; Lapointe, J.Y.; Cohen, E.A. Effects of Vpu expression on Xenopus oocyte membrane conductance. Virology 1998, 244, 39–49. [Google Scholar] [CrossRef]
- Premkumar, A.; Wilson, L.; Ewart, G.D.; Gage, P.W. Cation-selective ion channels formed by p7 of hepatitis C virus are blocked by hexamethylene amiloride. FEBS Lett. 2004, 557, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.; Mckinlay, C.; Gage, P.; Ewart, G. SARS coronavirus E protein forms cation-selective ion channels. Virology 2004, 330, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Castaño-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 2015, 485, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yang, R.; Wang, W.; Lee, I.; Zhang, R.; Zhang, W.; Sun, J.; Xu, B.; Meng, X. Computational Study of the Ion and Water Permeation and Transport Mechanisms of the SARS-CoV-2 Pentameric E Protein Channel. Front. Mol. Biosci. 2020, 7, 565797. [Google Scholar] [CrossRef] [PubMed]
- Verdiá-Báguena, C.; Nieto-Torres, J.L.; Alcaraz, A.; DeDiego, M.L.; Enjuanes, L.; Aguilella, V.M. Analysis of SARS-CoV E protein ion channel activity by tuning the protein and lipid charge. Biochim. Biophys. Acta (BBA)-Biomembr. 2013, 1828, 2026–2031. [Google Scholar] [CrossRef]
- Verdiá-Báguena, C.; Nieto-Torres, J.L.; Alcaraz, A.; DeDiego, M.L.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Coronavirus E protein forms ion channels with functionally and structurally-involved membrane lipids. Virology 2012, 432, 485–494. [Google Scholar] [CrossRef]
- Giorda, K.M.; Hebert, D.N. Viroporins Customize Host Cells for Efficient Viral Propagation. DNA Cell Biol. 2013, 32, 557–564. [Google Scholar] [CrossRef]
- Watanabe, T.; Watanabe, S.; Ito, H.; Kida, H.; Kawaoka, Y. Influenza A Virus Can Undergo Multiple Cycles of Replication without M2 Ion Channel Activity. J. Virol. 2001, 75, 5656–5662. [Google Scholar] [CrossRef]
- Henkel, J.R.; Weisz, O.A. Influenza Virus M2 Protein Slows Traffic along the Secretory Pathway: pH PERTURBATION OF ACIDIFIED COMPARTMENTS AFFECTS EARLY GOLGI TRANSPORT STEPS. J. Biol. Chem. 1998, 273, 6518–6524. [Google Scholar] [CrossRef]
- Ichinohe, T.; Pang, I.K.; Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 2010, 11, 404–410. [Google Scholar] [CrossRef]
- Hsu, K.; Han, J.; Shinlapawittayatorn, K.; Deschenes, I.; Marbán, E. Membrane Potential Depolarization as a Triggering Mechanism for Vpu-Mediated HIV-1 Release. Biophys. J. 2010, 99, 1718–1725. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Guatelli, J.C.; Stephens, E.B. The Vpu Protein: New Concepts in Virus Release and CD4 Down-Modulation. Curr. HIV Res. 2010, 8, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Magadán, J.G.; Bonifacino, J.S. Transmembrane Domain Determinants of CD4 Downregulation by HIV-1 Vpu. J. Virol. 2012, 86, 757. [Google Scholar] [CrossRef] [PubMed]
- Pujol, F.M.; Laketa, V.; Schmidt, F.; Mukenhirn, M.; Müller, B.; Boulant, S.; Grimm, D.; Keppler, O.T.; Fackler, O.T. HIV-1 Vpu Antagonizes CD317/Tetherin by Adaptor Protein-1-Mediated Exclusion from Virus Assembly Sites. J. Virol. 2016, 90, 6709–6723. [Google Scholar] [CrossRef]
- Strebel, K. HIV-1 Vpu—An ion channel in search of a job. Biochim. Biophys. Acta (BBA)-Biomembr. 2014, 1838, 1074–1081. [Google Scholar] [CrossRef]
- Jones, C.T.; Murray, C.L.; Eastman, D.K.; Tassello, J.; Rice, C.M. Hepatitis C Virus p7 and NS2 Proteins Are Essential for Production of Infectious Virus. J. Virol. 2007, 81, 8374–8383. [Google Scholar] [CrossRef]
- Gentzsch, J.; Brohm, C.; Steinmann, E.; Friesland, M.; Menzel, N.; Vieyres, G.; Perin, P.M.; Frentzen, A.; Kaderali, L.; Pietschmann, T. Hepatitis C Virus p7 is Critical for Capsid Assembly and Envelopment. PLoS Pathog. 2013, 9, e1003355. [Google Scholar] [CrossRef]
