
A Versatile Reporter Platform for Evaluating HDR- and NHEJ-Based Genome Editing in Airway Epithelial Cell Cultures Using an rAAV Vector


 , , ,
, , ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Lentiviral Vectors Production
2.2. rAAV Vector Production
2.3. PiggyBac (PB) Transfer Plasmid and Transposase Expression Plasmid
2.4. Cell Cultures
2.5. Lentiviral Vector Transduction
2.6. rAAV Vector Transduction
2.7. Flow Cytometry Analysis
3. Results
3.1. Homology-Independent Targeted Insertion in Primary Ferret Airway Epithelial Cultures
3.2. Generation of Gene Editing Reporter Cell Line Derived from CuFi-8 Cells
3.3. Evaluation of Homology-Independent Targeted Integration in CuFi-GER Cell Lines
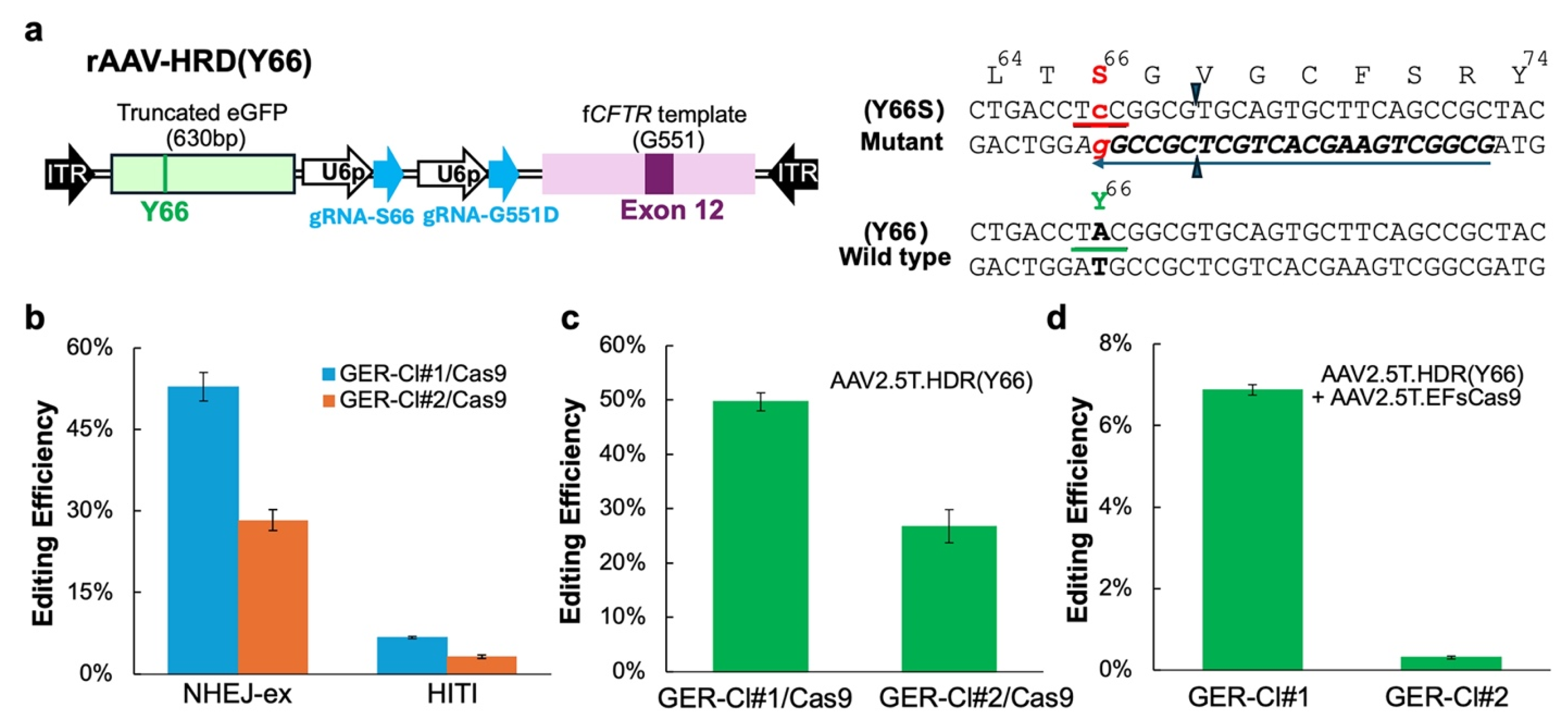
3.4. A Versatile Reporter Cell Line for Assessing NEHJ- and HDR-Based Gene Editing Efficiencies
3.5. Gene Editing in Well-Differentiated Polarized Airway Epithelial Cultures Derived from the CuFi-GER Cell Line
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| rAAV | Recombinant adeno-associated viral vector |
| NHEJ | Non-homologous end joining or homology-directed repair |
| NHEJ-ex | NHEJ-based excision |
| HDR | Homology-directed repair |
| HITI | Homology-independent targeted insertion |
| GER | Gene editing reporter |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| eGFP | Enhanced green fluorescent protein (derived from the Aequorea victoria jellyfish) |
| hrGFP | Humanized renilla green fluorescent protein (from the Renilla reniformis species) |
| DSB | Double-stranded DNA break |
| Blast | Blasticidin S |
| Puro | Puromycin |
| PBS | Phosphate-buffered saline |
| MOI | Multiplicity of infection |
| TEER | Transepithelial electrical resistance |
| Cre | Cre recombinase |
References
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Stoltz, D.A.; Meyerholz, D.K.; Welsh, M.J. Origins of cystic fibrosis lung disease. N. Engl. J. Med. 2015, 372, 1574–1575. [Google Scholar] [CrossRef] [PubMed]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Mall, M.A. The future of cystic fibrosis treatment: From disease mechanisms to novel therapeutic approaches. Lancet 2023, 402, 1185–1198. [Google Scholar] [CrossRef]
- Choi, S.H.; Engelhardt, J.F. Gene Therapy for Cystic Fibrosis: Lessons Learned and Paths Forward. Mol. Ther. 2021, 29, 428–430. [Google Scholar] [CrossRef]
- Yan, Z.; McCray, P.B., Jr.; Engelhardt, J.F. Advances in gene therapy for cystic fibrosis lung disease. Hum. Mol. Genet. 2019, 28, R88–R94. [Google Scholar] [CrossRef]
- Guggino, W.B.; Cebotaru, L. Gene Therapy for Cystic Fibrosis Paved the Way for the Use of Adeno-Associated Virus in Gene Therapy. Hum. Gene Ther. 2020, 31, 538–541. [Google Scholar] [CrossRef]
- Alton, E.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Calcedo, R.; et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015, 3, 684–691. [Google Scholar] [CrossRef]
