
Ten Previously Unassigned Human Cosavirus Genotypes Detected in Feces of Children with Non-Polio Acute Flaccid Paralysis in Nigeria in 2020

 ,
,  , , , , ,
, , , , ,  ,
,  , , , , and
, , , , and  add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Methods
2.1. Sample Collection and Preparation
2.2. Nucleic Acid Extraction, PCR Amplification, Library Preparation, and Sequencing
2.3. Sequence Assembly
2.4. Virus Typing and Recombination Analysis
2.5. Immuno-Informatic and Structural Analysis
3. Results
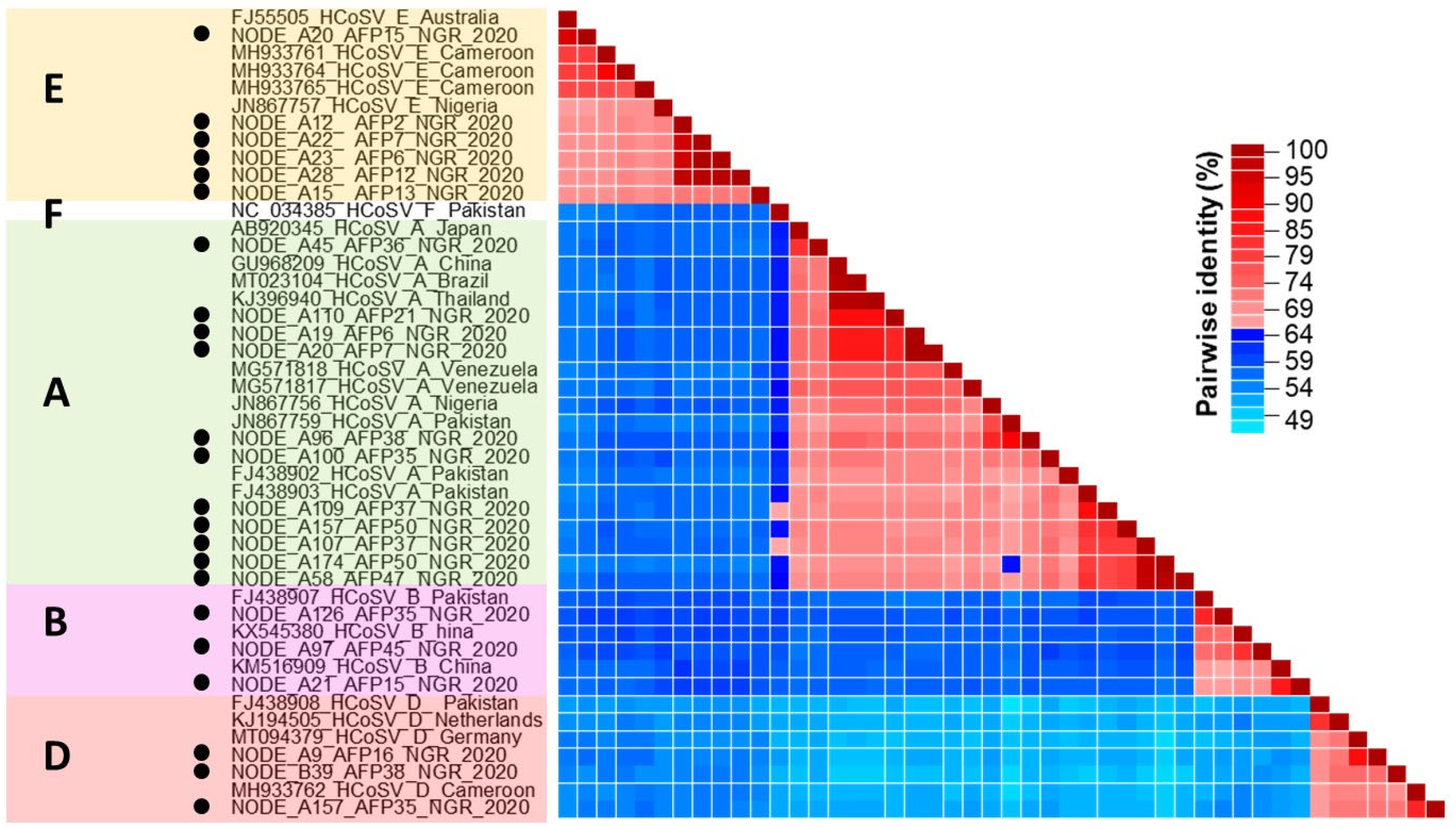
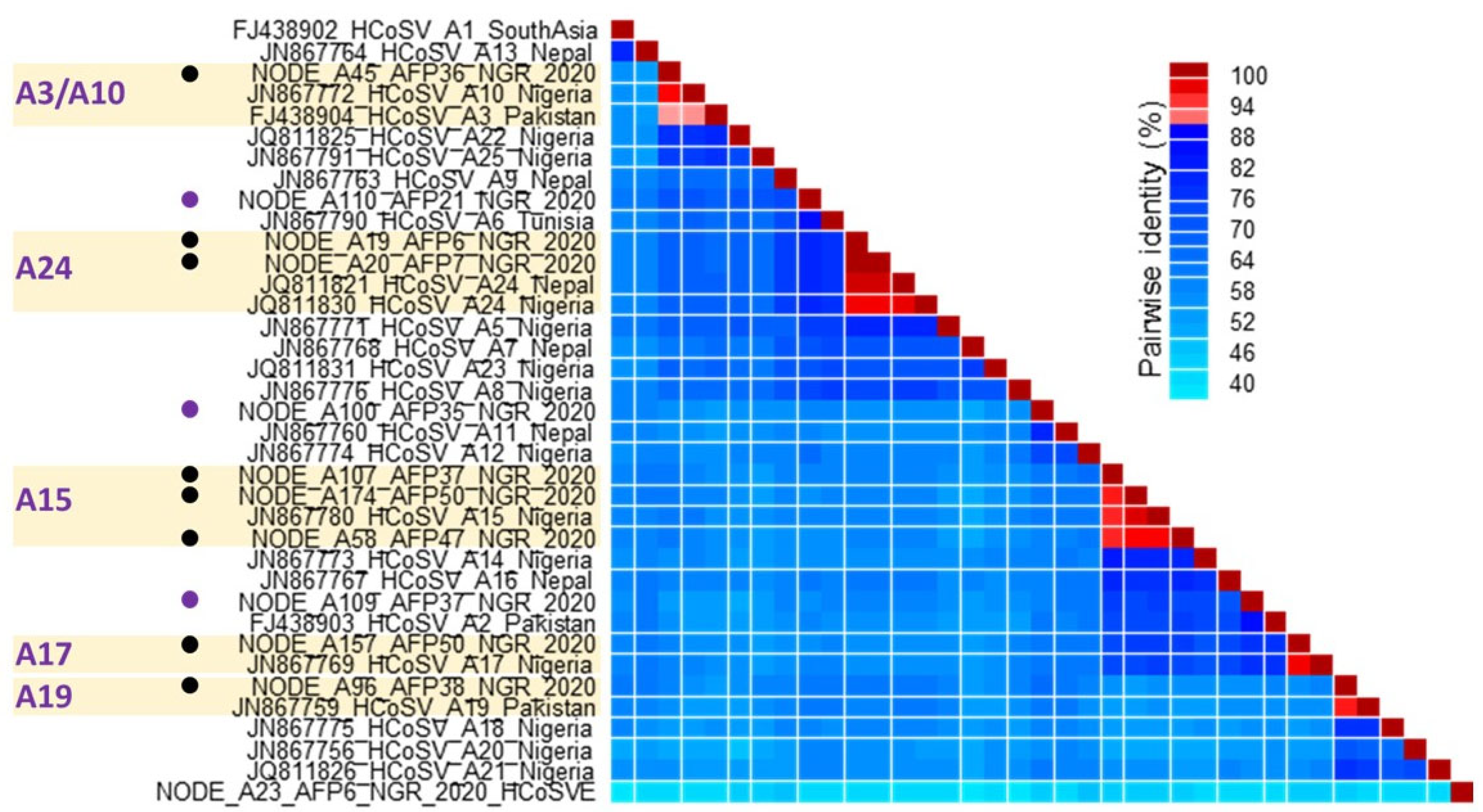
3.1. HCoSV Detection and Genotyping
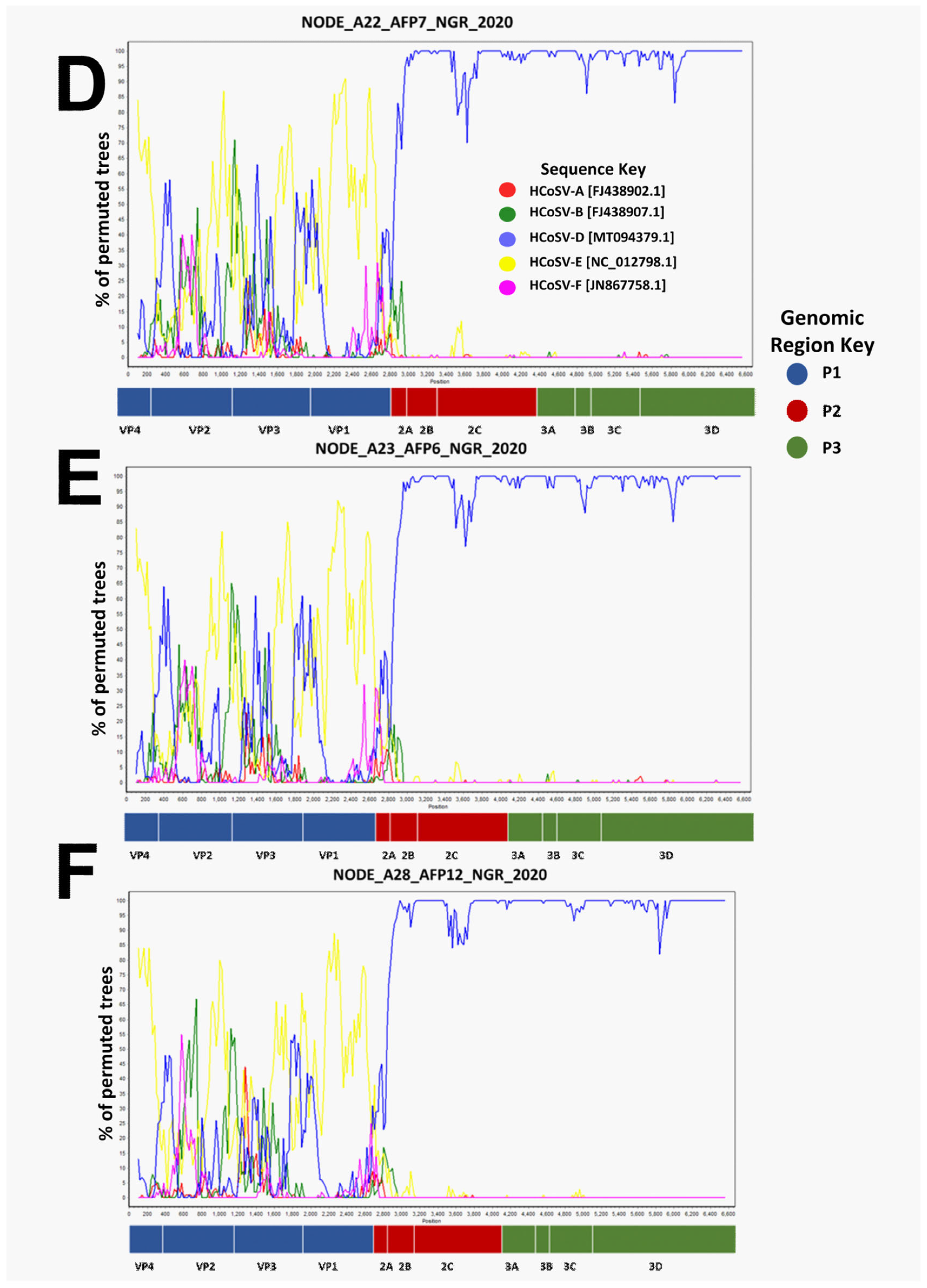
3.2. Recombination Analysis
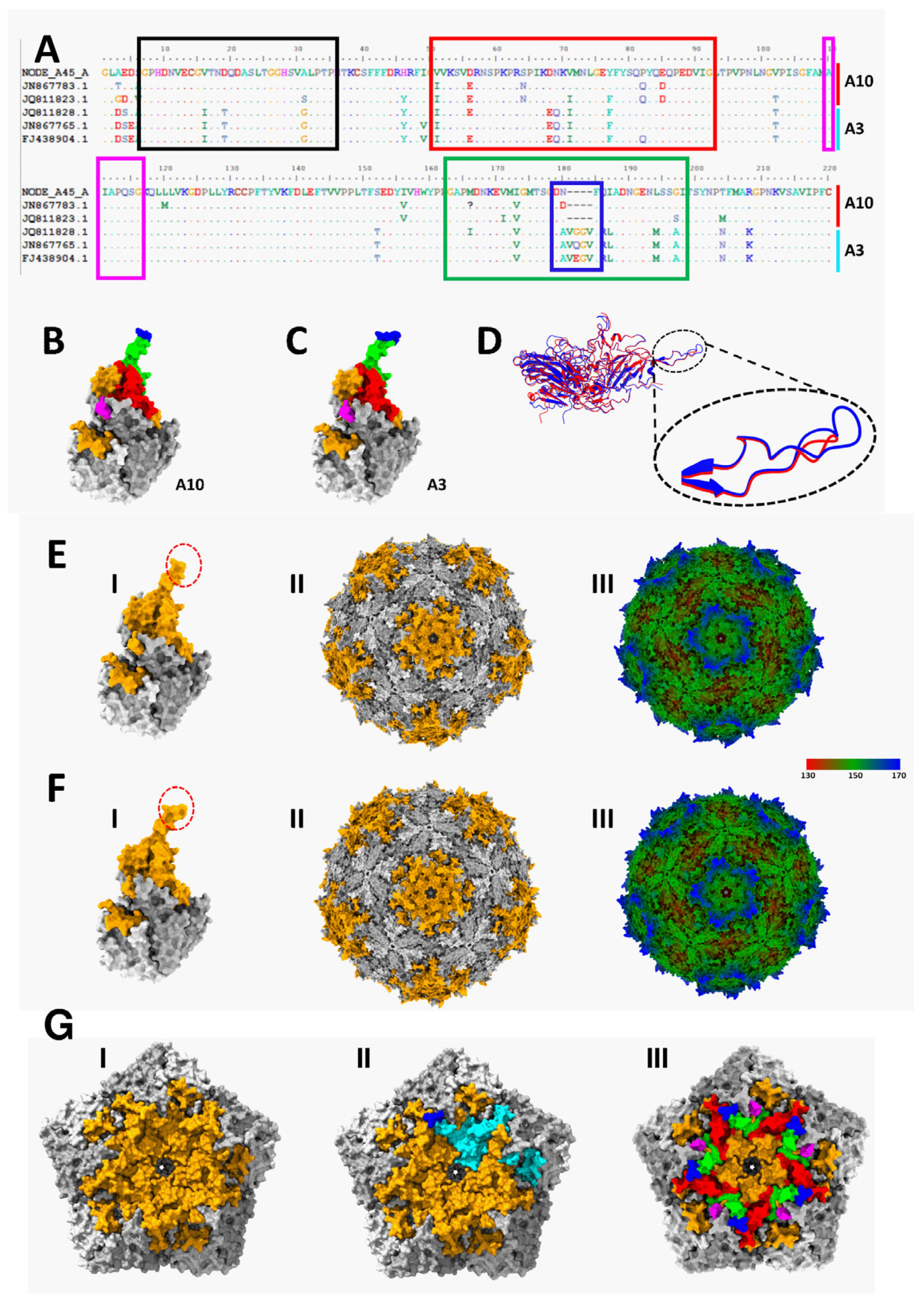
3.3. Immuno-Informatic and Structural Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woolhouse, M.F.; Scott, Z.; Hudson, R.; Howey, M. Chase-Topping. Human viruses: Discovery and emergence. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2864–2871. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Jovel, B.; Halloran, E.; Wine, J.; Patterson, G.; Ford, S.; O’Keefe, B.; Meng, D.; Song, Y.; Zhang, Z.; et al. Metagenomic analysis of microbiome in colon tissue from subjects with inflammatory bowel diseases reveals interplay of viruses and bacteria. Inflamm. Bowel Dis. 2015, 21, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Osunmakinde, C.O.; Selvarajan, R.; Sibanda, T.; Mamba, B.B.; Msagati, T.A.M. Overview of trends in the application of metagenomic techniques in the analysis of human enteric viral diversity in Africa’s environmental regimes. Viruses 2018, 10, 429. [Google Scholar] [CrossRef]
- Deaton, J.; Yu, F.B.; Quake, S.R. Mini-metagenomics and nucleotide composition aid the identification and host association of novel bacteriophage sequences. Adv. Biosyst. 2019, 3, e1900108. [Google Scholar] [CrossRef]
- Hamza, I.A.; Bibby, K. Critical issues in application of molecular methods to environmental virology. J. Virol. Methods 2019, 266, 11–24. [Google Scholar] [CrossRef]