- Sakai, A.; Claire, M.S.; Faulk, K.; Govindarajan, S.; Emerson, S.U.; Purcell, R.H.; Bukh, J. The p7 polypeptide of hepatitis C virus is critical for infectivity and contains functionally important genotype-specific sequences. Proc. Natl. Acad. Sci. USA 2003, 100, 11646–11651. [Google Scholar] [CrossRef]
- Griffin, S.; StGelais, C.; Owsianka, A.M.; Patel, A.H.; Rowlands, D.; Harris, M. Genotype-Dependent Sensitivity of Hepatitis C Virus to Inhibitors of the p7 Ion Channel. Hepatology 2008, 48, 1779–1790. [Google Scholar] [CrossRef]
- Cao, Y.; Yang, R.; Lee, I.; Zhang, W.; Sun, J.; Wang, W.; Meng, X. Characterization of the SARS-CoV-2 E Protein: Sequence, Structure, Viroporin, and Inhibitors. Protein Sci. 2021, 30, 1114–1130. [Google Scholar] [CrossRef]
- Madan, V.; Castelló, A.; Carrasco, L. Viroporins from RNA viruses induce caspase-dependent apoptosis. Cell. Microbiol. 2008, 10, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. β-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535.e14. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Triantafilou, M. Ion flux in the lung: Virus-induced inflammasome activation. Trends Microbiol. 2014, 22, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, R.; Igarashi, M.; Takada, A. Influenza A Virus M2 Protein: Roles from Ingress to Egress. Int. J. Mol. Sci. 2017, 18, 2649. [Google Scholar] [CrossRef] [PubMed]
- Beale, R.; Wise, H.; Stuart, A.; Ravenhill, B.J.; Digard, P.; Randow, F. A LC3-Interacting Motif in the Influenza A Virus M2 Protein Is Required to Subvert Autophagy and Maintain Virion Stability. Cell Host Microbe 2014, 15, 239–247. [Google Scholar] [CrossRef]
- McCown, M.F.; Pekosz, A. Distinct Domains of the Influenza A Virus M2 Protein Cytoplasmic Tail Mediate Binding to the M1 Protein and Facilitate Infectious Virus Production. J. Virol. 2006, 80, 8178–8189. [Google Scholar] [CrossRef]
- Cook, G.A.; Zhang, H.; Park, S.H.; Wang, Y.; Opella, S.J. Comparative NMR studies demonstrate profound differences between two viroporins: p7 of HCV and Vpu of HIV-1. Biochim. Biophys. Acta (BBA)-Biomembr. 2011, 1808, 554–560. [Google Scholar] [CrossRef]
- Dubé, M.; Bego, M.G.; Paquay, C.; Cohen, É.A. Modulation of HIV-1-host interaction: Role of the Vpu accessory protein. Retrovirology 2010, 7, 114. [Google Scholar] [CrossRef]
- Kueck, T.; Neil, S.J.D. A Cytoplasmic Tail Determinant in HIV-1 Vpu Mediates Targeting of Tetherin for Endosomal Degradation and Counteracts Interferon-Induced Restriction. PLoS Pathog. 2012, 8, e1002609. [Google Scholar] [CrossRef]
- Gargan, S.; Stevenson, N.J. Unravelling the Immunomodulatory Effects of Viral Ion Channels, towards the Treatment of Disease. Viruses 2021, 13, 2165. [Google Scholar] [CrossRef]
- Hsu, K.; Seharaseyon, J.; Dong, P.; Bour, S.; Marbán, E. Mutual Functional Destruction of HIV-1 Vpu and Host TASK-1 Channel. Mol. Cell 2004, 14, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Ashfaq, U.A.; Javed, T.; Rehman, S.; Nawaz, Z.; Riazuddin, S. An overview of HCV molecular biology, replication and immune responses. Virol. J. 2011, 8, 161. [Google Scholar] [CrossRef] [PubMed]
- Tedbury, P.; Welbourn, S.; Pause, A.; King, B.; Griffin, S.; Harris, M. The subcellular localization of the hepatitis C virus non-structural protein NS2 is regulated by an ion channel-independent function of the p7 protein. J. Gen. Virol. 2011, 92, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.R.; Ramos, B.; Nunes, A.; Ribeiro, D. Hepatitis C Virus: Evading the Intracellular Innate Immunity. J. Clin. Med. 2020, 9, 790. [Google Scholar] [CrossRef]
- Lopez, L.A.; Riffle, A.J.; Pike, S.L.; Gardner, D.; Hogue, B.G. Importance of Conserved Cysteine Residues in the Coronavirus Envelope Protein. J. Virol. 2008, 82, 3000–3010. [Google Scholar] [CrossRef]
- Li, Z.; Hao, P.; Zhao, Z.; Gao, W.; Huan, C.; Li, L.; Chen, X.; Wang, H.; Jin, N.; Luo, Z.-Q.; et al. The E3 ligase RNF5 restricts SARS-CoV-2 replication by targeting its envelope protein for degradation. Signal Transduct. Target. Ther. 2023, 8, 53. [Google Scholar] [CrossRef]
