
Discovery of a Rodent Hepacivirus in the Brazilian Amazon


 , ,
, ,  , , ,
, , ,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
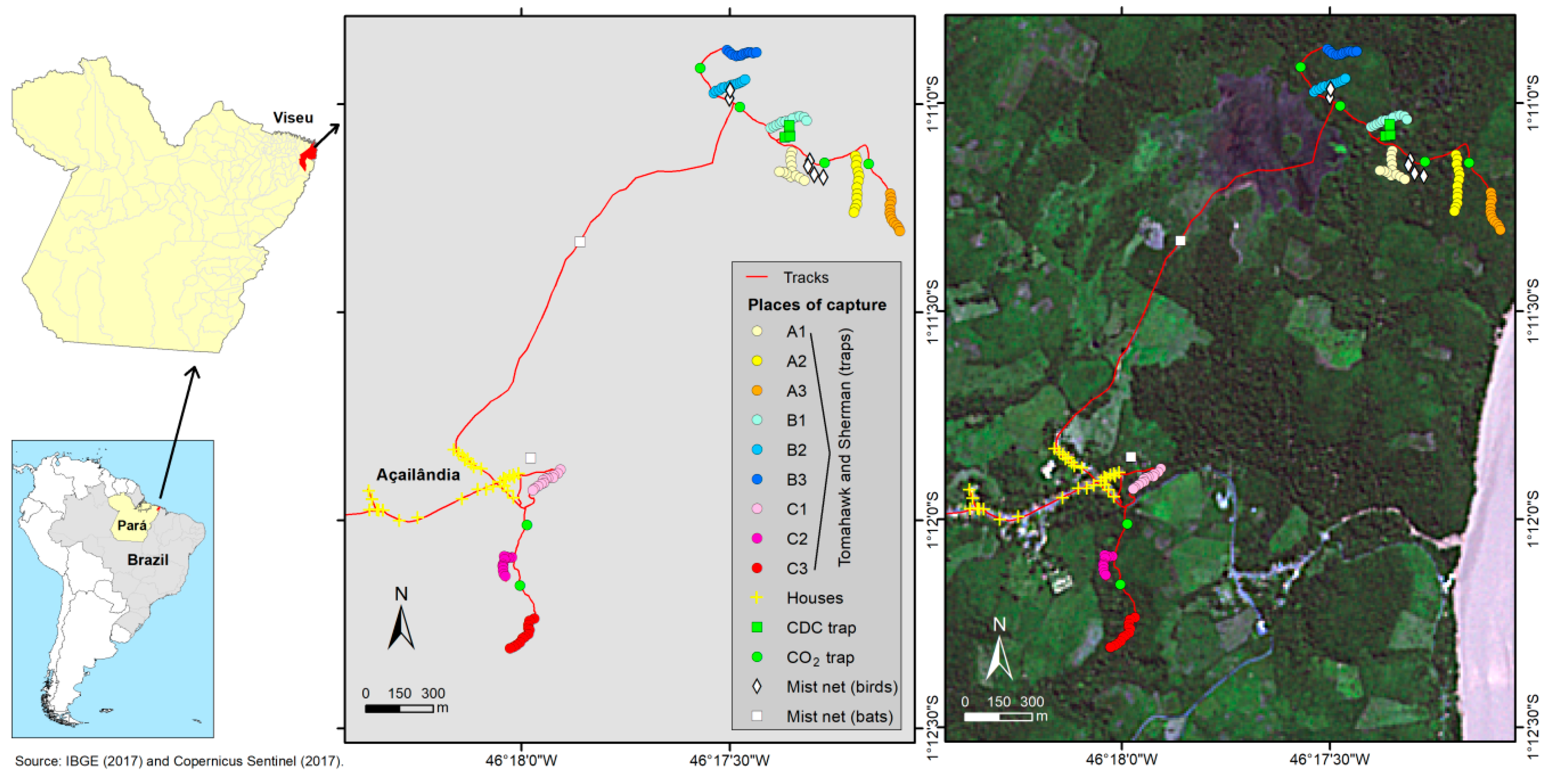
2.1. Sample Collection
2.2. RNA Extraction
2.3. cDNA Synthesis, Library Preparation, and Sequencing
2.4. Bioinformatic Analysis
2.4.1. Quality Control
2.4.2. Viral Diversity Assessment
2.4.3. Sequence Assembly and Contig Inspection
2.4.4. Phylogenetic Analysis
3. Results
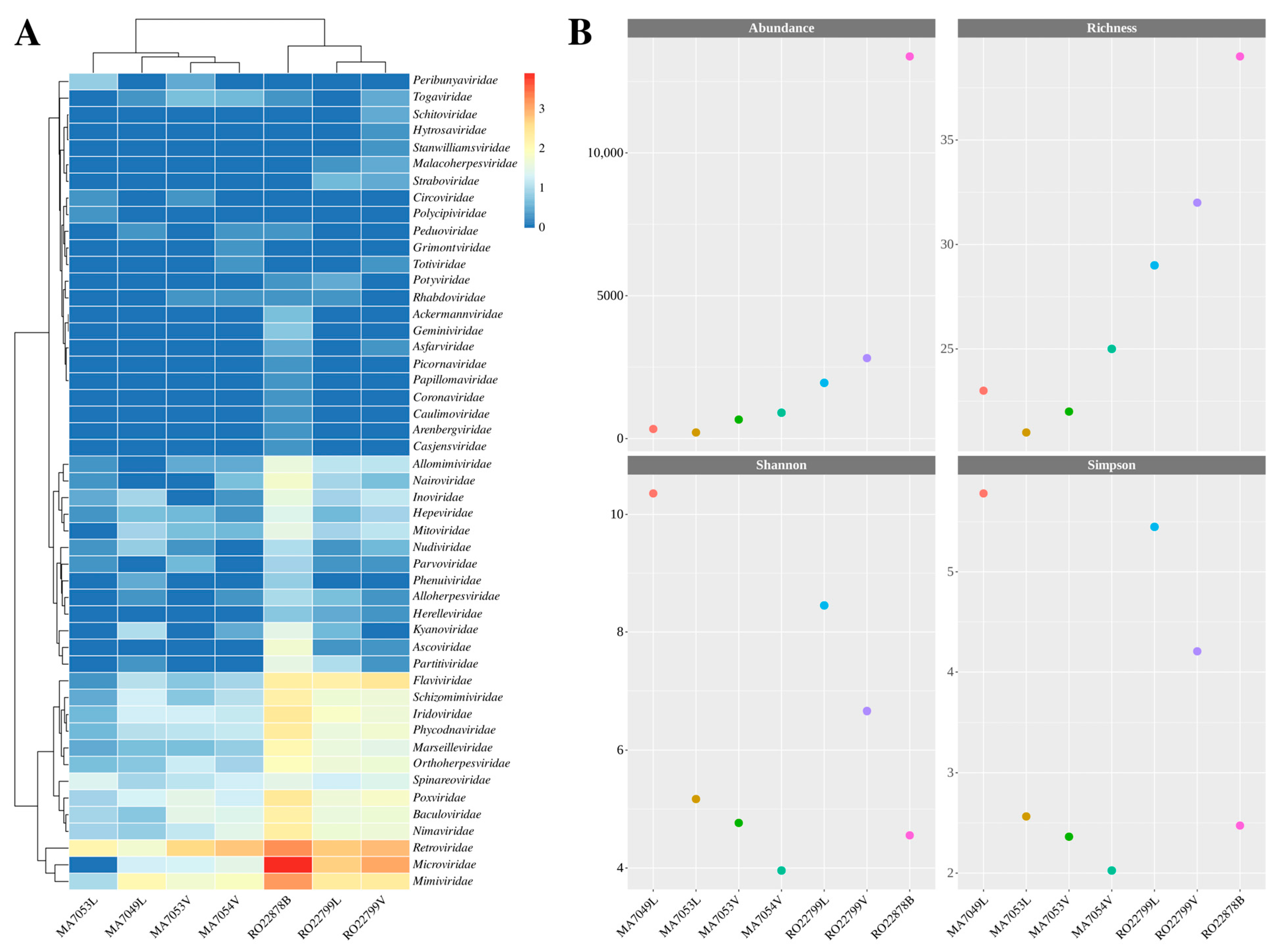
3.1. Data Processing and Viral Diversity
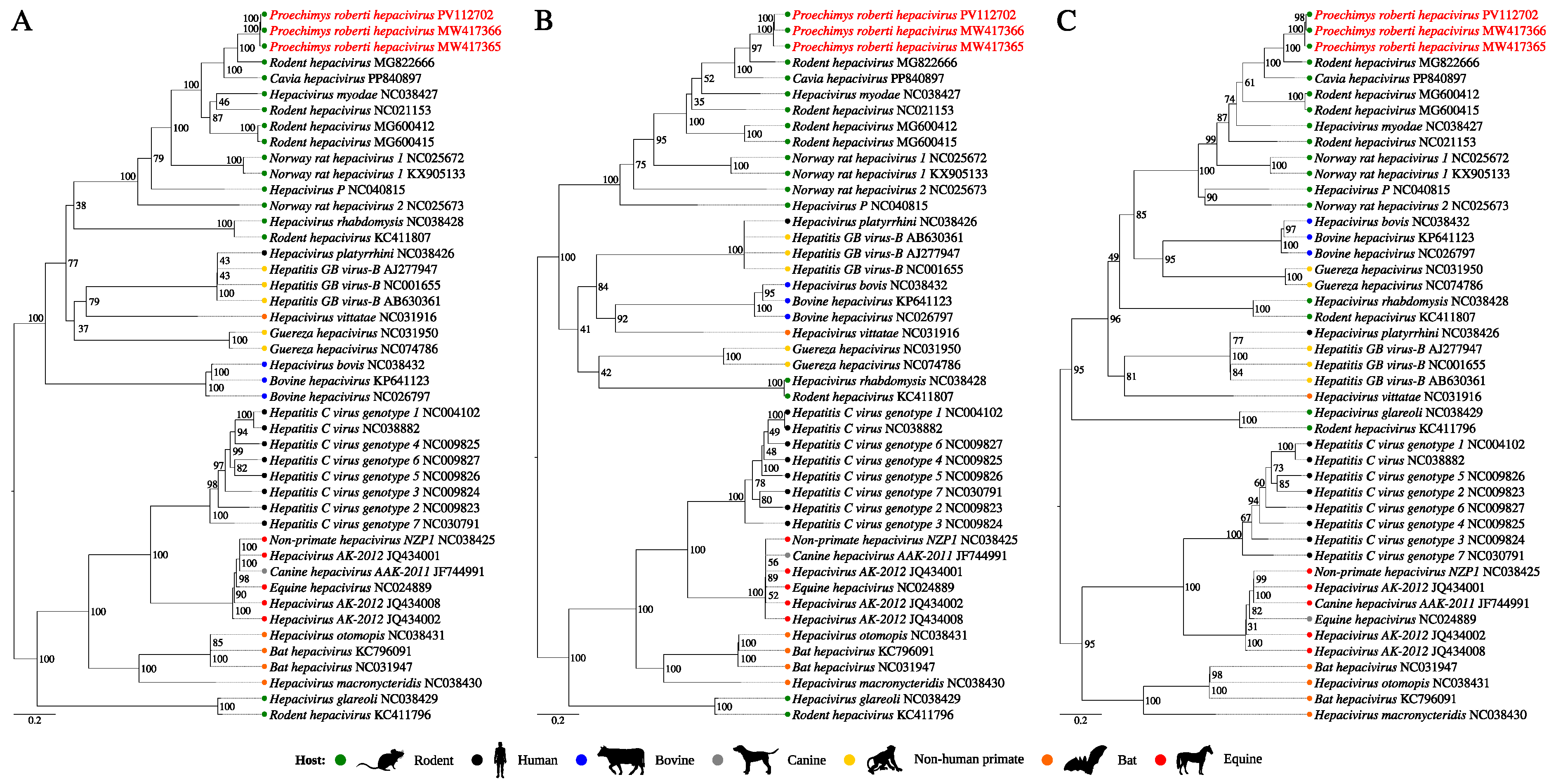
3.2. Inspection of Hepacivirus Sequences
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global Trends in Emerging Infectious Diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Hou, Y.; Li, Q.; Dian, Z.; Wang, B.; Xia, X. RNA virus diversity in rodents. Arch. Microbiol. 2024, 206, 9. [Google Scholar] [CrossRef]