- Li, R.; Zhu, L.; Cui, L.; Zhu, Y.G. Viral diversity and potential environmental risk in microplastic at watershed scale: Evidence from metagenomic analysis of plastisphere. Environ. Int. 2022, 161, 107146. [Google Scholar] [CrossRef]
- Finkbeiner, S.R.; Allred, A.F.; Tarr, P.I.; Klein, E.J.; Kirkwood, C.D.; Wang, D. Metagenomic analysis of human diarrhea: Viral detection and discovery. PLoS Pathog. 2008, 4, e1000011. [Google Scholar] [CrossRef]
- Conceicao-Neto, N.; Zeller, H.; Lefrere, P.; De-Bruyn, L.; Beller, W.; Deboutte, C.K.; Yinda, R.; Lavigne, P.; Maes, M.; Van-Ranst, E.; et al. Modular approach to customise sample preparation procedures for viral metagenomics: A reproducible protocol for virome analysis. Sci. Rep. 2015, 5, 16532. [Google Scholar] [CrossRef]
- Lojkic, I.; Bidin, M.; Prpic, J.; Simic, I.; Kresic, N.; Bedekovic, T. Faecal virome of red foxes from peri-urban areas. Comp. Immunol. Microbiol. Infect. Dis. 2016, 45, 10–15. [Google Scholar] [CrossRef]
- Lima, D.A.; Cibulski, S.P.; Finkler, F.; Teixeira, T.F.; Varela, A.P.M.; Cerva, C.; Loiko, M.R.; Scheffer, C.M.; Dos Santos, H.F.; Mayer, F.Q.; et al. Faecal virome of healthy chickens reveals a large diversity of the eukaryote viral community, including novel circular ssDNA viruses. J. Gen. Virol. 2017, 98, 690–703. [Google Scholar] [CrossRef]
- Duarte, M.A.; Silva, J.M.F.; Brito, C.R.; Teixeira, D.S.; Melo, F.L.; Ribeiro, B.; Nagata, T.; Campos, F.S. Faecal virome analysis of wild animals from Brazil. Viruses 2019, 11, 803. [Google Scholar] [CrossRef] [PubMed]
- Lima, D.A.; Cibulski, S.P.; Tochetto, C.; Varela, A.P.M.; Finkler, F.; Teixeira, T.F.; Loiko, M.R.; Cerva, C.; Junqueira, D.M.; Mayer, F.Q.; et al. The intestinal virome of malabsorption syndrome-affected and unaffected broilers through shotgun metagenomics. Virus Res. 2019, 261, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Victoria, J.; Simmonds, P.; Slikas, E.; Chieochansin, T.; Naeem, A.; Shaukat, S.; Sharif, S.; Alam, M.M.; Angez, M.; et al. A highly prevalent and genetically diversified picornaviridae genus in south asian children. Proc. Natl. Acad. Sci. USA 2008, 105, 20482–20487. [Google Scholar] [CrossRef] [PubMed]