- Mortola, E.; Roy, P. Efficient assembly and release of SARS coronavirus-like particles by a heterologous expression system. FEBS Lett. 2004, 576, 174–178. [Google Scholar] [CrossRef]
- Hogue, B.G.; Machamer, C.E. Coronavirus Structural Proteins and Virus Assembly. In Nidoviruses; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2007; pp. 179–200. [Google Scholar] [CrossRef]
- Teoh, K.-T.; Siu, Y.-L.; Chan, W.-L.; Schlüter, M.A.; Liu, C.-J.; Peiris, J.S.M.; Bruzzone, R.; Margolis, B.; Nal, B. The SARS Coronavirus E Protein Interacts with PALS1 and Alters Tight Junction Formation and Epithelial Morphogenesis. Mol. Biol. Cell 2010, 21, 3838–3852. [Google Scholar] [CrossRef]
- De Maio, F.; Lo Cascio, E.; Babini, G.; Sali, M.; Della Longa, S.; Tilocca, B.; Roncada, P.; Arcovito, A.; Sanguinetti, M.; Scambia, G.; et al. Improved binding of SARS-CoV-2 Envelope protein to tight junction-associated PALS1 could play a key role in COVID-19 pathogenesis. Microbes Infect. 2020, 22, 592–597. [Google Scholar] [CrossRef]
- Chai, J.; Cai, Y.; Pang, C.; Wang, L.; McSweeney, S.; Shanklin, J.; Liu, Q. Structural basis for SARS-CoV-2 envelope protein recognition of human cell junction protein PALS1. Nat. Commun. 2021, 12, 3433. [Google Scholar] [CrossRef]
- Jimenez-Guardeño, J.M.; Nieto-Torres, J.L.; DeDiego, M.L.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Enjuanes, L. The PDZ-Binding Motif of Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Is a Determinant of Viral Pathogenesis. PLoS Pathog. 2014, 10, e1004320. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xiong, Z.; Zhang, S.; Yan, Y.; Nguyen, J.; Ng, B.; Lu, H.; Brendese, J.; Yang, F.; Wang, H.; et al. Bcl-xL inhibits T-cell apoptosis induced by expression of SARS coronavirus E protein in the absence of growth factors. Biochem. J. 2005, 392, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Yalcinkaya, M.; Liu, W.; Islam, M.N.; Kotini, A.G.; Gusarova, G.A.; Fidler, T.P.; Papapetrou, E.P.; Bhattacharya, J.; Wang, N.; Tall, A.R. Modulation of the NLRP3 inflammasome by Sars-CoV-2 Envelope protein. Sci. Rep. 2021, 11, 24432. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.-D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.W.; Savani, R.C.; Zaki, H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. eLife 2021, 10, e68563. [Google Scholar] [CrossRef]
- Netland, J.; DeDiego, M.L.; Zhao, J.; Fett, C.; Álvarez, E.; Nieto-Torres, J.L.; Enjuanes, L.; Perlman, S. Immunization with an attenuated severe acute respiratory syndrome coronavirus deleted in E protein protects against lethal respiratory disease. Virology 2010, 399, 120–128. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Álvarez, E.; Almazán, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.-J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. J. Virol. 2007, 81, 1701–1713. [Google Scholar] [CrossRef]
- Regla-Nava, J.A.; Nieto-Torres, J.L.; Jimenez-Guardeño, J.M.; Fernandez-Delgado, R.; Fett, C.; Castaño-Rodríguez, C.; Perlman, S.; Enjuanes, L.; DeDiego, M.L. Severe Acute Respiratory Syndrome Coronaviruses with Mutations in the E Protein Are Attenuated and Promising Vaccine Candidates. J. Virol. 2015, 89, 3870–3887. [Google Scholar] [CrossRef]
- Mi, S.; Li, Y.; Yan, J.; Gao, G.F. Na+/K+-ATPase β1 subunit interacts with M2 proteins of influenza A and B viruses and affects the virus replication. Sci. China Life Sci. 2010, 53, 1098–1105. [Google Scholar] [CrossRef]
- Ji, H.-L.; Song, W.; Gao, Z.; Su, X.-F.; Nie, H.-G.; Jiang, Y.; Peng, J.-B.; He, Y.-X.; Liao, Y.; Zhou, Y.-J.; et al. SARS-CoV proteins decrease levels and activity of human ENaC via activation of distinct PKC isoforms. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2009, 296, L372–L383. [Google Scholar] [CrossRef]
