

HIV, Inflammation, and Immunometabolism: A Model of the Inflammatory Theory of Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
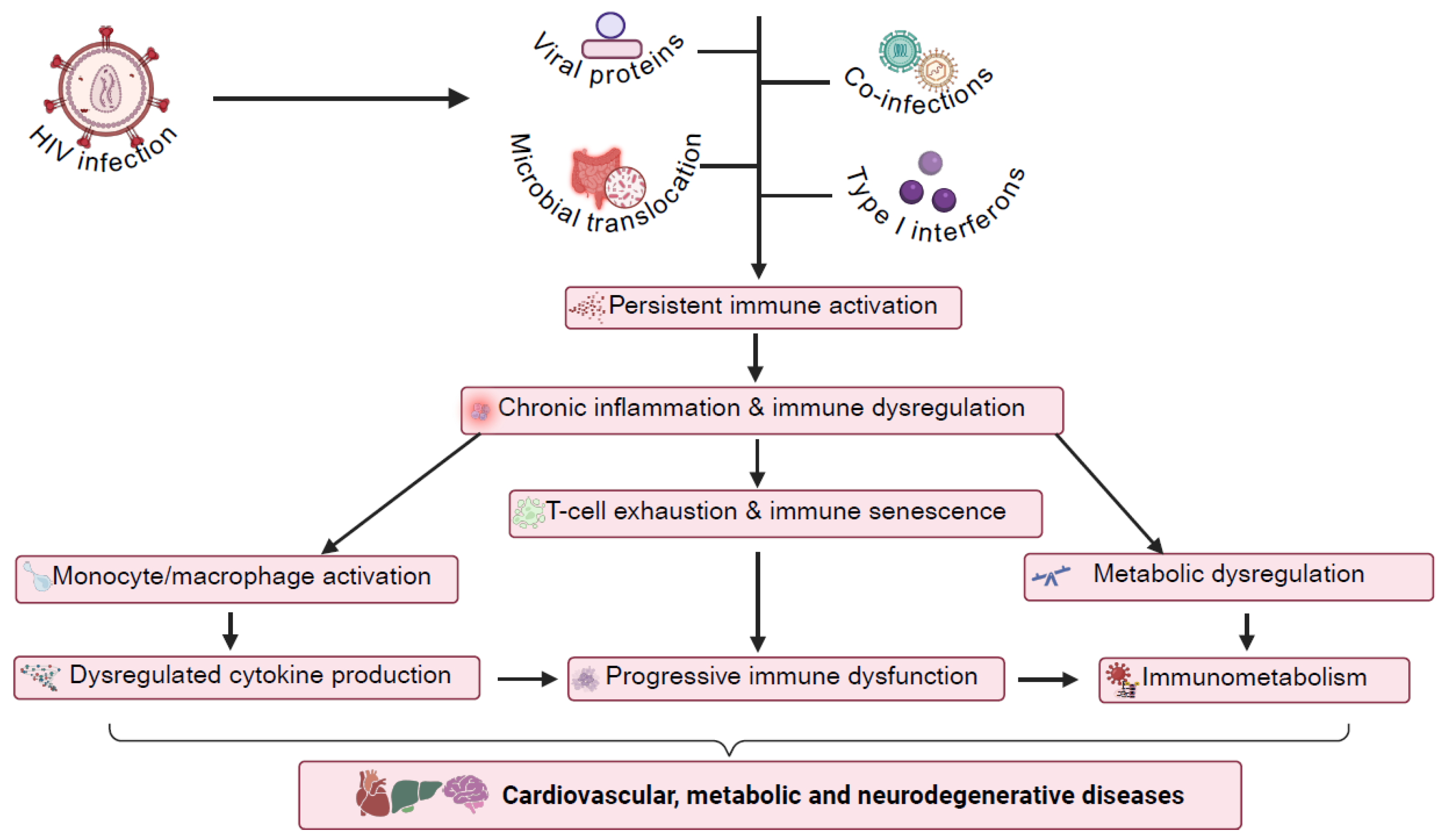
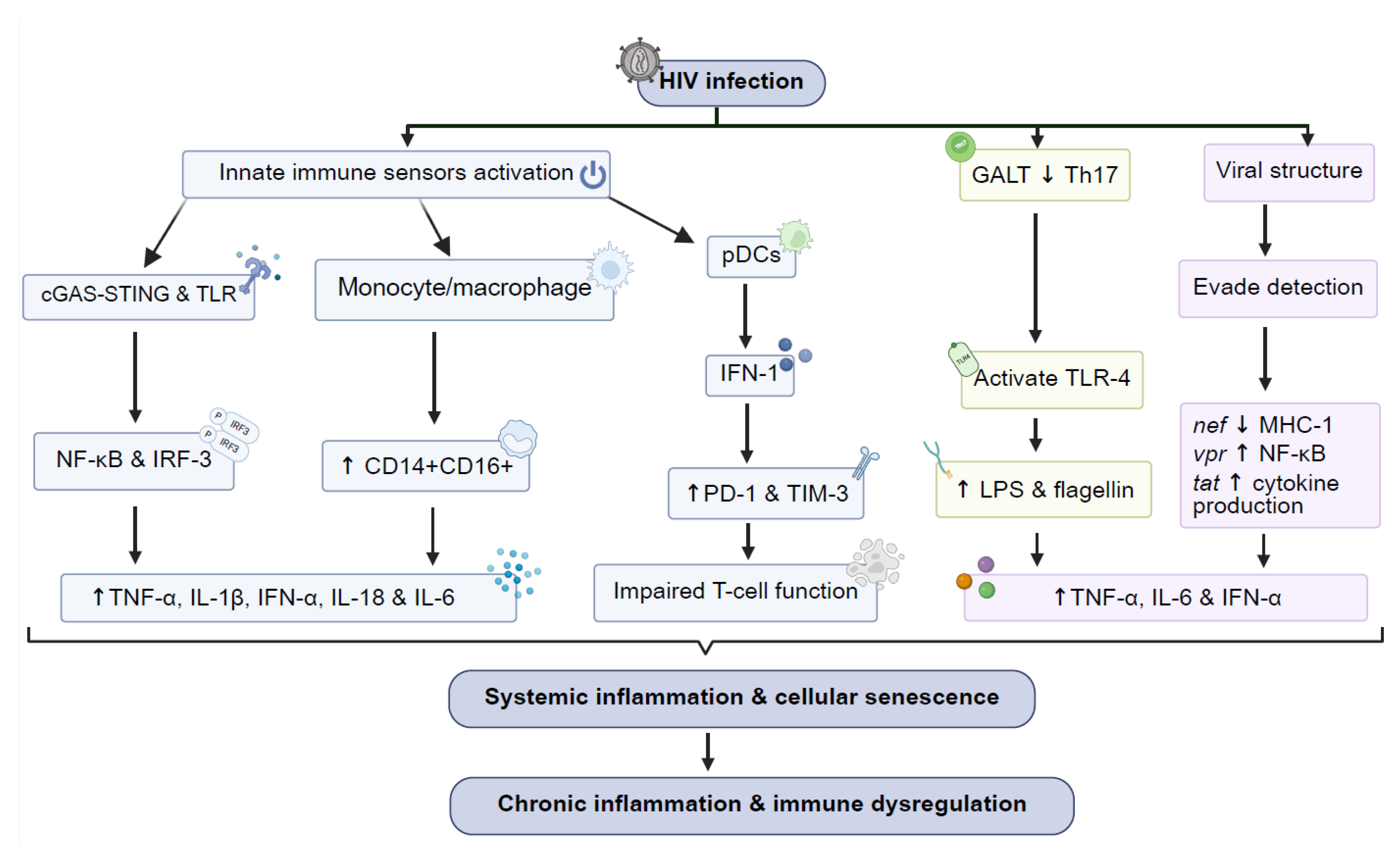
2. Mechanisms of Chronic Immune Activation in HIV
- Pre-ART vs. ART era changes
- Cellular and Molecular Pathways
3. Immune Cell Dysfunction and Senescence in HIV
- Imbalance Between Pro- and Anti-Inflammatory Responses
- HIV-Induced Senescence and the Senescence-Associated Secretory Phenotype
4. Immunometabolic Reprogramming in HIV Infection
- I.
- Metabolic Reprogramming of Immune Cells in HIV
- II.
- Glycolysis, Chronic Inflammation, and the Inflammatory Theory of Disease
- III.
- The Role of Lipid Metabolism in HIV-Driven Inflammation
5. Gut Microbiome, Immune Dysregulation, and Metabolite Changes
6. Chronic Inflammation and Immune Activation in HIV-Associated Comorbidities: Focus on CVDs and HANDs
- Cardiovascular Diseases (CVDs) during HIV Infection
- HIV-Associated Neurocognitive Disorders (HANDs)
7. Potential Therapeutic Targets to Address HIV-Related Immunometabolism
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Abudukelimu, A.; Barberis, M.; Redegeld, F.A.; Sahin, N.; Westerhoff, H.V. Predictable Irreversible Switching Between Acute and Chronic Inflammation. Front. Immunol. 2018, 9, 1596. [Google Scholar] [CrossRef] [PubMed]
- Pahwa, R.; Goyal, A.; Jialal, I. Chronic Inflammation. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Teer, E.; Joseph, D.E.; Glashoff, R.H.; Faadiel Essop, M. Monocyte/Macrophage-Mediated Innate Immunity in HIV-1 Infection: From Early Response to Late Dysregulation and Links to Cardiovascular Diseases Onset. Virol. Sin. 2021, 36, 565–576. [Google Scholar] [CrossRef]
- Antar, S.A.; Ashour, N.A.; Marawan, M.E.; Al-Karmalawy, A.A. Fibrosis: Types, Effects, Markers, Mechanisms for Disease Progression, and Its Relation with Oxidative Stress, Immunity, and Inflammation. Int. J. Mol. Sci. 2023, 24, 4004. [Google Scholar] [CrossRef]
- Hunter, P. The inflammation theory of disease. The growing realization that chronic inflammation is crucial in many diseases opens new avenues for treatment. EMBO Rep. 2012, 13, 968–970. [Google Scholar] [CrossRef]
- Hsue, P.Y.; Deeks, S.G.; Hunt, P.W. Immunologic basis of cardiovascular disease in HIV-infected adults. J. Infect. Dis. 2012, 205 (Suppl. S3), S375–S382. [Google Scholar] [CrossRef]
- Zicari, S.; Sessa, L.; Cotugno, N.; Ruggiero, A.; Morrocchi, E.; Concato, C.; Rocca, S.; Zangari, P.; Manno, E.C.; Palma, P. Immune Activation, Inflammation, and Non-AIDS Co-Morbidities in HIV-Infected Patients under Long-Term ART. Viruses 2019, 11, 200. [Google Scholar] [CrossRef]
- Gannon, P.; Khan, M.Z.; Kolson, D.L. Current understanding of HIV-associated neurocognitive disorders pathogenesis. Curr. Opin. Neurol. 2011, 24, 275–283. [Google Scholar] [CrossRef]
- Mu, W.; Patankar, V.; Kitchen, S.; Zhen, A. Examining Chronic Inflammation, Immune Metabolism, and T Cell Dysfunction in HIV Infection. Viruses 2024, 16, 219. [Google Scholar] [CrossRef]
- Klatt, N.R.; Funderburg, N.T.; Brenchley, J.M. Microbial translocation, immune activation, and HIV disease. Trends Microbiol. 2013, 21, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Raehtz, K.D.; Barrenas, F.; Xu, C.; Busman-Sahay, K.; Valentine, A.; Law, L.; Ma, D.; Policicchio, B.B.; Wijewardana, V.; Brocca-Cofano, E.; et al. African green monkeys avoid SIV disease progression by preventing intestinal dysfunction and maintaining mucosal barrier integrity. PLoS Pathog. 2020, 16, e1008333. [Google Scholar] [CrossRef] [PubMed]
- Pandrea, I.V.; Gautam, R.; Ribeiro, R.M.; Brenchley, J.M.; Butler, I.F.; Pattison, M.; Rasmussen, T.; Marx, P.A.; Silvestri, G.; Lackner, A.A.; et al. Acute loss of intestinal CD4+ T cells is not predictive of simian immunodeficiency virus virulence. J. Immunol. 2007, 179, 3035–3046. [Google Scholar] [CrossRef]