- Stöcker, A.; Breno, F.; de Carvalho, D.; Souza, T.; Cristina, M.; Ribeiro, E.; Martins, N.; Luciana, O.; Araujo, J.; Ivan, C.; et al. Cosavirus infection in persons with and without gastroenteritis, Brazil. Emerg. Infect. Dis. 2012, 18, 656–659. [Google Scholar] [CrossRef]
- Okitsu, S.; Khamrin, P.; Thongprachum, A.; Nishimura, S.; Kalesaran, A.F.C.; Takanashi, S.; Shimizu, H.; Hayakawa, S.; Mizuguchi, M.; Ushijima, H. Detection and molecular characterization of human cosavirus in a pediatric patient with acute gastroenteritis, Japan. Infect. Genet. Evol. 2014, 28, 125–129. [Google Scholar] [CrossRef]
- Yang, Y.; Ju, A.; Xu, X.; Cao, X.; Tao, Y. A novel type of cosavirus from children with nonpolio acute flaccid paralysis. Virol. J. 2016, 13, 169. [Google Scholar] [CrossRef]
- Vizzi, E.R.; Fernández, L.A.; Angulo, R.; Blanco; Pérez, C. Human cosavirus infection in HIV subjects with diarrhoea: Persistent detection associated with fatal outcome. J. Clin. Virol. 2021, 139, 104825. [Google Scholar] [CrossRef]
- Blinkova, O.; Rosario, K.; Li, L.; Kapoor, A.; Slikas, B.; Bernardin, F.; Breitbart, M.; Delwart, E. Frequent detection of highly 423 diverse variants of cardiovirus, cosavirus, bocavirus, and circovirus in sewage samples collected in the United States. J. Clin. Microbiol. 2009, 47, 3507–3513. [Google Scholar] [CrossRef]
- Moghaddam, F.S.; Ghaderi, M.; Parsania, M.; Mozhgani, S.-H.; Arjmand, R. First human cosavirus detection from cerebrospinal fluid in hospitalized children with aseptic meningitis and encephalitis in Iran. Pediatr. Infect. Dis. J. 2021, 40, e459–e461. [Google Scholar] [CrossRef]
- Osundare, F.A.; Opaleye, O.O.; Akindele, A.A.; Adedokun, S.A.; Akanbi, O.A.; Bock, C.T.; Diedrich, S.; Böttcher, S. Detection and characterization of human enteroviruses, human cosaviruses, and a new human parechovirus type in healthy individuals in Osun State, Nigeria, 2016/2017. Viruses 2019, 11, 1037. [Google Scholar] [CrossRef]