- Jiang, Q.; Engelhardt, J.F. Cellular heterogeneity of CFTR expression and function in the lung: Implications for gene therapy of cystic fibrosis. Eur. J. Hum. Genet. 1998, 6, 12–31. [Google Scholar] [CrossRef]
- Tang, Y.; Yan, Z.; Engelhardt, J.F. Viral Vectors, Animal Models, and Cellular Targets for Gene Therapy of Cystic Fibrosis Lung Disease. Hum. Gene Ther. 2020, 31, 524–537. [Google Scholar] [CrossRef]
- Lei, L.; Traore, S.; Romano Ibarra, G.S.; Karp, P.H.; Rehman, T.; Meyerholz, D.K.; Zabner, J.; Stoltz, D.A.; Sinn, P.L.; Welsh, M.J.; et al. CFTR-rich ionocytes mediate chloride absorption across airway epithelia. J. Clin. Investig. 2023, 133, e171268. [Google Scholar] [CrossRef]
- Okuda, K.; Dang, H.; Kobayashi, Y.; Carraro, G.; Nakano, S.; Chen, G.; Kato, T.; Asakura, T.; Gilmore, R.C.; Morton, L.C.; et al. Secretory Cells Dominate Airway CFTR Expression and Function in Human Airway Superficial Epithelia. Am. J. Respir. Crit. Care Med. 2021, 203, 1275–1289. [Google Scholar] [CrossRef]
- Reihill, J.A.; Douglas, L.E.J.; Martin, S.L. Modulation of Ion Transport to Restore Airway Hydration in Cystic Fibrosis. Genes 2021, 12, 453. [Google Scholar] [CrossRef]
- King, N.E.; Suzuki, S.; Barilla, C.; Hawkins, F.J.; Randell, S.H.; Reynolds, S.D.; Stripp, B.R.; Davis, B.R. Correction of Airway Stem Cells: Genome Editing Approaches for the Treatment of Cystic Fibrosis. Hum. Gene Ther. 2020, 31, 956–972. [Google Scholar] [CrossRef]
- Cox, D.B.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Aslesh, T.; Erkut, E.; Yokota, T. Restoration of dystrophin expression and correction of Duchenne muscular dystrophy by genome editing. Expert Opin. Biol. Ther. 2021, 21, 1049–1061. [Google Scholar] [CrossRef]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Chen, P.J.; Liu, D.R. Prime editing for precise and highly versatile genome manipulation. Nat. Rev. Genet. 2023, 24, 161–177. [Google Scholar] [CrossRef]
- Healey, N. Next-generation CRISPR-based gene-editing therapies tested in clinical trials. Nat. Med. 2024, 30, 2380–2381. [Google Scholar] [CrossRef]
- Yan, Z.; Vorhies, K.; Feng, Z.; Park, S.Y.; Choi, S.H.; Zhang, Y.; Winter, M.; Sun, X.; Engelhardt, J.F. Recombinant Adeno-Associated Virus-Mediated Editing of the G551D Cystic Fibrosis Transmembrane Conductance Regulator Mutation in Ferret Airway Basal Cells. Hum. Gene Ther. 2022, 33, 1023–1036. [Google Scholar] [CrossRef]
- Suzuki, S.; Crane, A.M.; Anirudhan, V.; Barilla, C.; Matthias, N.; Randell, S.H.; Rab, A.; Sorscher, E.J.; Kerschner, J.L.; Yin, S.; et al. Highly Efficient Gene Editing of Cystic Fibrosis Patient-Derived Airway Basal Cells Results in Functional CFTR Correction. Mol. Ther. 2020, 28, 1684–1695. [Google Scholar] [CrossRef]
- Vaidyanathan, S.; Salahudeen, A.A.; Sellers, Z.M.; Bravo, D.T.; Choi, S.S.; Batish, A.; Le, W.; Baik, R.; de la O, S.; Kaushik, M.P.; et al. High-Efficiency, Selection-free Gene Repair in Airway Stem Cells from Cystic Fibrosis Patients Rescues CFTR Function in Differentiated Epithelia. Cell Stem Cell 2020, 26, 161–171.e4. [Google Scholar] [CrossRef]