- Lazrak, A.; Iles, K.E.; Liu, G.; Noah, D.L.; Noah, J.W.; Matalon, S. Influenza virus M2 protein inhibits epithelial sodium channels by increasing reactive oxygen species. FASEB J. 2009, 23, 3829–3842. [Google Scholar] [CrossRef] [PubMed]
- Acharya, R.; Carnevale, V.; Fiorin, G.; Levine, B.G.; Polishchuk, A.L.; Balannik, V.; Samish, I.; Lamb, R.A.; Pinto, L.H.; DeGrado, W.F.; et al. Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc. Natl. Acad. Sci. USA 2010, 107, 15075–15080. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Mrse, A.A.; Nevzorov, A.A.; Mesleh, M.F.; Oblatt-Montal, M.; Montal, M.; Opella, S.J. Three-dimensional Structure of the Channel-forming Trans-membrane Domain of Virus Protein “u” (Vpu) from HIV-1. J. Mol. Biol. 2003, 333, 409–424. [Google Scholar] [CrossRef] [PubMed]
- OuYang, B.; Xie, S.; Berardi, M.J.; Zhao, X.; Dev, J.; Yu, W.; Sun, B.; Chou, J.J. Unusual architecture of the p7 channel from hepatitis C virus. Nature 2013, 498, 521–525. [Google Scholar] [CrossRef]
- Mandala, V.S.; McKay, M.J.; Shcherbakov, A.A.; Dregni, A.J.; Kolocouris, A.; Hong, M. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nat. Struct. Mol. Biol. 2020, 27, 1202–1208. [Google Scholar] [CrossRef]
- UCSF ChimeraX Home Page. Available online: https://www.rbvi.ucsf.edu/chimerax/ (accessed on 8 April 2025).
- Button, J.M.; Qazi, S.A.; Wang, J.C.-Y.; Mukhopadhyay, S. Revisiting an old friend: New findings in alphavirus structure and assembly. Curr. Opin. Virol. 2020, 45, 25–33. [Google Scholar] [CrossRef]
- Schnell, J.R.; Chou, J.J. Structure and Mechanism of the M2 Proton Channel of Influenza A Virus. Nature 2008, 451, 591–595. [Google Scholar] [CrossRef]
- Stouffer, A.L.; Acharya, R.; Salom, D.; Levine, A.S.; Di Costanzo, L.; Soto, C.S.; Tereshko, V.; Nanda, V.; Stayrook, S.; DeGrado, W.F. Structural basis for the function and inhibition of an influenza virus proton channel. Nature 2008, 451, 596–599. [Google Scholar] [CrossRef]
- Wang, C.; Lamb, R.A.; Pinto, L.H. Activation of the M2 ion channel of influenza virus: A role for the transmembrane domain histidine residue. Biophys. J. 1995, 69, 1363–1371. [Google Scholar] [CrossRef]
- Cordes, F.S.; Tustian, A.D.; Sansom, M.S.P.; Watts, A.; Fischer, W.B. Bundles consisting of extended transmembrane segments of Vpu from HIV-1: Computer simulations and conductance measurements. Biochemistry 2002, 41, 7359–7365. [Google Scholar] [CrossRef]
- Lopez, C.F.; Montal, M.; Blasie, J.K.; Klein, M.L.; Moore, P.B. Molecular dynamics investigation of membrane-bound bundles of the channel-forming transmembrane domain of viral protein U from the human immunodeficiency virus HIV-1. Biophys. J. 2002, 83, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.-X.; Sharpe, S.; Ghirlando, R.; Yau, W.-M.; Tycko, R. Oligomerization state and supramolecular structure of the HIV-1 Vpu protein transmembrane segment in phospholipid bilayers. Protein Sci. 2010, 19, 1877–1896. [Google Scholar] [CrossRef] [PubMed]
- Padhi, S.; Khan, N.; Jameel, S.; Priyakumar, U.D. Molecular Dynamics Simulations Reveal the HIV-1 Vpu Transmembrane Protein to Form Stable Pentamers. PLoS ONE 2013, 8, e79779. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, T.; Routh, A.; Judge, P.J.; Lam, Y.H.; Fischer, D.; Watts, A.; Fischer, W.B. Biophysical characterization of Vpu from HIV-1 suggests a channel-pore dualism. Proteins Struct. Funct. Bioinform. 2008, 70, 1488–1497. [Google Scholar] [CrossRef]
- Montserret, R.; Saint, N.; Vanbelle, C.; Salvay, A.G.; Simorre, J.-P.; Ebel, C.; Sapay, N.; Renisio, J.-G.; Böckmann, A.; Steinmann, E.; et al. NMR Structure and Ion Channel Activity of the p7 Protein from Hepatitis C Virus. J. Biol. Chem. 2010, 285, 31446–31461. [Google Scholar] [CrossRef]
- Cook, G.A.; Dawson, L.A.; Tian, Y.; Opella, S.J. Three-Dimensional Structure and Interaction Studies of Hepatitis C Virus p7 in 1,2-Dihexanoyl-sn-glycero-3-phosphocholine by Solution Nuclear Magnetic Resonance. Biochemistry 2013, 52, 5295–5303. [Google Scholar] [CrossRef]