- Holtz, L.R.; Finkbeiner, S.R.; Kirkwood, C.D.; Wang, D. Identification of a novel picornavirus related to cosaviruses in a child with acute diarrhea. Virol. J. 2008, 5, 159. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, A.C.; Luchs, A.; de Pádua Milagres, F.A.; Komninakis, S.V.; Gill, D.E.; Lobato, M.C.A.B.S.; Brustulin, R.; das Chagas, R.T.; Dos Santos, M.d.F.N.; Soares, C.V.d.D.A.; et al. Near full length genome of a recombinant (e/d) cosavirus strain from a rural area in the central region of Brazil. Sci. Rep. 2018, 8, 12304. [Google Scholar] [CrossRef] [PubMed]
- Daprà, V.; Galliano, I.; Montanari, P.; Zaniol, E.; Calvi, C.; Alliaudi, C.; Bergallo, M. Bufavirus, cosavirus, and salivirus in diarrheal Italian infants. Intervirology 2021, 64, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Kapusinszky, B.; Phan, T.G.; Kapoor, A.; Delwart, E. Genetic diversity of the genus cosavirus in the family picornaviridae: A new species, recombination, and 26 new genotypes. PLoS ONE 2012, 7, e36685. [Google Scholar] [CrossRef]
- Cotten, M.; Oude Munnink, B.; Canuti, M.; Deijs, M.; Watson, S.J.; Kellam, P.; van der Hoek, L. Full genome virus detection in fecal samples using sensitive nucleic acid preparation, deep sequencing, and a novel iterative sequence classification algorithm. PLoS ONE 2014, 9, e93269. [Google Scholar] [CrossRef]
- International Committee for Taxonomy of Viruses. 2023. Available online: https://ictv.global/report/chapter/picornaviridae/picornaviridae/cosavirus (accessed on 25 May 2025).
- World Health Organization. Polio Laboratory Manual, 4th ed.; WHO: Geneva, Switzerland, 2004. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 5. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Cosavirus in the Picornavirus Pages. Available online: https://www.picornaviridae.com/caphthovirinae/cosavirus/cosavirus.htm (accessed on 25 May 2025).