- Plaeger, S.F.; Collins, B.S.; Musib, R.; Deeks, S.G.; Read, S.; Embry, A. Immune activation in the pathogenesis of treated chronic HIV disease: A workshop summary. AIDS Res. Hum. Retroviruses 2012, 28, 469–477. [Google Scholar] [CrossRef]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef]
- Iyer, S.S.; Cheng, G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef]
- Paiardini, M.; Muller-Trutwin, M. HIV-associated chronic immune activation. Immunol. Rev. 2013, 254, 78–101. [Google Scholar] [CrossRef]
- Deeks, S.G.; Tracy, R.; Douek, D.C. Systemic effects of inflammation on health during chronic HIV infection. Immunity 2013, 39, 633–645. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar] [CrossRef]
- Deeks, S.G.; Kitchen, C.M.; Liu, L.; Guo, H.; Gascon, R.; Narvaez, A.B.; Hunt, P.; Martin, J.N.; Kahn, J.O.; Levy, J.; et al. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood 2004, 104, 942–947. [Google Scholar] [CrossRef]
- Mogensen, T.H.; Melchjorsen, J.; Larsen, C.S.; Paludan, S.R. Innate immune recognition and activation during HIV infection. Retrovirology 2010, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Utay, N.S.; Hunt, P.W. Role of immune activation in progression to AIDS. Curr. Opin. HIV AIDS 2016, 11, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Hileman, C.O.; Funderburg, N.T. Inflammation, Immune Activation, and Antiretroviral Therapy in HIV. Curr. HIV/AIDS Rep. 2017, 14, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Cohn, L.B.; Chomont, N.; Deeks, S.G. The Biology of the HIV-1 Latent Reservoir and Implications for Cure Strategies. Cell Host Microbe 2020, 27, 519–530. [Google Scholar] [CrossRef]
- Banica, L.; Vlaicu, O.; Jipa, R.; Abagiu, A.; Nicolae, I.; Neaga, E.; Otelea, D.; Paraschiv, S. Exhaustion and senescence of CD4 and CD8 T cells that express co-stimulatory molecules CD27 and CD28 in subjects that acquired HIV by drug use or by sexual route. Germs 2021, 11, 66–77. [Google Scholar] [CrossRef]
- Nabatanzi, R.; Cose, S.; Joloba, M.; Jones, S.R.; Nakanjako, D. Effects of HIV infection and ART on phenotype and function of circulating monocytes, natural killer, and innate lymphoid cells. AIDS Res. Ther. 2018, 15, 7. [Google Scholar] [CrossRef]
- Kuller, L.H.; Tracy, R.; Belloso, W.; De Wit, S.; Drummond, F.; Lane, H.C.; Ledergerber, B.; Lundgren, J.; Neuhaus, J.; Nixon, D.; et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008, 5, e203. [Google Scholar] [CrossRef]
- Ntsekhe, M.; Baker, J.V. Cardiovascular Disease Among Persons Living with HIV: New Insights into Pathogenesis and Clinical Manifestations in a Global Context. Circulation 2023, 147, 83–100. [Google Scholar] [CrossRef]
- Yu, B.; Pasipanodya, E.; Montoya, J.L.; Moore, R.C.; Gianella, S.; McCutchan, A.; Ellis, R.; Heaton, R.K.; Jeste, D.V.; Moore, D.J.; et al. Metabolic Syndrome and Neurocognitive Deficits in HIV Infection. J. Acquir. Immune Defic. Syndr. 2019, 81, 95–101. [Google Scholar] [CrossRef]
- Ngo, C.; Garrec, C.; Tomasello, E.; Dalod, M. The role of plasmacytoid dendritic cells (pDCs) in immunity during viral infections and beyond. Cell. Mol. Immunol. 2024, 21, 1008–1035. [Google Scholar] [CrossRef]
- Moreno-Fernandez, M.E.; Presicce, P.; Chougnet, C.A. Homeostasis and function of regulatory T cells in HIV/SIV infection. J. Virol. 2012, 86, 10262–10269. [Google Scholar] [CrossRef] [PubMed]
- Rocco, J.; Mellors, J.W.; Macatangay, B.J. Regulatory T cells: The ultimate HIV reservoir? J. Virus Erad. 2018, 4, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Spudich, S. HIV and neurocognitive dysfunction. Curr. HIV/AIDS Rep. 2013, 10, 235–243. [Google Scholar] [CrossRef]
- Saikh, K.U.; Anam, K.; Sultana, H.; Ahmed, R.; Kumar, S.; Srinivasan, S.; Ahmed, H. Targeting Myeloid Differentiation Primary Response Protein 88 (MyD88) and Galectin-3 to Develop Broad-Spectrum Host-Mediated Therapeutics against SARS-CoV-2. Int. J. Mol. Sci. 2024, 25, 8421. [Google Scholar] [CrossRef]