- Foster, T.L.; Thompson, G.S.; Kalverda, A.P.; Kankanala, J.; Bentham, M.; Wetherill, L.F.; Thompson, J.; Barker, A.M.; Clarke, D.; Noerenberg, M.; et al. Structure-guided design affirms inhibitors of hepatitis C virus p7 as a viable class of antivirals targeting virion release. Hepatology 2014, 59, 408–422. [Google Scholar] [CrossRef]
- OuYang, B.; Chou, J.J. The minimalist architectures of viroporins and their therapeutic implications. Biochim. Biophys. Acta (BBA)-Biomembr. 2014, 1838, 1058–1067. [Google Scholar] [CrossRef]
- Oestringer, B.P.; Bolivar, J.H.; Claridge, J.K.; Almanea, L.; Chipot, C.; Dehez, F.; Holzmann, N.; Schnell, J.R.; Zitzmann, N. Hepatitis C virus sequence divergence preserves p7 viroporin structural and dynamic features. Sci. Rep. 2019, 9, 8383. [Google Scholar] [CrossRef]
- Cook, G.A.; Opella, S.J. Secondary structure, dynamics, and architecture of the p7 membrane protein from hepatitis C virus by NMR spectroscopy. Biochim. Biophys. Acta (BBA)-Biomembr. 2011, 1808, 1448–1453. [Google Scholar] [CrossRef]
- Weis, N.; Bollerup, S.; Sund, J.D.; Glamann, J.B.; Vinten, C.; Jensen, L.R.; Sejling, C.; Kledal, T.N.; Rosenkilde, M.M. Amantadine for COVID-19 treatment (ACT) study: A randomized, double-blinded, placebo-controlled clinical trial. Clin. Microbiol. Infect. 2023, 29, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Luscombe, C.A.; Huang, Z.; Murray, M.G.; Miller, M.; Wilkinson, J.; Ewart, G.D. A novel Hepatitis C virus p7 ion channel inhibitor, BIT225, inhibits bovine viral diarrhea virus in vitro and shows synergism with recombinant interferon-α-2b and nucleoside analogues. Antivir. Res. 2010, 86, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Luscombe, C.A.; Avihingsanon, A.; Supparatpinyo, K.; Gatechompol, S.; Han, W.M.; Ewart, G.D.; Thomson, A.S.; Miller, M.; Becker, S.; Murphy, R.L. Human Immunodeficiency Virus Type 1 Vpu Inhibitor, BIT225, in Combination with 3-Drug Antiretroviral Therapy: Inflammation and Immune Cell Modulation. J. Infect. Dis. 2021, 223, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Ewart, G.; Bobardt, M.; Bentzen, B.H.; Yan, Y.; Thomson, A.; Klumpp, K.; Becker, S.; Rosenkilde, M.M.; Miller, M.; Gallay, P. Post-infection treatment with the E protein inhibitor BIT225 reduces disease severity and increases survival of K18-hACE2 transgenic mice infected with a lethal dose of SARS-CoV-2. PLoS Pathog. 2023, 19, e1011328. [Google Scholar] [CrossRef]
- StGelais, C.; Foster, T.L.; Verow, M.; Atkins, E.; Fishwick, C.W.G.; Rowlands, D.; Harris, M.; Griffin, S. Determinants of Hepatitis C Virus p7 Ion Channel Function and Drug Sensitivity Identified In Vitro. J. Virol. 2009, 83, 7970–7981. [Google Scholar] [CrossRef]
- van Soest, H.; van der Schaar, P.J.; Koek, G.H.; de Vries, R.A.; van Ooteghem, N.A.; van Hoek, B.; Drenth, J.P.H.; Vrolijk, J.M.; Lieverse, R.J.; Houben, P.; et al. No beneficial effects of amantadine in treatment of chronic hepatitis C patients. Dig. Liver Dis. 2010, 42, 496–502. [Google Scholar] [CrossRef]
- StGelais, C.; Tuthill, T.J.; Clarke, D.S.; Rowlands, D.J.; Harris, M.; Griffin, S. Inhibition of hepatitis C virus p7 membrane channels in a liposome-based assay system. Antivir. Res. 2007, 76, 48–58. [Google Scholar] [CrossRef]
- Steinmann, E.; Whitfield, T.; Kallis, S.; Dwek, R.A.; Zitzmann, N.; Pietschmann, T.; Bartenschlager, R. Antiviral effects of amantadine and iminosugar derivatives against hepatitis C virus. Hepatology 2007, 46, 330–338. [Google Scholar] [CrossRef]
- Foster, T.L.; Verow, M.; Wozniak, A.L.; Bentham, M.J.; Thompson, J.; Atkins, E.; Weinman, S.A.; Fishwick, C.; Foster, R.; Harris, M.; et al. Resistance mutations define specific antiviral effects for inhibitors of the hepatitis C virus p7 ion channel. Hepatology 2011, 54, 79–90. [Google Scholar] [CrossRef]
- Pawlotsky, J.-M.; Chevaliez, S.; McHutchison, J.G. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007, 132, 1979–1998. [Google Scholar] [CrossRef]