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Montiel, G.D.; Santoyo, R.N.; Ho, P.; Carrillo, T.M.; Iii, C.L.B.; Johnson, J.E.; Reddy, V.S. VIPERdb v3.0: A structure-based data analytics platform for viral capsids. Nucleic Acids Res. 2021, 49, D809–D816. [Google Scholar] [CrossRef]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef]
- Lobo, P.S.; Cardoso, J.F.; Barata, R.R.; Lemos, P.S.; Guerra, S.F.; Soares, L.S.; Nunes, M.R.; Mascarenhas, J.D. Near-complete genome of cosavirus A from a child hospitalized with acute gastroenteritis, Brazil. Infect. Genet. Evol. 2020, 85, 104555. [Google Scholar] [CrossRef]
- Okitsu, S.; Khamrin, P.; Hanaoka, N.; Thongprachum, A.; Takanashi, S.; Fujimoto, T.; Mizuguchi, M.; Shimizu, H.; Hayakawa, S.; Maneekarn, N.; et al. Cosavirus (family Picornaviridae) in pigs in Thailand and Japan. Arch. Virol. 2016, 161, 159–163. [Google Scholar] [CrossRef]
- López, G.R.; Martinez, L.M.; Freyre, L.; Freire, M.C.; Vladimirsky, S.; Rabossi, A.; Cisterna, D.M. Persistent Detection of Cosavirus and SaffoldCardiovirus in Riachuelo River, Argentina. Food Environ. Virol. 2021, 13, 64–73. [Google Scholar] [CrossRef]
- Schneider, J.; Engler, M.; Hofmann, J.; Selinka, H.; Jones, T.; Drosten, C.; Diedrich, S.; Corman, V.; Böttcher, S. Molecular detection of cosaviruses in a patient with acute flaccid paralysis and in sewage samples in Germany. Virus Res. 2021, 297, 198285. [Google Scholar] [CrossRef]
- Yu, J.M.; Ao, Y.Y.; Li, L.L.; Duan, Z.J. Identification of a novel cosavirus species in faeces of children and its relationship with acute gastroenteritis in China. Clin. Microbiol. Infect. 2017, 23, 550–554. [Google Scholar] [CrossRef]
- Rezig, D.; Lamari, A.; Touzi, H.; Meddeb, Z.; Triki, H. Typing of Human Cosaviruses by sequencing of full VP1: Update on global genetic diversity and identification of possible new genotypes circulating in Tunisia, North Africa. Infect. Genet. Evol. 2020, 78, 104115. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cui, X.; Cai, X.; An, T. Recombination in positive-strand RNA viruses. Front. Microbiol. 