- Meier, A.; Alter, G.; Frahm, N.; Sidhu, H.; Li, B.; Bagchi, A.; Teigen, N.; Streeck, H.; Stellbrink, H.J.; Hellman, J.; et al. MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded Toll-like receptor ligands. J. Virol. 2007, 81, 8180–8191. [Google Scholar] [CrossRef]
- Douek, D.C. Immune activation, HIV persistence, and the cure. Top. Antivir. Med. 2016, 21, 128. [Google Scholar]
- Wang, Q.; Shan, L. Role of the CARD8 inflammasome in HIV pathogenesis. Cell Insight 2024, 3, 100193. [Google Scholar] [CrossRef]
- Wang, Q.; Clark, K.M.; Tiwari, R.; Raju, N.; Tharp, G.K.; Rogers, J.; Harris, R.A.; Raveendran, M.; Bosinger, S.E.; Burdo, T.H.; et al. The CARD8 inflammasome dictates HIV/SIV pathogenesis and disease progression. Cell 2024, 187, 1223–1237.e1216. [Google Scholar] [CrossRef]
- Miller, E.; Bhardwaj, N. Dendritic cell dysregulation during HIV-1 infection. Immunol. Rev. 2013, 254, 170–189. [Google Scholar] [CrossRef]
- Scagnolari, C.; Antonelli, G. Type I interferon and HIV: Subtle balance between antiviral activity, immunopathogenesis and the microbiome. Cytokine Growth Factor Rev. 2018, 40, 19–31. [Google Scholar] [CrossRef]
- Prabakaran, P.; Dimitrov, A.S.; Fouts, T.R.; Dimitrov, D.S. Structure and function of the HIV envelope glycoprotein as entry mediator, vaccine immunogen, and target for inhibitors. Adv. Pharmacol. 2007, 55, 33–97. [Google Scholar] [CrossRef] [PubMed]
- Chen, B. Molecular Mechanism of HIV-1 Entry. Trends Microbiol. 2019, 27, 878–891. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Zhao, J.; Wang, A.; Viswanath, R.; Harma, H.; Little, R.F.; Yarchoan, R.; Stramer, S.L.; Nyambi, P.N.; Lee, S.; et al. Characterization of immune responses to capsid protein p24 of human immunodeficiency virus type 1 and implications for detection. Clin. Vaccine Immunol. 2010, 17, 124420131251. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Picado, J.; Zurakowski, R.; Buzon, M.J.; Stevenson, M. Episomal HIV-1 DNA and its relationship to other markers of HIV-1 persistence. Retrovirology 2018, 15, 15. [Google Scholar] [CrossRef]
- Yin, X.; Langer, S.; Zhang, Z.; Herbert, K.M.; Yoh, S.; Konig, R.; Chanda, S.K. Sensor Sensibility-HIV-1 and the Innate Immune Response. Cells 2020, 9, 254. [Google Scholar] [CrossRef]
- Kok, Y.L.; Vongrad, V.; Chaudron, S.E.; Shilaih, M.; Leemann, C.; Neumann, K.; Kusejko, K.; Di Giallonardo, F.; Kuster, H.; Braun, D.L.; et al. HIV-1 integration sites in CD4+ T cells during primary, chronic, and late presentation of HIV-1 infection. JCI Insight 2021, 6, e143940. [Google Scholar] [CrossRef]
- Tekeste, S.S.; Wilkinson, T.A.; Weiner, E.M.; Xu, X.; Miller, J.T.; Le Grice, S.F.; Clubb, R.T.; Chow, S.A. Interaction between Reverse Transcriptase and Integrase Is Required for Reverse Transcription during HIV-1 Replication. J. Virol. 2015, 89, 12058–12069. [Google Scholar] [CrossRef]
- Buffalo, C.Z.; Iwamoto, Y.; Hurley, J.H.; Ren, X. How HIV Nef Proteins Hijack Membrane Traffic to Promote Infection. J. Virol. 2019, 93, 10-1128. [Google Scholar] [CrossRef]
- Wonderlich, E.R.; Leonard, J.A.; Collins, K.L. HIV immune evasion disruption of antigen presentation by the HIV Nef protein. Adv. Virus Res. 2011, 80, 103–127. [Google Scholar] [CrossRef]
- Vaughan, S.; Jat, P.S. Deciphering the role of nuclear factor-kappaB in cellular senescence. Aging 2011, 3, 913–919. [Google Scholar] [CrossRef]
- Khan, I.A.; Worrad, A.H.; Singh, M.V.; Maggirwar, S.B.; Singh, V.B. Human immunodeficiency virus-1 Tat exerts its neurotoxic effects by downregulating Sonic hedgehog signaling. J. Neurovirol. 2022, 28, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.W.; Sereti, I. Residual immune dysfunction under antiretroviral therapy. Semin. Immunol. 2021, 51, 101471. [Google Scholar] [CrossRef] [PubMed]
- Nou, E.; Lo, J.; Grinspoon, S.K. Inflammation, immune activation, and cardiovascular disease in HIV. AIDS 2016, 30, 1495–1509. [Google Scholar] [CrossRef] [PubMed]