- Williams, E.P.; Spruill-Harrell, B.M.; Taylor, M.K.; Lee, J.; Nywening, A.V.; Yang, Z.; Nichols, J.H.; Camp, J.V.; Owen, R.D.; Jonsson, C.B. Common Themes in Zoonotic Spillover and Disease Emergence: Lessons Learned from Bat- and Rodent-Borne RNA Viruses. Viruses 2021, 13, 1509. [Google Scholar] [CrossRef]
- Li, Y.; Tang, C.; Zhang, Y.; Li, Z.; Wang, G.; Peng, R.; Huang, Y.; Hu, X.; Xin, H.; Feng, B.; et al. Diversity and independent evolutionary profiling of rodent-borne viruses in Hainan, a tropical island of China. Virol. Sin. 2023, 38, 651–662. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses. Family: Flaviviridae. Genus: Hepacivirus. Available online: https://ictv.global/report/chapter/flaviviridae/flaviviridae/hepacivirus (accessed on 11 January 2025).
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef]
- Kapoor, A.; Simmonds, P.; Gerold, G.; Qaisar, N.; Jain, K.; Henriquez, J.A.; Firth, C.; Hirschberg, D.L.; Rice, C.M.; Shields, S.; et al. Characterization of a Canine Homolog of Hepatitis C Virus. Proc. Natl. Acad. Sci. USA 2011, 108, 11608–11613. [Google Scholar] [CrossRef]
- Quan, P.-L.; Firth, C.; Conte, J.M.; Williams, S.H.; Zambrana-Torrelio, C.M.; Anthony, S.J.; Ellison, J.A.; Gilbert, A.T.; Kuzmin, I.V.; Niezgoda, M.; et al. Bats Are a Major Natural Reservoir for Hepaciviruses and Pegiviruses. Proc. Natl. Acad. Sci. USA 2013, 110, 8194–8199. [Google Scholar] [CrossRef]
- Pybus, O.G.; Thézé, J. Hepacivirus cross-species transmission and the origins of the hepatitis C virus. Curr. Opin. Virol. 2016, 16, 1–7. [Google Scholar] [CrossRef]
- Li, Y.Q.; Ghafari, M.; Holbrook, A.J.; Boonen, I.; Amor, N.; Catalano, S.; Webster, J.P.; Li, Y.Y.; Li, H.T.; Vergote, V.; et al. The evolutionary history of hepaciviruses. bioRxiv 2023. [Google Scholar] [CrossRef]
- Brazilian National Institute for Space Research. Legal Amazon Deforestation Rates. Available online: https://terrabrasilis.dpi.inpe.br/app/dashboard/deforestation/biomes/legal_amazon/increments (accessed on 21 January 2025).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and Accurate Filtering of Ribosomal RNAs in Metatranscriptomic Data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive Protein Alignments at Tree-of-Life Scale Using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 12 January 2025).
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. InterPro in 2022. Nucleic Acids Res. 2023, 51, D418–D427. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.-I.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 13 January 2025).
- Inkscape. Available online: https://inkscape.org/release/inkscape-1.1/ (accessed on 13 January 2025).