- Krishnamurthy, S.; Traore, S.; Cooney, A.L.; Brommel, C.M.; Kulhankova, K.; Sinn, P.L.; Newby, G.A.; Liu, D.R.; McCray, P.B. Functional correction of CFTR mutations in human airway epithelial cells using adenine base editors. Nucleic Acids Res. 2021, 49, 10558–10572. [Google Scholar] [CrossRef]
- Bulcaen, M.; Kortleven, P.; Liu, R.B.; Maule, G.; Dreano, E.; Kelly, M.; Ensinck, M.M.; Thierie, S.; Smits, M.; Ciciani, M.; et al. Prime editing functionally corrects cystic fibrosis-causing CFTR mutations in human organoids and airway epithelial cells. Cell Rep. Med. 2024, 5, 101544. [Google Scholar] [CrossRef]
- Zhou, Z.P.; Yang, L.L.; Cao, H.; Chen, Z.R.; Zhang, Y.; Wen, X.Y.; Hu, J. In Vitro Validation of a CRISPR-Mediated CFTR Correction Strategy for Preclinical Translation in Pigs. Hum. Gene Ther. 2019, 30, 1101–1116. [Google Scholar] [CrossRef]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef]
- Alysandratos, K.D.; Herriges, M.J.; Kotton, D.N. Epithelial Stem and Progenitor Cells in Lung Repair and Regeneration. Annu. Rev. Physiol. 2021, 83, 529–550. [Google Scholar] [CrossRef]
- Rawlins, E.L.; Hogan, B.L. Ciliated epithelial cell lifespan in the mouse trachea and lung. Am. J. Physiol. Lung. Cell Mol. Physiol. 2008, 295, L231–L234. [Google Scholar] [CrossRef]
- Excoffon, K.J.; Koerber, J.T.; Dickey, D.D.; Murtha, M.; Keshavjee, S.; Kaspar, B.K.; Zabner, J.; Schaffer, D.V. Directed evolution of adeno-associated virus to an infectious respiratory virus. Proc. Natl. Acad. Sci. USA 2009, 106, 3865–3870. [Google Scholar] [CrossRef]
- Cooney, A.L.; Brommel, C.M.; Traore, S.; Newby, G.A.; Liu, D.R.; McCray, P.B., Jr.; Sinn, P.L. Reciprocal mutations of lung-tropic AAV capsids lead to improved transduction properties. Front. Genome Ed. 2023, 5, 1271813. [Google Scholar] [CrossRef]
- Yu, M.; Sun, X.; Tyler, S.R.; Liang, B.; Swatek, A.M.; Lynch, T.J.; He, N.; Yuan, F.; Feng, Z.; Rotti, P.G.; et al. Highly Efficient Transgenesis in Ferrets Using CRISPR/Cas9-Mediated Homology-Independent Insertion at the ROSA26 Locus. Sci. Rep. 2019, 9, 1971. [Google Scholar] [CrossRef]
- Zabner, J.; Karp, P.; Seiler, M.; Phillips, S.L.; Mitchell, C.J.; Saavedra, M.; Welsh, M.; Klingelhutz, A.J. Development of cystic fibrosis and noncystic fibrosis airway cell lines. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 284, L844–L854. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef]
- Choi, S.H.; Reeves, R.E.; Romano Ibarra, G.S.; Lynch, T.J.; Shahin, W.S.; Feng, Z.; Gasser, G.N.; Winter, M.C.; Evans, T.I.A.; Liu, X.; et al. Detargeting Lentiviral-Mediated CFTR Expression in Airway Basal Cells Using miR-106b. Genes 2020, 11, 1169. [Google Scholar] [CrossRef]
- Liu, X.; Luo, M.; Guo, C.; Yan, Z.; Wang, Y.; Lei-Butters, D.C.; Engelhardt, J.F. Analysis of adeno-associated virus progenitor cell transduction in mouse lung. Mol. Ther. 2009, 17, 285–293. [Google Scholar] [CrossRef]