- Davies, W.L.; Grunert, R.R.; Haff, R.F.; McGahen, J.W.; Neumayer, E.M.; Paulshock, M.; Watts, J.C.; Wood, T.R.; Hermann, E.C.; Hoffmann, C.E. Antiviral Activity of 1-Adamantanamine (Amantadine). Science 1964, 144, 862–863. [Google Scholar] [CrossRef] [PubMed]
- Kendal, A.P.; Klenk, H.D. Amantadine inhibits an early, M2 protein-dependent event in the replication cycle of avian influenza (H7) viruses. Arch. Virol. 1991, 119, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Takeuchi, K.; Pinto, L.H.; Lamb, R.A. Ion channel activity of influenza A virus M2 protein: Characterization of the amantadine block. J. Virol. 1993, 67, 5585–5594. [Google Scholar] [CrossRef] [PubMed]
- Ewart, G.D.; Mills, K.; Cox, G.B.; Gage, P.W. Amiloride derivatives block ion channel activity and enhancement of virus-like particle budding caused by HIV-1 protein Vpu. Eur. Biophys. J. 2002, 31, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Römer, W.; Lam, Y.H.; Fischer, D.; Watts, A.; Fischer, W.B.; Göring, P.; Wehrspohn, R.B.; Gösele, U.; Steinem, C. Channel Activity of a Viral Transmembrane Peptide in Micro-BLMs: Vpu1-32 from HIV-1. J. Am. Chem. Soc. 2004, 126, 16267–16274. [Google Scholar] [CrossRef]
- Park, S.H.; Opella, S.J. Conformational changes induced by a single amino acid substitution in the trans-membrane domain of Vpu: Implications for HIV-1 susceptibility to channel blocking drugs. Protein Sci. 2007, 16, 2205–2215. [Google Scholar] [CrossRef]
- Khoury, G.; Ewart, G.; Luscombe, C.; Miller, M.; Wilkinson, J. Antiviral Efficacy of the Novel Compound BIT225 against HIV-1 Release from Human Macrophages. Antimicrob. Agents Chemother. 2010, 54, 835–845. [Google Scholar] [CrossRef]
- Wilson, L.; Gage, P.; Ewart, G. Hexamethylene amiloride blocks E protein ion channels and inhibits coronavirus replication. Virology 2006, 353, 294–306. [Google Scholar] [CrossRef]
- Toft-Bertelsen, T.L.; Jeppesen, M.G.; Tzortzini, E.; Xue, K.; Giller, K.; Becker, S.; Mujezinovic, A.; Bentzen, B.H.; Andreas, L.B.; Kolocouris, A.; et al. Amantadine inhibits known and novel ion channels encoded by SARS-CoV-2 in vitro. Commun. Biol. 2021, 4, 1347. [Google Scholar] [CrossRef]
- Hong, M.; DeGrado, W.F. Structural basis for proton conduction and inhibition by the influenza M2 protein. Protein Sci. 2012, 21, 1620–1633. [Google Scholar] [CrossRef]
- Elliott, J. Consensus on amantadine use in influenza A. JAMA 1979, 242, 2383–2387. [Google Scholar] [CrossRef] [PubMed]
- Hay, A.J.; Wolstenholme, A.J.; Skehel, J.J.; Smith, M.H. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 1985, 4, 3021–3024. [Google Scholar] [CrossRef] [PubMed]
- Bright, R.A.; Shay, D.K.; Shu, B.; Cox, N.J.; Klimov, A.I. Adamantane Resistance Among Influenza A Viruses Isolated Early During the 2005-2006 Influenza Season in the United States. JAMA 2006, 295, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Ohigashi, Y.; Ma, C.; Jing, X.; Balannick, V.; Pinto, L.H.; Lamb, R.A. An amantadine-sensitive chimeric BM2 ion channel of influenza B virus has implications for the mechanism of drug inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 18775–18779. [Google Scholar] [CrossRef] [PubMed]
- Pielak, R.M.; Oxenoid, K.; Chou, J.J. Structural Investigation of Rimantadine Inhibition of the AM2-BM2 Chimera Channel of Influenza Viruses. Structure 2011, 19, 1655–1663. [Google Scholar] [CrossRef]
- Torres, J.; Maheswari, U.; Parthasarathy, K.; Ng, L.; Liu, D.X.; Gong, X. Conductance and amantadine binding of a pore formed by a lysine-flanked transmembrane domain of SARS coronavirus envelope protein. Protein Sci. 2007, 16, 2065–2071. [Google Scholar] [CrossRef]
- Hout, D.R.; Gomez, L.M.; Pacyniak, E.; Miller, J.-M.; Hill, M.S.; Stephens, E.B. A single amino acid substitution within the transmembrane domain of the human immunodeficiency virus type 1 Vpu protein renders simian–human immunodeficiency virus (SHIVKU-1bMC33) susceptible to rimantadine. Virology 2006, 348, 449–461. [Google Scholar] [CrossRef]