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Gibb, R.; Redding, D.W.; Friant, S.; Jones, K.E. Towards a ‘people and nature’ paradigm for biodiversity and infectious disease. Philos. Trans. R Soc. Lond. B Biol. Sci. 2025, 380, 20230259. [Google Scholar] [CrossRef]
- Ellwanger, J.H.; Kulmann-Leal, B.; Kaminski, V.L.; Valverde-Villegas, J.M.; Veiga, A.B.G.D.; Spilki, F.R.; Fearnside, P.M.; Caesar, L.; Giatti, L.L.; Wallau, G.L.; et al. Beyond diversity loss and climate change: Impacts of Amazon deforestation on infectious diseases and public health. An. Acad. Bras. Cienc. 2020, 92, e20191375. [Google Scholar] [CrossRef] [PubMed]
- French, R.K.; Anderson, S.; Cain, K.; Digby, A.; Greene, T.C.; Miskelly, C.M.; Muller, C.G.; Taylor, M.W.; Recovery Team, K.; Geoghegan, J.L.; et al. Diversity and cross-species transmission of viruses in a remote island ecosystem: Implications for wildlife conservation. Virus Evol. 2024, 11, veae113. [Google Scholar] [CrossRef] [PubMed]
- Van Brussel, K.; Holmes, E.C. Zoonotic disease and virome diversity in bats. Curr. Opin. Virol. 2021, 52, 192–202. [Google Scholar] [CrossRef]
- Wu, Z.; Lu, L.; Du, J.; Yang, L.; Ren, X.; Liu, B.; Jiang, J.; Yang, J.; Dong, J.; Sun, L.; et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 2018, 6, 178. [Google Scholar] [CrossRef]
- He, X.; Wang, X.; Fan, G.; Li, F.; Wu, W.; Wang, Z.; Fu, M.; Wei, X.; Ma, S.; Ma, X. Metagenomic analysis of viromes in tissues of wild Qinghai vole from the eastern Tibetan Plateau. Sci. Rep. 2022, 12, 17239. [Google Scholar] [CrossRef]
- Monteiro, A.F.M.; da Silva, F.S.; Cruz, A.C.R.; da Silva, S.P.; Queiroz, A.L.N.; Casseb, L.M.N.; Martins, L.C.; Medeiros, D.B.A. Viral diversity in wild rodents in the regions of Canaã de Carajás and Curionopólis, State of Pará, Brazil. Front. Microbiol. 2025, 15, 1502462. [Google Scholar] [CrossRef]
- Ritsch, M.; Brait, N.; Harvey, E.; Marz, M.; Lequime, S. Endogenous viral elements: Insights into data availability and accessibility. Virus Evol. 2024, 10, veae099. [Google Scholar] [CrossRef]
- Kirchberger, P.C.; Martinez, Z.A.; Ochman, H. Organizing the Global Diversity of Microviruses. mBio 2022, 13, e0058822. [Google Scholar] [CrossRef]
- Kalafati, E.; Papanikolaou, E.; Marinos, E.; Anagnou, N.P.; Pappa, K.I. Mimiviruses: Giant viruses with novel and intriguing features (Review). Mol. Med. Rep. 2022, 25, 207. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Lukashev, A.N.; Gmyl, A.; Coutard, B.; Adam, A.; Ritz, D.; Leijten, L.M.; van Riel, D.; et al. Evidence for Novel Hepaciviruses in Rodents. PLoS Pathog. 2013, 9, e1003438. [Google Scholar] [CrossRef]
- Hartlage, A.S.; Cullen, J.M.; Kapoor, A. The Strange, Expanding World of Animal Hepaciviruses. Annu. Rev. Virol. 2016, 3, 53–75. [Google Scholar] [CrossRef] [PubMed]