2022, 13, 870759. [Google Scholar] [CrossRef] [PubMed]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, J.D.; Dominguez-Bello, M.G.; Contreras, M.; Lander, O.; Caballero-Arias, H.; Deng, X.; Noya-Alarcon, O.; Delwart, E. Complex virome in feces from Amerindian children in isolated Amazonian villages. Nat. Commun. 2018, 9, 4270. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contig ID | Genome Length | Coding Sequence | Mean Depth of Coverage | Species | Accession; Query Cover (%); Pairwise Identity (%); Country; Year | |
|---|---|---|---|---|---|---|
| Best Hit 1 | Best Hit 2 | |||||
| NODE_A12_AFP2_NGR_2020 | 7527 | Complete | 51× | HCoSV E | JN867757.1; 77; 91.1; Nigeria; 2007 | MH933764.1;77: 90.73; Cameroon; 2014 |
| NODE_A19_AFP6_NGR_2020 | 7761 | Complete | 464× | HCoSV A | MG571817.1; 94; 88.3; Venezuela; 2015 | MG571823.1; 94; 84.1; VENEZUELA; 2015 |
| NODE_A23_AFP6_NGR_2020 | 6662 | Complete | 131× | HCoSV E | MG571820; 74; 83.9; Venezuela; 2015 | MH933764.1; 66; 91; Cameroon; 2014 |
| NODE_A20_AFP7_NGR_2020 | 7785 | Complete | 288× | HCoSV A | MG571817.1;94;88.3; Venezuela; 2015 | MG571823.1; 94; 84.1; VENEZUELA; 2015 |
| NODE_A22_AFP7_NGR_2020 | 7475 | Complete | 80× | HCoSV E | MG571820; 77; 83.9; Venezuela; 2015 | MH933764.1; 69; 90.9 Cameroon; 2014 |
| NODE_A28_AFP12_NGR_2020 | 7490 | Complete | 138× | HCoSV E | JN867757.1; 77; 91.1; Nigeria; 2007 | MH933764.1; 69; 92.3 Cameroon; 2014 |
| NODE_A15_AFP13_NGR_2020 | 7763 | Complete | 1029× | HCoSV E | JN867757.1; 76; 91.5; Nigeria; 2007 | MH933764.1; 71; 90.3 Cameroon; 2014 |
| NODE_A20_AFP15_NGR_2020 | 7194 | Complete | 222× | HCoSV E | MH933764.1; 100; 84.8; Cameroon; 2014 | MH933765.1; 100; 83.74; Cameroon; 2014 |
| NODE_A21_AFP15_NGR_2020 | 7149 | Complete | 945× | HCoSV B | KM516909.1;99;79.4; China; 2012 | GU968209.1; 28; 84.51; China; 2009 |
| NODE_A9_AFP16_NGR_2020 | 6610 | Complete | 80× | HCoSV D | MT094379.1; 99; 84.6; Germany; 2018 | MH933762.1; 72; 87.5;Cameroon; 2014 |