- Nishiyama, J.; Mikuni, T.; Yasuda, R. Virus-Mediated Genome Editing via Homology-Directed Repair in Mitotic and Postmitotic Cells in Mammalian Brain. Neuron 2017, 96, 755–768.e5. [Google Scholar] [CrossRef]
- Yan, Z.; Lei-Butters, D.C.; Keiser, N.W.; Engelhardt, J.F. Distinct transduction difference between adeno-associated virus type 1 and type 6 vectors in human polarized airway epithelia. Gene Ther. 2013, 20, 328–337. [Google Scholar] [CrossRef]
- Yan, Z.; Sun, X.; Evans, I.A.; Tyler, S.R.; Song, Y.; Liu, X.; Sui, H.; Engelhardt, J.F. Postentry processing of recombinant adeno-associated virus type 1 and transduction of the ferret lung are altered by a factor in airway secretions. Hum. Gene Ther. 2013, 24, 786–796. [Google Scholar] [CrossRef]
- Zabner, J.; Smith, J.J.; Karp, P.H.; Widdicombe, J.H.; Welsh, M.J. Loss of CFTR chloride channels alters salt absorption by cystic fibrosis airway epithelia in vitro. Mol. Cell 1998, 2, 397–403. [Google Scholar] [CrossRef]
- Duan, D.; Yue, Y.; Yan, Z.; Yang, J.; Engelhardt, J.F. Endosomal processing limits gene transfer to polarized airway epithelia by adeno-associated virus. J. Clin. Investig. 2000, 105, 1573–1587. [Google Scholar] [CrossRef]
- Yan, Z.; Lei-Butters, D.C.; Liu, X.; Zhang, Y.; Zhang, L.; Luo, M.; Zak, R.; Engelhardt, J.F. Unique biologic properties of recombinant AAV1 transduction in polarized human airway epithelia. J. Biol. Chem. 2006, 281, 29684–29692. [Google Scholar] [CrossRef]
- Hao, S.; Zhang, X.; Ning, K.; Feng, Z.; Park, S.Y.; Aksu Kuz, C.; McFarlin, S.; Richart, D.; Cheng, F.; Zhang, E.Y.; et al. Identification of host essential factors for recombinant AAV transduction of the polarized human airway epithelium. J. Virol. 2023, 97, e0133023. [Google Scholar] [CrossRef]
- Miyazaki, J.; Takaki, S.; Araki, K.; Tashiro, F.; Tominaga, A.; Takatsu, K.; Yamamura, K. Expression vector system based on the chicken beta-actin promoter directs efficient production of interleukin-5. Gene 1989, 79, 269–277. [Google Scholar]
- Park, S.Y.; Feng, Z.; Choi, S.H.; Zhang, X.; Tang, Y.; Gasser, G.N.; Richart, D.; Yuan, F.; Qiu, J.; Engelhardt, J.F.; et al. Recombinant Adeno-Associated Virus Vector Mediated Gene Editing in Proliferating and Polarized Cultures of Human Airway Epithelial Cells. Hum. Gene Ther. 2025. [Google Scholar] [CrossRef]
- Ning, K.; Kuz, C.A.; Cheng, F.; Feng, Z.; Yan, Z.; Qiu, J. Adeno-Associated Virus Monoinfection Induces a DNA Damage Response and DNA Repair That Contributes to Viral DNA Replication. mBio 2023, 14, e0352822. [Google Scholar] [CrossRef]
- Ning, K.; Zhao, J.; Feng, Z.; Park, S.Y.; McFarlin, S.; Cheng, F.; Yan, Z.; Wang, J.; Qiu, J. N6-methyladenosine modification of a parvovirus-encoded small noncoding RNA facilitates viral DNA replication through recruiting Y-family DNA polymerases. Proc. Natl. Acad. Sci. USA 2024, 121, e2320782121. [Google Scholar] [CrossRef]