- Schmid, J.; Rasche, A.; Eibner, G.; Jeworowski, L.; Page, R.A.; Corman, V.M.; Drosten, C.; Sommer, S. Ecological drivers of Hepacivirus infection in a neotropical rodent inhabiting landscapes with various degrees of human environmental change. Oecologia 2018, 188, 289–302. [Google Scholar] [CrossRef] [PubMed]
- de Souza, W.M.; Fumagalli, M.J.; Sabino-Santos, G., Jr.; Motta Maia, F.G.; Modha, S.; Teixeira Nunes, M.R.; Murcia, P.R.; Moraes Figueiredo, L.T. A Novel Hepacivirus in Wild Rodents from South America. Viruses 2019, 11, 297. [Google Scholar] [CrossRef] [PubMed]
- Birlem, G.E.; Sita, A.; Gularte, J.S.; de Souza da Silva, D.; Demoliner, M.; de Almeida, P.R.; Fleck, J.D.; Spilki, F.R.; Dos Santos Higino, S.S.; de Azevedo, S.S.; et al. Detection of a novel hepacivirus in wild cavies (Cavia aperea aperea). Arch. Virol. 2024, 170, 19. [Google Scholar] [CrossRef]
- Wolfisberg, R.; Holmbeck, K.; Billerbeck, E.; Thorselius, C.E.; Batista, M.N.; Fahnøe, U.; Lundsgaard, E.A.; Kennedy, M.J.; Nielsen, L.; Rice, C.M.; et al. Molecular Determinants of Mouse Adaptation of Rat Hepacivirus. J. Virol. 2023, 97, e0181222. [Google Scholar] [CrossRef]
- Li, L.; Giannitti, F.; Low, J.; Keyes, C.; Ullmann, L.S.; Deng, X.; Aleman, M.; Pesavento, P.A.; Pusterla, N.; Delwart, E. Exploring the virome of diseased horses. J. Gen. Virol. 2015, 96, 2721–2733. [Google Scholar] [CrossRef]
- Altan, E.; Li, Y.; Sabino-Santos, G., Jr.; Sawaswong, V.; Barnum, S.; Pusterla, N.; Deng, X.; Delwart, E. Viruses in Horses with Neurologic and Respiratory Diseases. Viruses 2019, 11, 942. [Google Scholar] [CrossRef]
- Weksler, M.; Bonvicino, C.R.; Otazu, I.B.; Silva, J.S. Status of Proechimys roberti and P. oris (Rodentia: Echimyidae) from Eastern Amazonia and Central Brazil. J. Mammal. 2001, 82, 109–122. [Google Scholar] [CrossRef]
- Patton, J.L.; Leite, R.N. Genus Proechimys. In Mammals of South America; Patton, J.L., Pardiñas, U.F.J., D’Elía, G., Eds.; The University of Chicago Press: Chicago, IL, USA, 2015; Volume 2, pp. 950–989. [Google Scholar]
- Miranda, C.L.; Silva, M.N.F. Roedores equimídeos da Amazônia brasileira: Composição, distribuição geográfica e diagnoses. In Pequenos Mamíferos Não-Voadores da Amazônia Brasileira; Mendes-Oliveira, A.C., Miranda, C.L., Eds.; Sociedade Brasileira de Mastozoologia: Rio de Janeiro, Brazil, 2015; pp. 187–212. [Google Scholar]
- Cordeiro, H.C.; Melo, F.T.; Furtado, A.P.; Giese, E.G.; Maldonado, A., Jr.; dos Santos, J.N. Squamasnema amazonica n. gen. n. sp. (Heligmonellinae): A new parasite of Proechimys roberti (Rodentia: Echimyidae) in the Brazilian Amazon. Acta Trop. 2015, 148, 46–50. [Google Scholar] [CrossRef]
- Serrano, P.C.; Durette-Desset, M.C.; Digiani, M.C. Pudicinae (Nematoda) coparasitic in Proechimys roberti (Rodentia: Echimyidae) from the Brazilian Amazonia: Description of a new species of Pudica, redescription of Pudica evandroi (Travassos) and updated key to the species of the genus. An. Acad. Bras. Cienc. 2019, 91, e20180714. [Google Scholar] [CrossRef]