- Chavda, V.P.; Feehan, J.; Apostolopoulos, V. Inflammation: The Cause of All Diseases. Cells 2024, 13, 1906. [Google Scholar] [CrossRef]
- Mazzuti, L.; Turriziani, O.; Mezzaroma, I. The Many Faces of Immune Activation in HIV-1 Infection: A Multifactorial Interconnection. Biomedicines 2023, 11, 159. [Google Scholar] [CrossRef]
- Wiertsema, S.P.; van Bergenhenegouwen, J.; Garssen, J.; Knippels, L.M.J. The Interplay between the Gut Microbiome and the Immune System in the Context of Infectious Diseases throughout Life and the Role of Nutrition in Optimizing Treatment Strategies. Nutrients 2021, 13, 886. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Hunt, P.W.; Landay, A.L.; Sinclair, E.; Martinson, J.A.; Hatano, H.; Emu, B.; Norris, P.J.; Busch, M.P.; Martin, J.N.; Brooks, C.; et al. A low T regulatory cell response may contribute to both viral control and generalized immune activation in HIV controllers. PLoS ONE 2011, 6, e15924. [Google Scholar] [CrossRef]
- Caruso, M.P.; Falivene, J.; Holgado, M.P.; Zurita, D.H.; Laufer, N.; Castro, C.; Nico, A.; Maeto, C.; Salido, J.; Perez, H.; et al. Impact of HIV-ART on the restoration of Th17 and Treg cells in blood and female genital mucosa. Sci. Rep. 2019, 9, 1978. [Google Scholar] [CrossRef]
- Omenetti, S.; Pizarro, T.T. The Treg/Th17 Axis: A Dynamic Balance Regulated by the Gut Microbiome. Front. Immunol. 2015, 6, 639. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, X.; Wan, Y.Y. Intricacies of TGF-beta signaling in Treg and Th17 cell biology. Cell. Mol. Immunol. 2023, 20, 1002–1022. [Google Scholar] [CrossRef] [PubMed]
- Esmail Nia, G.; Mohammadi, M.; Sharifizadeh, M.; Ghalamfarsa, G.; Bolhassani, A. The role of T regulatory cells in the immunopathogenesis of HIV: Clinical implications. Braz. J. Infect. Dis. 2024, 28, 103866. [Google Scholar] [CrossRef] [PubMed]
- Atehortua, L.; Baig, M.; Morris, J.; Trentman, S.; Davidson, W.S.; Fichtenbaum, C.J.; Chougnet, C.A. Impaired response of memory Treg to high density lipoproteins is associated with intermediate/high cardiovascular disease risk in persons with HIV. Front. Immunol. 2023, 14, 1146624. [Google Scholar] [CrossRef] [PubMed]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder--pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef]
- Eggena, M.P.; Barugahare, B.; Jones, N.; Okello, M.; Mutalya, S.; Kityo, C.; Mugyenyi, P.; Cao, H. Depletion of regulatory T cells in HIV infection is associated with immune activation. J. Immunol. 2005, 174, 4407–4414. [Google Scholar] [CrossRef]
- Boasso, A. Type I interferon in HIV treatment: From antiviral drug to therapeutic target. HIV Ther. 2009, 3, 269–282. [Google Scholar] [CrossRef]
- Teer, E.; Mukonowenzou, N.C.; Essop, M.F. The Role of Sustained Type I Interferon Secretion in Chronic HIV Pathogenicity: Implications for Viral Persistence, Immune Activation, and Immunometabolism. Viruses 2025, 17, 139. [Google Scholar] [CrossRef]
- Kahan, S.M.; Wherry, E.J.; Zajac, A.J. T cell exhaustion during persistent viral infections. Virology 2015, 479–480, 180–193. [Google Scholar] [CrossRef]
- Sanjabi, S.; Zenewicz, L.A.; Kamanaka, M.; Flavell, R.A. Anti-inflammatory and pro-inflammatory roles of TGF-beta, IL-10, and IL-22 in immunity and autoimmunity. Curr. Opin. Pharmacol. 2009, 9, 447–453. [Google Scholar] [CrossRef]
- Nasi, M.; De Biasi, S.; Gibellini, L.; Bianchini, E.; Pecorini, S.; Bacca, V.; Guaraldi, G.; Mussini, C.; Pinti, M.; Cossarizza, A. Ageing and inflammation in patients with HIV infection. Clin. Exp. Immunol. 2017, 187, 44–52. [Google Scholar] [CrossRef]
- Angin, M.; Kwon, D.S.; Streeck, H.; Wen, F.; King, M.; Rezai, A.; Law, K.; Hongo, T.C.; Pyo, A.; Piechocka-Trocha, A.; et al. Preserved function of regulatory T cells in chronic HIV-1 infection despite decreased numbers in blood and tissue. J. Infect. Dis. 2012, 205, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Torres, C. HIV-associated cellular senescence: A contributor to accelerated aging. Ageing Res. Rev. 2017, 36, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.; Landay, A. Early immune senescence in HIV disease. Curr. HIV/AIDS Rep. 2010, 7, 4–10. [Google Scholar] [CrossRef]