| NODE_A110_AFP21_NGR_2020 | 7678 | Complete | 78× | HCoSV A | MG571817; 97; 88.3; Venezuela; 2015 | MG571823.1; 95; 84.1; Venezuela; 2015 |
| NODE_A100_AFP35_NGR_2020 | 7747 | Complete | 264× | HCoSV A | MG571817; 85; 88.1; Venezuela; 2015 | FJ438903.1; 81; 87.4; Pakistan |
| NODE_A126_AFP35_NGR_2020 | 6723 | Complete | 38× | HCoSV B | FJ438907; 99; 74.8; Pakistan | MH933763.1; 38; 72.3; Cameroon; 2014 |
| NODE_A157_AFP35_NGR_2020 | 5983 | Partial | 27× | HCoSV D | MH933762.1; 98; 85.2; Cameroon; 2014 | MT094379.1; 93; 87.8; Germany; 2018 |
| NODE_A45_AFP36_NGR_2020 | 7794 | Complete | 320× | HCoSV A | AB920345; 93; 82.6; Japan; 2012 | FJ438904.1: 82: 83.81; Pakistan |
| NODE_A107_AFP37_NGR_2020 | 6668 | Complete | 369× | HCoSV A | FJ438903; 95; 87.6; Pakistan | KJ396940; 85; 90.6; Thailand; 2011 |
| NODE_A109_AFP37_NGR_2020 | 6613 | Complete | 181× | HCoSV A | FJ438903; 99; 83.1; Pakistan | MG571817.1; 89; 86.5; Venezuela; 2015 |
| NODE_A96_AFP38_NGR_2020 | 7388 | Complete | 300× | HCoSV A | JN867759; 89; 84.7; Pakistan; 2006 | GU968209; 87; 84.8; China; 2009 |
| NODE_B39_AFP38_NGR_2020 | 6735 | Complete | 369× | HCoSV D | MT094379.1; 96; 87.3; Germany; 2018 | KJ194505.1; 86; 90.4; Netherlands; 1994 |
| NODE_A97_AFP45_NGR_2020 | 6618 | Partial | 1097× | HCoSV B | KX545380; 97; 76.1; China; 2014 | MH933763; 50; 72.4;Cameroon; 2014 |
| NODE_A58_AFP47_NGR_2020 | 7756 | Complete | 216× | HCoSV A | FJ438903; 94; 82.9; Pakistan | MT023104.1; 82; 87.6; Brazil; 2017 |
| NODE_A157_AFP50_NGR_2020 | 5257 | Partial | 135× | HCoSV A | FJ438903; 99; 79.6; Pakistan | MG571818.1; 71; 85.6; Venezuela; 2015 |
| NODE_A174_AFP50_NGR_2020 | 4931 | Partial | 141× | HCoSV A | FJ438903; 99; 79; Pakistan | GU968209.1; 79; 84.7; China; 2009 |
| S/N | Sample Pool ID | Number of Samples in Pool | Number of Variants | HCoSV Genotype(s) |
|---|---|---|---|---|
| 1 | AFP2 | 6 | 1 | E-New |
| 2 | AFP6 | 5 | 2 | A24, E-New |
| 3 | AFP7 | 5 | 2 | A24, E-New |
| 4 | AFP12 | 1 | 1 | E-New |
| 5 | AFP13 | 5 | 1 | E-New |
| 6 | AFP15 | 7 | 2 | B-New, E1 |
| 7 | AFP16 | 5 | 1 | D-New |