- Tran, N.D.; Liu, X.; Yan, Z.; Abbote, D.; Jiang, Q.; Kmiec, E.B.; Sigmund, C.D.; Engelhardt, J.F. Efficiency of chimeraplast gene targeting by direct nuclear injection using a GFP recovery assay. Mol. Ther. 2003, 7, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.J.; Foti, S.B.; Schwartz, J.W.; Bachaboina, L.; Taylor-Blake, B.; Coleman, J.; Ehlers, M.D.; Zylka, M.J.; McCown, T.J.; Samulski, R.J. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum. Gene Ther. 2011, 22, 1143–1153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.N.; Karp, P.; Gerard, C.J.; Pastor, E.; Laux, D.; Munson, K.; Yan, Z.; Liu, X.; Godwin, S.; Thomas, C.P.; et al. Dual therapeutic utility of proteasome modulating agents for pharmaco-gene therapy of the cystic fibrosis airway. Mol. Ther. 2004, 10, 990–1002. [Google Scholar] [CrossRef] [PubMed]
- Urban, N.; Cheung, T.H. Stem cell quiescence: The challenging path to activation. Development 2021, 148, dev165084. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, X.; Lin, Y.; Zeng, Y. Roles of airway basal stem cells in lung homeostasis and regenerative medicine. Respir. Res. 2022, 23, 122. [Google Scholar] [CrossRef]
- Yu, C.; Liu, Y.; Ma, T.; Liu, K.; Xu, S.; Zhang, Y.; Liu, H.; La Russa, M.; Xie, M.; Ding, S.; et al. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem. Cell 2015, 16, 142–147. [Google Scholar] [CrossRef]
- Stack, J.T.; Rayner, R.E.; Nouri, R.; Suarez, C.J.; Kim, S.H.; Kanke, K.L.; Vetter, T.A.; Cormet-Boyaka, E.; Vaidyanathan, S. DNA-PKcs inhibition improves sequential gene insertion of the full-length CFTR cDNA in airway stem cells. Mol. Ther. Nucleic Acids 2024, 35, 102339. [Google Scholar] [CrossRef]
- Canny, M.D.; Moatti, N.; Wan, L.C.K.; Fradet-Turcotte, A.; Krasner, D.; Mateos-Gomez, P.A.; Zimmermann, M.; Orthwein, A.; Juang, Y.C.; Zhang, W.; et al. Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR-Cas9 genome-editing efficiency. Nat. Biotechnol. 2018, 36, 95–102. [Google Scholar] [CrossRef]
- Liang, S.Q.; Walkey, C.J.; Martinez, A.E.; Su, Q.; Dickinson, M.E.; Wang, D.; Lagor, W.R.; Heaney, J.D.; Gao, G.; Xue, W. AAV5 delivery of CRISPR-Cas9 supports effective genome editing in mouse lung airway. Mol. Ther. 2022, 30, 238–243. [Google Scholar] [CrossRef]
- Thomas, S.P.; Domm, J.M.; van Vloten, J.P.; Xu, L.; Vadivel, A.; Yates, J.G.E.; Pei, Y.; Ingrao, J.; van Lieshout, L.P.; Jackson, S.R.; et al. A promoterless AAV6.2FF-based lung gene editing platform for the correction of surfactant protein B deficiency. Mol. Ther. 2023, 31, 3457–3477. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhou, J.; Lei, J.; Mo, G.; Wu, Y.; Liu, H.; Pang, Z.; Du, M.; Zhou, Z.; Paek, C.; et al. Engineering of a compact, high-fidelity EbCas12a variant that can be packaged with its crRNA into an all-in-one AAV vector delivery system. PLoS Biol. 2024, 22, e3002619. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Yang, S.; Hu, X.; Yin, D.; Dai, Y.; Qian, X.; Wang, D.; Pan, X.; Hong, J.; Sun, X.; et al. Lentiviral delivery of co-packaged Cas9 mRNA and a Vegfa-targeting guide RNA prevents wet age-related macular degeneration in mice. Nat. Biomed. Eng. 2021, 5, 144–156. [Google Scholar] [CrossRef]