- Soper, A.; Kimura, I.; Nagaoka, S.; Konno, Y.; Yamamoto, K.; Koyanagi, Y.; Sato, K. Type I Interferon Responses by HIV-1 Infection: Association with Disease Progression and Control. Front. Immunol. 2017, 8, 1823. [Google Scholar] [CrossRef]
- Apetroaei, M.-M.; Baliou, S.; Ioannou, P.; Fragkiadaki, P.; Ștefan, G.; Nedea, M.I.; Burcea-Dragomiroiu, G.-T.-A.; Velescu, B.Ș.; Docea, A.O.; Udeanu, D.I.; et al. The Hallmarks of Ageing in Human Immunodeficiency Virus Infection and the Impact of Antiretroviral Therapy on Telomeres: A Molecular Perspective. Curr. Issues Mol. Biol. 2025, 47, 273. [Google Scholar] [CrossRef]
- Wing, E.J. HIV and aging. Int. J. Infect. Dis. 2016, 53, 61–68. [Google Scholar] [CrossRef]
- Chi, H. Immunometabolism at the intersection of metabolic signaling, cell fate, and systems immunology. Cell. Mol. Immunol. 2022, 19, 299–302. [Google Scholar] [CrossRef]
- Teer, E.; Joseph, D.E.; Dominick, L.; Glashoff, R.H.; Essop, M.F. Expansion of GARP-Expressing CD4+CD25−FoxP3+ T Cells and SATB1 Association with Activation and Coagulation in Immune Compromised HIV-1-Infected Individuals in South Africa. Virol. Sin. 2021, 36, 1133–1143. [Google Scholar] [CrossRef]
- Chan, Y.T.; Cheong, H.C.; Tang, T.F.; Rajasuriar, R.; Cheng, K.K.; Looi, C.Y.; Wong, W.F.; Kamarulzaman, A. Immune Checkpoint Molecules and Glucose Metabolism in HIV-Induced T Cell Exhaustion. Biomedicines 2022, 10, 2809. [Google Scholar] [CrossRef]
- Palmer, C.S.; Cherry, C.L.; Sada-Ovalle, I.; Singh, A.; Crowe, S.M. Glucose Metabolism in T Cells and Monocytes: New Perspectives in HIV Pathogenesis. eBioMedicine 2016, 6, 31–41. [Google Scholar] [CrossRef]
- Yi, J.S.; Cox, M.A.; Zajac, A.J. T-cell exhaustion: Characteristics, causes and conversion. Immunology 2010, 129, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [PubMed]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef]
- Fenwick, C.; Joo, V.; Jacquier, P.; Noto, A.; Banga, R.; Perreau, M.; Pantaleo, G. T-cell exhaustion in HIV infection. Immunol. Rev. 2019, 292, 149–163. [Google Scholar] [CrossRef]
- Khaitan, A.; Unutmaz, D. Revisiting immune exhaustion during HIV infection. Curr. HIV/AIDS Rep. 2011, 8, 4–11. [Google Scholar] [CrossRef]
- Alzahrani, J.; Hussain, T.; Simar, D.; Palchaudhuri, R.; Abdel-Mohsen, M.; Crowe, S.M.; Mbogo, G.W.; Palmer, C.S. Inflammatory and immunometabolic consequences of gut dysfunction in HIV: Parallels with IBD and implications for reservoir persistence and non-AIDS comorbidities. eBioMedicine 2019, 46, 522–531. [Google Scholar] [CrossRef]
- Soto-Heredero, G.; Gomez de Las Heras, M.M.; Gabande-Rodriguez, E.; Oller, J.; Mittelbrunn, M. Glycolysis—A key player in the inflammatory response. FEBS J. 2020, 287, 3350–3369. [Google Scholar] [CrossRef]
- Butterfield, T.R.; Landay, A.L.; Anzinger, J.J. Dysfunctional Immunometabolism in HIV Infection: Contributing Factors and Implications for Age-Related Comorbid Diseases. Curr. HIV/AIDS Rep. 2020, 17, 125–137. [Google Scholar] [CrossRef]
- Rohm, T.V.; Meier, D.T.; Olefsky, J.M.; Donath, M.Y. Inflammation in obesity, diabetes, and related disorders. Immunity 2022, 55, 31–55. [Google Scholar] [CrossRef]
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216. [Google Scholar] [CrossRef]