- Plugge, B.; Gazzarrini, S.; Nelson, M.; Cerana, R.; Van, J.L.; Etten, D.C.; DiFrancesco, D.; Moroni, A.; Thiel, G. A Potassium Channel Protein Encoded by Chlorella Virus PBCV-1. Science 2000, 287, 1641–1644. [Google Scholar] [CrossRef]
- Rosenberg, M.R.; Weaver, L.M.; Casarotto, M.G. Probing interactions of Vpu from HIV-1 with amiloride-based compounds. Biochim. Biophys. Acta (BBA)-Biomembr. 2016, 1858, 733–739. [Google Scholar] [CrossRef]
- Ewart, G.D.; Nasr, N.; Naif, H.; Cox, G.B.; Cunningham, A.L.; Gage, P.W. Potential New Anti-Human Immunodeficiency Virus Type 1 Compounds Depress Virus Replication in Cultured Human Macrophages. Antimicrob. Agents Chemother. 2004, 48, 2325–2330. [Google Scholar] [CrossRef]
- Kim, C.G.; Lemaitre, V.; Watts, A.; Fischer, W.B. Drug–protein interaction with Vpu from HIV-1: Proposing binding sites for amiloride and one of its derivatives. Anal. Bioanal. Chem. 2006, 386, 2213–2217. [Google Scholar] [CrossRef] [PubMed]
- Clinical Programs—Biotron Limited. Available online: https://www.biotron.com.au/technology/clinical-trials/ (accessed on 8 April 2025).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Deletion/Mutation | Virus | Phenotype | References | |
|---|---|---|---|---|
| Deletion/mutation of palmitoylation sites in 6K | SINV | Sites in TF also affected | Decrease in virus yield Formation of multicored particles Deformation of budding virus particles Change in level of TF in virus particles | [61,71,79] |
| Deletion of 6K | SFV | Less efficient polyprotein cleavage Defect in budding and lower viral titer (host/cell-line-dependent) Reduced membrane fusion capacity in liposomal fusion assay No effect on glycoprotein production and heterodimerization No effect on specific infectivity and particle morphology | [32,81,82] | |
| 6K chimeras | SINV | SINV 6K replaced with RRV 6K | SINV (RR6K) chimera has lower titer (10% of WT) No effect on glycoprotein heterodimerization and trafficking | [83] |
| SINV | SINV 6K-E1 replaced with RRV 6K-E1 | SINV (RR6K-E1) has much lower titer (10−7 of WT) Defects in glycoprotein heterodimerization and interaction with nucleocapsids No effect on glycoprotein processing and trafficking Revertants have mutations in E2 that improve virus yield | [83,84] | |
| SINV | 6K channel replaced with M2, Vpu, and p7 | Defects of 6K deletion rescued partially in M2, Vpu, and p7 chimeras | [13] | |
| Deletion of 6K and TF | SINV | Residues 24–45 deleted in 6K (no TF produced) | 2 log reduction in viral titer Defects in glycoprotein processing and trafficking 6K Q21L (∆24–45) revertant rescues processing defect (by enhancing membrane association and resolving signalase cleavage defect) but does not affect budding 6K protein co-expression in trans does not rescue 6K mutation/deletion-associated defects | [80,85] |
| Mutations in 6K interfacial domains | SINV | 9(YLW→AAA)11 and 18(FWV→AAA)20 | Lower membrane permeabilization and toxicity in E. coli and BHK cells | [63] |
| Deletion of TF | SFV | 6K unaffected | ~56% reduction in growth relative to WT SFV (plaque assay in BHK cells) | [67] |
| Deletion of TF | SINV | 6K unaffected | Budding defect and reduction in viral titer No effect on genome synthesis and specific infectivity No effect on expression and fusogenicity of glycoproteins Reduced mortality in mouse model | [35] |
| Deletion of 6K | CHIKV | 6K deletion mutant protects against high dose of CHIKV after single immunization and elicits humoral and cellular responses | [86] | |
| Deletion of 6K | SAV | Loss of viability Viral proteins translated but do not reach PM for budding | [73] | |