| 8 | AFP21 | 5 | 1 | A-New |
| 9 | AFP35 | 5 | 3 | A-New, B-New, D-New |
| 10 | AFP36 | 4 | 1 | A3/A10 |
| 11 | AFP37 | 5 | 2 | A-New, A15 |
| 12 | AFP38 | 5 | 2 | A19, D3 |
| 13 | AFP45 | 2 | 1 | B-New |
| 14 | AFP47 | 2 | 1 | A15 |
| 15 | AFP50 | 5 | 2 | A15, A17 |
| Total | 67 | 23 | 17 (including 10 new genotypes) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ajileye, T.G.; Akinleye, T.E.; Faleye, T.O.C.; De Coninck, L.; George, U.E.; Onoja, A.B.; Agbaje, S.T.; Ifeorah, I.M.; Olayinka, O.A.; Oni, E.I.; et al. Ten Previously Unassigned Human Cosavirus Genotypes Detected in Feces of Children with Non-Polio Acute Flaccid Paralysis in Nigeria in 2020. Viruses 2025, 17, 844. https://doi.org/10.3390/v17060844
Ajileye TG, Akinleye TE, Faleye TOC, De Coninck L, George UE, Onoja AB, Agbaje ST, Ifeorah IM, Olayinka OA, Oni EI, et al. Ten Previously Unassigned Human Cosavirus Genotypes Detected in Feces of Children with Non-Polio Acute Flaccid Paralysis in Nigeria in 2020. Viruses. 2025; 17(6):844. https://doi.org/10.3390/v17060844
Chicago/Turabian StyleAjileye, Toluwani Goodnews, Toluwanimi Emmanuel Akinleye, Temitope O. C. Faleye, Lander De Coninck, Uwem Etop George, Anyebe Bernard Onoja, Sheriff Tunde Agbaje, Ijeoma Maryjoy Ifeorah, Oluseyi Adebowale Olayinka, Elijah Igbekele Oni, and et al. 2025. "Ten Previously Unassigned Human Cosavirus Genotypes Detected in Feces of Children with Non-Polio Acute Flaccid Paralysis in Nigeria in 2020" Viruses 17, no. 6: 844. https://doi.org/10.3390/v17060844
APA StyleAjileye, T. G., Akinleye, T. E., Faleye, T. O. C., De Coninck, L., George, U. E., Onoja, A. B., Agbaje, S. T., Ifeorah, I. M., Olayinka, O. A., Oni, E. I., Oragwa, A. O., Popoola, B. O., Olayinka, O. T., Osasona, O. G., George, O. A., Ajayi, P. G., Suleiman, A. A., Muhammad, A. I., Komolafe, I., ... Adewumi, M. O. (2025). Ten Previously Unassigned Human Cosavirus Genotypes Detected in Feces of Children with Non-Polio Acute Flaccid Paralysis in Nigeria in 2020. Viruses, 17(6), 844. https://doi.org/10.3390/v17060844