- Ahmed, D.; Roy, D.; Cassol, E. Examining Relationships between Metabolism and Persistent Inflammation in HIV Patients on Antiretroviral Therapy. Mediat. Inflamm. 2018, 2018, 6238978. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, R.; Varacallo, M.A. Biochemistry, Glycolysis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Boutens, L.; Hooiveld, G.J.; Dhingra, S.; Cramer, R.A.; Netea, M.G.; Stienstra, R. Unique metabolic activation of adipose tissue macrophages in obesity promotes inflammatory responses. Diabetologia 2018, 61, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Soedono, S.; Julietta, V.; Nawaz, H.; Cho, K.W. Dynamic Roles and Expanding Diversity of Adipose Tissue Macrophages in Obesity. J. Obes. Metab. Syndr. 2024, 33, 193–212. [Google Scholar] [CrossRef]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef]
- Hu, T.; Liu, C.H.; Lei, M.; Zeng, Q.; Li, L.; Tang, H.; Zhang, N. Metabolic regulation of the immune system in health and diseases: Mechanisms and interventions. Signal Transduct. Target. Ther. 2024, 9, 268. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Amengual, J.; Barrett, T.J. Monocytes and macrophages in atherogenesis. Curr. Opin. Lipidol. 2019, 30, 401–408. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Meyers, A.K.; Zhu, X. The NLRP3 Inflammasome: Metabolic Regulation and Contribution to Inflammaging. Cells 2020, 9, 1808. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brule, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; An, X.; Yang, C.; Sun, W.; Ji, H.; Lian, F. The crucial role and mechanism of insulin resistance in metabolic disease. Front. Endocrinol. 2023, 14, 1149239. [Google Scholar] [CrossRef] [PubMed]
- Dirajlal-Fargo, S.; Funderburg, N. HIV and cardiovascular disease: The role of inflammation. Curr. Opin. HIV AIDS 2022, 17, 286–292. [Google Scholar] [CrossRef]
- Mandal, N.; Grambergs, R.; Mondal, K.; Basu, S.K.; Tahia, F.; Dagogo-Jack, S. Role of ceramides in the pathogenesis of diabetes mellitus and its complications. J. Diabetes Complicat. 2021, 35, 107734. [Google Scholar] [CrossRef]
- Funderburg, N.T.; Mehta, N.N. Lipid Abnormalities and Inflammation in HIV Inflection. Curr. HIV/AIDS Rep. 2016, 13, 218–225. [Google Scholar] [CrossRef]
- Zhang, J.; Jin, H.L.; Jian, F.B.; Feng, S.L.; Zhu, W.T.; Li, L.H.; Yuan, Z.W. Evaluation of lipid metabolism imbalance in HIV-infected patients with metabolic disorders using high-performance liquid chromatography-tandem mass spectrometry. Clin. Chim. Acta 2022, 526, 30–42. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Douek, D.C. HIV infection and the gastrointestinal immune system. Mucosal Immunol. 2008, 1, 23–30. [Google Scholar] [CrossRef]
- Moretti, S.; Schietroma, I.; Sberna, G.; Maggiorella, M.T.; Sernicola, L.; Farcomeni, S.; Giovanetti, M.; Ciccozzi, M.; Borsetti, A. HIV-1-Host Interaction in Gut-Associated Lymphoid Tissue (GALT): Effects on Local Environment and Comorbidities. Int. J. Mol. Sci. 2023, 24, 12193. [Google Scholar] [CrossRef]
- Sandler, N.G.; Douek, D.C. Microbial translocation in HIV infection: Causes, consequences and treatment opportunities. Nat. Rev. Microbiol. 2012, 10, 655–666. [Google Scholar] [CrossRef]
- Zevin, A.S.; McKinnon, L.; Burgener, A.; Klatt, N.R. Microbial translocation and microbiome dysbiosis in HIV-associated immune activation. Curr. Opin. HIV AIDS 2016, 11, 182–190. [Google Scholar] [CrossRef] [PubMed]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef] [PubMed]
- Sereti, I.; Verburgh, M.L.; Gifford, J.; Lo, A.; Boyd, A.; Verheij, E.; Verhoeven, A.; Wit, F.; Schim van der Loeff, M.F.; Giera, M.; et al. Impaired gut microbiota-mediated short-chain fatty acid production precedes morbidity and mortality in people with HIV. Cell Rep. 2023, 42, 113336. [Google Scholar] [CrossRef] [PubMed]
- Teer, E.; Dominick, L.; Mukonowenzou, N.C.; Essop, M.F. HIV-Related Myocardial Fibrosis: Inflammatory Hypothesis and Crucial Role of Immune Cells Dysregulation. Cells 2022, 11, 2825. [Google Scholar] [CrossRef]
- Teer, E.; Mukonowenzou, N.C.; Essop, M.F. The Role of Immunometabolism in HIV-1 Pathogenicity: Links to Immune Cell Responses. Viruses 2022, 14, 1813. [Google Scholar] [CrossRef]
- Teer, E.; Essop, M.F. HIV and Cardiovascular Disease: Role of Immunometabolic Perturbations. Physiology 2018, 33, 74–82. [Google Scholar] [CrossRef]