| Deletion of 6K and TF | RRV | No effect of in-frame deletion on polyprotein processing | Mutant virus defective in virion release but not particle production in BHK-21 cells RRV(∆6K) more sensitive to pH and temperature changes than WT RRV Lower viral titers and milder disease outcomes in mice compared to WT RRV Mice immunized with RRV(∆6K) show faster viral elimination upon secondary infection with WT RRV | [37] |
| Deletion of 6K and TF | SINV | Except signal peptide | Approximately 4 log reduction in titer Reduced E2 surface expression at 6 and 12hpi (rescued by HIV-1 Vpu) Impaired CPV-II formation | [13] |
| Deletion of 6K | GETV (Getah virus) | 1–2 log reduction in viral titer 24hpi Increase in number of cell-associated viruses Hampered E2 glycoprotein trafficking to PM Decreased thermostability at 52 °C and 56 °C Milder disease outcomes in vivo No effect on viral entry | [87] |
| Properties of TF | Virus/Cell System | Details | References |
|---|---|---|---|
| −1 PRF | Multiple viruses | TF is a product of −1 PRF in alphavirus 6K sequences | [66,67,77] |
| RNA secondary structure | Multiple viruses | Species-specific diversity exists in RNA secondary structures downstream of the PRF slip site | [77] |
| Palmitoylation | SINV | TF palmitoylation affects its subcellular localization and incorporation into the budding virion | [71,99] |
| Oligomerization | Ectopic expression of CHIKV TF in 293T cells | TF cysteine residues are involved in oligomerization TF palmitoylation reduces its oligomerization | [95] |
| Ion conductance | Maltose-binding protein-tagged CHIKV TF incorporated in lipid bilayer | CHIKV TF conducts ions in lipid bilayer membranes in vitro | [89] |
| Effect on assembly and budding | SINV, SFV | TF affects viral budding and particle morphology | [35,67] |
| Effect on entry and fusion | NA | ||
| Protein–protein interactions | Ectopic expression of CHIKV proteins in 293T cells | TF can interact with most CHIKV proteins except E3 TF interacts with nsp1 and nsp3 in a palmitoylation-dependent manner Potential interaction with human Scribble protein | [95,100] |
| Effect on IFN response | SINV | TF antagonizes IFN response Mechanism unknown | [101] |
| Effect on virulence in vivo | SINV | TF promotes infection in mice | [35] |
| Viroporin | Length | Membrane-Spanning Domains | Oligomeric State | Inhibitor | References |
|---|---|---|---|---|---|
| 6K | 55aa (SINV) | 1–2 TMD | Unknown | HMA | [13] |
| 61aa (CHIKV) | Amantadine | [88] | |||
| p7 | 63aa | 2TMD | Hexamer | Amantadine/ Adamantanes | [15,42,163,221,222,223] |
| HMA | [15,145,163,211] | ||||
| Long acyl-chain iminosugar derivatives | [43,224,225,226] | ||||
| BIT225 | [218] | ||||
| M2 | 97aa | 1TMD | Tetramer | Amantadine | [40,56,204,227,228,229] |
| Vpu | 81aa | 1TMD | Pentamer | Amiloride derivatives (HMA) | [230,231] |
| Rimantadine | [232] | ||||
| BIT225 | [233] | ||||
| SARS-CoV and SARS-CoV-2 E | 75–76aa | 1TMD | Pentamer | Amiloride derivatives (HMA, EIPA, DMA) | [38,234] |
| Amantadine | [235] | ||||
| BIT225 | [220] | ||||
| BE-33 | [36] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Negi, V.; Miller, A.S.; Kuhn, R.J. Advances in Viroporin Function and Structure: A Comparative Analysis of Alphavirus 6K with Well-Characterized Viroporins. Viruses 2025, 17, 868. https://doi.org/10.3390/v17060868
Negi V, Miller AS, Kuhn RJ. Advances in Viroporin Function and Structure: A Comparative Analysis of Alphavirus 6K with Well-Characterized Viroporins. Viruses. 2025; 17(6):868. https://doi.org/10.3390/v17060868
Chicago/Turabian StyleNegi, Vashi, Andrew S. Miller, and Richard J. Kuhn. 2025. "Advances in Viroporin Function and Structure: A Comparative Analysis of Alphavirus 6K with Well-Characterized Viroporins" Viruses 17, no. 6: 868. https://doi.org/10.3390/v17060868
APA StyleNegi, V., Miller, A. S., & Kuhn, R. J. (2025). Advances in Viroporin Function and Structure: A Comparative Analysis of Alphavirus 6K with Well-Characterized Viroporins. Viruses, 17(6), 868. https://doi.org/10.3390/v17060868