- Li, A.; Lee, C.M.; Hurley, A.E.; Jarrett, K.E.; De Giorgi, M.; Lu, W.; Balderrama, K.S.; Doerfler, A.M.; Deshmukh, H.; Ray, A.; et al. A Self-Deleting AAV-CRISPR System for In Vivo Genome Editing. Mol. Ther. Methods Clin. Dev. 2019, 12, 111–122. [Google Scholar] [CrossRef]
- Ibraheim, R.; Tai, P.W.L.; Mir, A.; Javeed, N.; Wang, J.; Rodriguez, T.C.; Namkung, S.; Nelson, S.; Khokhar, E.S.; Mintzer, E.; et al. Self-inactivating, all-in-one AAV vectors for precision Cas9 genome editing via homology-directed repair in vivo. Nat. Commun. 2021, 12, 6267. [Google Scholar] [CrossRef]
- Wei, T.; Sun, Y.; Cheng, Q.; Chatterjee, S.; Traylor, Z.; Johnson, L.T.; Coquelin, M.L.; Wang, J.; Torres, M.J.; Lian, X.; et al. Lung SORT LNPs enable precise homology-directed repair mediated CRISPR/Cas genome correction in cystic fibrosis models. Nat. Commun. 2023, 14, 7322. [Google Scholar] [CrossRef]
- Li, B.; Manan, R.S.; Liang, S.Q.; Gordon, A.; Jiang, A.; Varley, A.; Gao, G.; Langer, R.; Xue, W.; Anderson, D. Combinatorial design of nanoparticles for pulmonary mRNA delivery and genome editing. Nat. Biotechnol. 2023, 41, 1410–1415. [Google Scholar] [CrossRef]
- Crudele, J.M.; Chamberlain, J.S. Cas9 immunity creates challenges for CRISPR gene editing therapies. Nat. Commun. 2018, 9, 3497. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.Y.; Feng, Z.; Zhang, X.; Tang, Y.; Richart, D.; Vorhies, K.E.; Qiu, J.; Engelhardt, J.F.; Yan, Z. A Versatile Reporter Platform for Evaluating HDR- and NHEJ-Based Genome Editing in Airway Epithelial Cell Cultures Using an rAAV Vector. Viruses 2025, 17, 821. https://doi.org/10.3390/v17060821
Park SY, Feng Z, Zhang X, Tang Y, Richart D, Vorhies KE, Qiu J, Engelhardt JF, Yan Z. A Versatile Reporter Platform for Evaluating HDR- and NHEJ-Based Genome Editing in Airway Epithelial Cell Cultures Using an rAAV Vector. Viruses. 2025; 17(6):821. https://doi.org/10.3390/v17060821
Chicago/Turabian StylePark, Soo Yeun, Zehua Feng, Xiujuan Zhang, Yinghua Tang, Donovan Richart, Kai E. Vorhies, Jianming Qiu, John F. Engelhardt, and Ziying Yan. 2025. "A Versatile Reporter Platform for Evaluating HDR- and NHEJ-Based Genome Editing in Airway Epithelial Cell Cultures Using an rAAV Vector" Viruses 17, no. 6: 821. https://doi.org/10.3390/v17060821
APA StylePark, S. Y., Feng, Z., Zhang, X., Tang, Y., Richart, D., Vorhies, K. E., Qiu, J., Engelhardt, J. F., & Yan, Z. (2025). A Versatile Reporter Platform for Evaluating HDR- and NHEJ-Based Genome Editing in Airway Epithelial Cell Cultures Using an rAAV Vector. Viruses, 17(6), 821. https://doi.org/10.3390/v17060821