- Dominick, L.L. An Evaluation of the Role of Platelet Activation in HIV-Related Cardiovascular Diseases Onset. Master’s Thesis, Stellenbosch University, Stellenbosch, South Africa, 2021. [Google Scholar]
- Teer, E.; Joseph, D.E.; Driescher, N.; Nell, T.A.; Dominick, L.; Midgley, N.; Deshpande, G.; Page, M.J.; Pretorius, E.; Woudberg, N.J.; et al. HIV and cardiovascular diseases risk: Exploring the interplay between T-cell activation, coagulation, monocyte subsets, and lipid subclass alterations. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1146–H1157. [Google Scholar] [CrossRef]
- MohanKumar, S.M.J.; Murugan, A.; Palaniyappan, A.; MohanKumar, P.S. Role of cytokines and reactive oxygen species in brain aging. Mech. Ageing Dev. 2023, 214, 111855. [Google Scholar] [CrossRef]
- Joseph, S.B.; Gianella, S.; Burdo, T.H.; Cinque, P.; Gisslen, M.; Letendre, S.; Nath, A.; Morgello, S.; Ndhlovu, L.C.; Spudich, S. Biotypes of Central Nervous System Complications in People with Human Immunodeficiency Virus: Virology, Immunology, and Neuropathology. J. Infect. Dis. 2023, 227, S3–S15. [Google Scholar] [CrossRef]
- Lopez-Atalaya, J.P.; Bhojwani-Cabrera, A.M. Type I interferon signalling and interferon-responsive microglia in health and disease. FEBS J. 2025, 1–20. [Google Scholar] [CrossRef]
- Sil, S.; Periyasamy, P.; Thangaraj, A.; Niu, F.; Chemparathy, D.T.; Buch, S. Advances in the Experimental Models of HIV-Associated Neurological Disorders. Curr. HIV/AIDS Rep. 2021, 18, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Fert, A.; Richard, J.; Marchand, L.R.; Planas, D.; Routy, J.P.; Chomont, N.; Finzi, A.; Ancuta, P. Metformin Enhances Antibody-Mediated Recognition of HIV-Infected CD4+ T-Cells by Decreasing Viral Release. bioRxiv 2024. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, Z.; Sun, H.; Ouyang, X.; Han, Y.; Yu, H.; Wu, N.; Xie, Y.; Su, B. Regulation of CD8+ T memory and exhaustion by the mTOR signals. Cell. Mol. Immunol. 2023, 20, 1023–1039. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Fitch, K.V.; Fulda, E.S.; Grinspoon, S.K. Statins for primary cardiovascular disease prevention among people with HIV: Emergent directions. Curr. Opin. HIV AIDS 2022, 17, 293–300. [Google Scholar] [CrossRef]
- Zhou, Q.; Liao, J.K. Statins and cardiovascular diseases: From cholesterol lowering to pleiotropy. Curr. Pharm. Des. 2009, 15, 467–478. [Google Scholar] [CrossRef]
- Eckard, A.R.; McComsey, G.A. The role of statins in the setting of HIV infection. Curr. HIV/AIDS Rep. 2015, 12, 305–312. [Google Scholar] [CrossRef]
- Zivkovic, S.; Maric, G.; Cvetinovic, N.; Lepojevic-Stefanovic, D.; Bozic Cvijan, B. Anti-Inflammatory Effects of Lipid-Lowering Drugs and Supplements-A Narrative Review. Nutrients 2023, 15, 1517. [Google Scholar] [CrossRef]
- Grinspoon, S.K.; Fitch, K.V.; Zanni, M.V.; Fichtenbaum, C.J.; Umbleja, T.; Aberg, J.A.; Overton, E.T.; Malvestutto, C.D.; Bloomfield, G.S.; Currier, J.S.; et al. Pitavastatin to Prevent Cardiovascular Disease in HIV Infection. N. Engl. J. Med. 2023, 389, 687–699. [Google Scholar] [CrossRef]
- d’Ettorre, G.; Ceccarelli, G.; Giustini, N.; Serafino, S.; Calantone, N.; De Girolamo, G.; Bianchi, L.; Bellelli, V.; Ascoli-Bartoli, T.; Marcellini, S.; et al. Probiotics Reduce Inflammation in Antiretroviral Treated, HIV-Infected Individuals: Results of the “Probio-HIV” Clinical Trial. PLoS ONE 2015, 10, e0137200. [Google Scholar] [CrossRef]
- Al-Habsi, N.; Al-Khalili, M.; Haque, S.A.; Elias, M.; Olqi, N.A.; Al Uraimi, T. Health Benefits of Prebiotics, Probiotics, Synbiotics, and Postbiotics. Nutrients 2024, 16, 3955. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, C.; Reale, M.; Costantini, E. Microbiota and Probiotics in Health and HIV Infection. Nutrients 2017, 9, 615. [Google Scholar] [CrossRef]
 : oxidative phosphorylation; and
: oxidative phosphorylation; and  : glycolysis. Image created using Biorender.