- Lima, M.F.; Silvestre, M.D.P.S.A.; Santos, E.C.D.; Martins, L.C.; Quaresma, J.A.S.; de Barros, B.C.V.; Silva, M.J.A.; Lima, L.N.G.C. The Presence of Mycobacterium leprae in Wild Rodents. Microorganisms 2022, 10, 1114. [Google Scholar] [CrossRef]
- Anggakusuma; Brown, R.J.P.; Banda, D.H.; Todt, D.; Vieyres, G.; Steinmann, E.; Pietschmann, T. Hepacivirus NS3/4A Proteases Interfere with MAVS Signaling in both Their Cognate Animal Hosts and Humans: Implications for Zoonotic Transmission. J. Virol. 2016, 90, 10670–10681. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Host Species | Collection Date | Total Reads | Fastp | SortMeRNA | Removed | Validated (%) |
|---|---|---|---|---|---|---|---|
| MA7049L 1 | Marmosa murina | 27 November 2014 | 23,083,368 | 20,080,020 | 1,155,010 | 21,928,358 | 5.0 |
| MA7053L | Philander opossum | 27 November 2014 | 34,585,464 | 30,206,256 | 1,052,840 | 33,532,624 | 3.0 |
| MA7053V 2 | 38,751,852 | 34,305,968 | 2,507,726 | 36,244,126 | 6.5 | ||
| MA7054V | M. demerarae | 27 November 2014 | 33,483,380 | 29,036,538 | 6,645,268 | 26,838,112 | 19.8 |
| RO22799L | Proechimys roberti | 28 November 2014 | 29,624,298 | 25,665,772 | 927,080 | 28,697,218 | 3.1 |
| RO22799V | 38,632,322 | 33,938,920 | 1,602,564 | 37,029,758 | 4.1 | ||
| RO22878B 3 | P. roberti | 20 August 2015 | 33,642,686 | 29,172,270 | 3,652,558 | 29,990,128 | 10.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prestes, N.G.O.; Hernández, L.H.A.; da Silva, F.S.; da Paz, T.Y.B.; Aragão, A.O.; de Barros, B.C.V.; Guimarães, R.J.P.S.; Ramos, R.T.J.; Casseb, L.M.N.; da Silva, S.P.; et al. Discovery of a Rodent Hepacivirus in the Brazilian Amazon. Viruses 2025, 17, 830. https://doi.org/10.3390/v17060830
Prestes NGO, Hernández LHA, da Silva FS, da Paz TYB, Aragão AO, de Barros BCV, Guimarães RJPS, Ramos RTJ, Casseb LMN, da Silva SP, et al. Discovery of a Rodent Hepacivirus in the Brazilian Amazon. Viruses. 2025; 17(6):830. https://doi.org/10.3390/v17060830
Chicago/Turabian StylePrestes, Nelielma G. Oliveira, Leonardo H. Almeida Hernández, Fábio Silva da Silva, Thito Y. Bezerra da Paz, Andressa O. Aragão, Bruno C. Veloso de Barros, Ricardo J. P. S. Guimarães, Rommel T. J. Ramos, Lívia Medeiros Neves Casseb, Sandro Patroca da Silva, and et al. 2025. "Discovery of a Rodent Hepacivirus in the Brazilian Amazon" Viruses 17, no. 6: 830. https://doi.org/10.3390/v17060830
APA StylePrestes, N. G. O., Hernández, L. H. A., da Silva, F. S., da Paz, T. Y. B., Aragão, A. O., de Barros, B. C. V., Guimarães, R. J. P. S., Ramos, R. T. J., Casseb, L. M. N., da Silva, S. P., Vasconcelos, P. F. d. C., & Cruz, A. C. R. (2025). Discovery of a Rodent Hepacivirus in the Brazilian Amazon. Viruses, 17(6), 830. https://doi.org/10.3390/v17060830