: oxidative phosphorylation; and : glycolysis. Image created using Biorender.
: glycolysis. Image created using Biorender.
: oxidative phosphorylation; and : glycolysis. Image created using Biorender.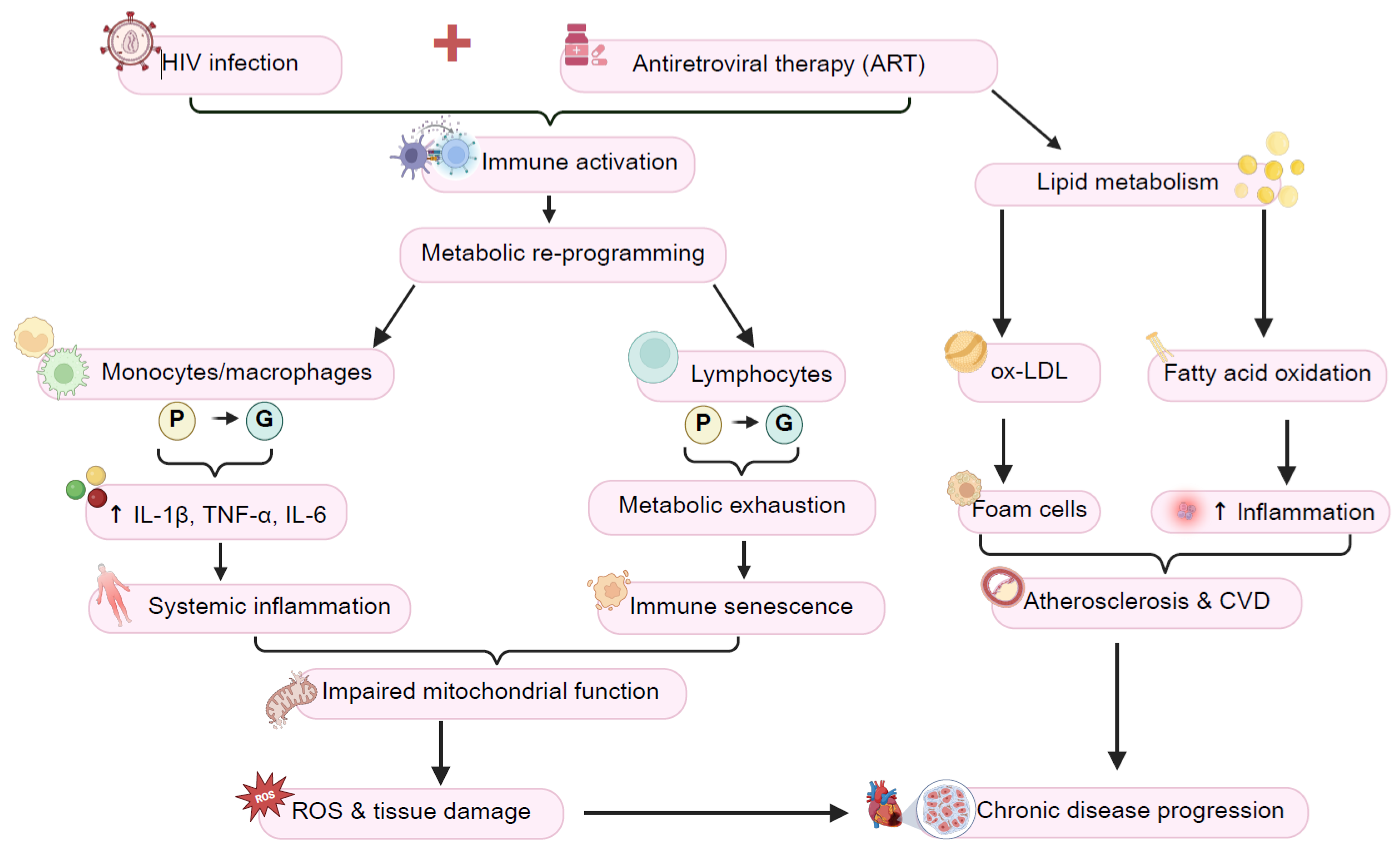
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teer, E.; Mukonowenzou, N.C.; Essop, M.F. HIV, Inflammation, and Immunometabolism: A Model of the Inflammatory Theory of Disease. Viruses 2025, 17, 839. https://doi.org/10.3390/v17060839
Teer E, Mukonowenzou NC, Essop MF. HIV, Inflammation, and Immunometabolism: A Model of the Inflammatory Theory of Disease. Viruses. 2025; 17(6):839. https://doi.org/10.3390/v17060839
Chicago/Turabian StyleTeer, Eman, Nyasha C. Mukonowenzou, and M. Faadiel Essop. 2025. "HIV, Inflammation, and Immunometabolism: A Model of the Inflammatory Theory of Disease" Viruses 17, no. 6: 839. https://doi.org/10.3390/v17060839
APA StyleTeer, E., Mukonowenzou, N. C., & Essop, M. F. (2025). HIV, Inflammation, and Immunometabolism: A Model of the Inflammatory Theory of Disease. Viruses, 17(6), 839. https://doi.org/10.3390/v17060839