
Facilitating Cross-border Viral Sequencing Through Nucleic Acid Sample Transport Using Dry Cards

 , , , , and
, , , , and
Abstract
1. Introduction
2. Methods
2.1. Sample Collection and Processing in Local Laboratory
2.2. Loading DNA Product to FTA Card and Transportation
2.3. Nucleic Acid Elution and Sequencing
2.4. Data Analysis
3. Results
3.1. Nucleic Acid Recovery from FTA Card
3.2. Sequencing Performance
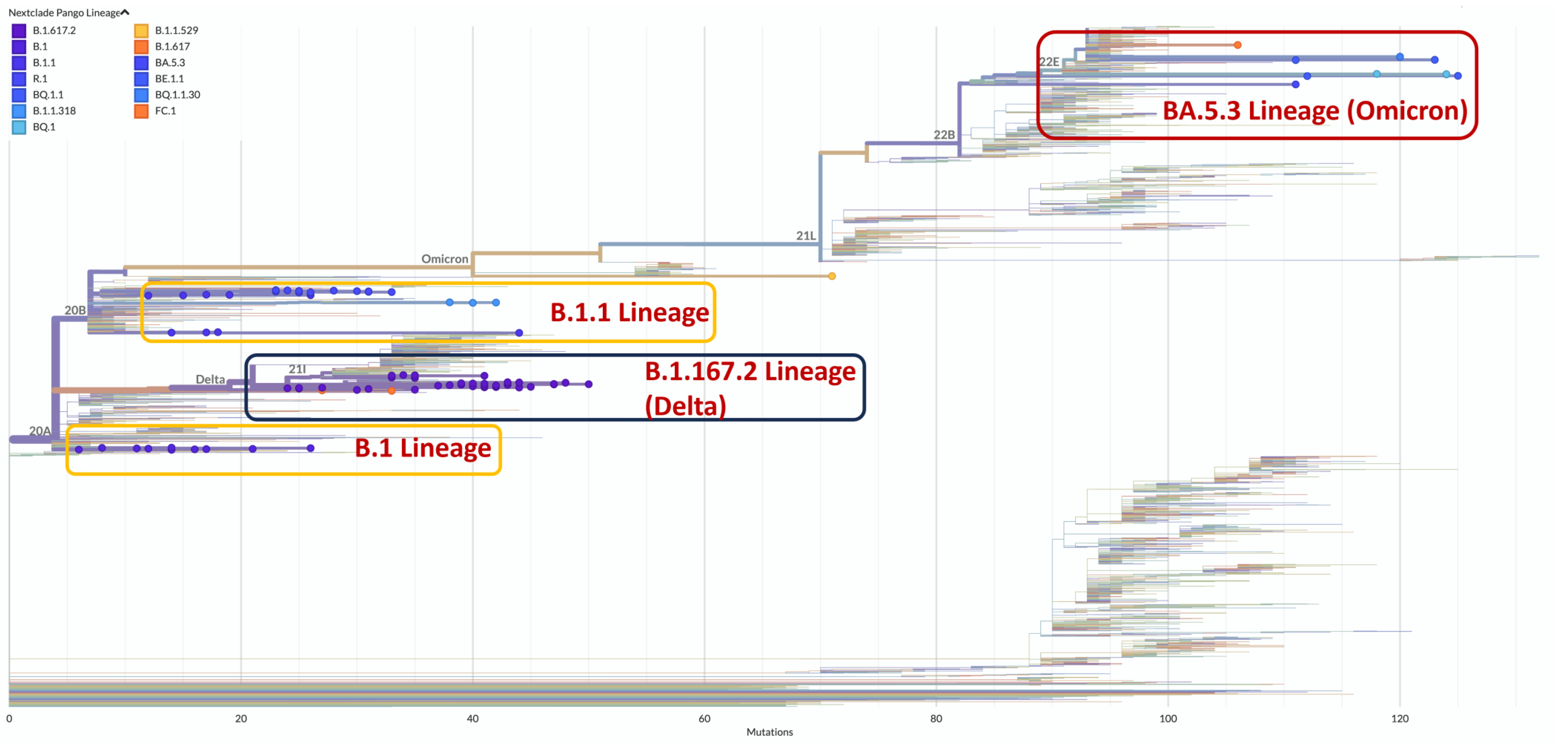
3.3. SARS-CoV-2 Lineage Classification
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Ladner, J.T.; Sahl, J.W. Towards a post-pandemic future for global pathogen genome sequencing. PLoS Biol. 2023, 21, e3002225. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; San, J.E.; Cotten, M.; Moir, M.; Tegomoh, B.; Mboowa, G.; Martin, D.P.; Baxter, C.; Lambisia, A.W.; Diallo, A.; et al. The evolving SARS-CoV-2 epidemic in Africa: Insights from rapidly expanding genomic surveillance. Science 2022, 378, eabq5358. [Google Scholar] [CrossRef] [PubMed]
- Beyerl, J.; Rubio-Acero, R.; Castelletti, N.; Paunovic, I.; Kroidl, I.; Khan, Z.N.; Bakuli, A.; Tautz, A.; Oft, J.; Hoelscher, M.; et al. A dried blood spot protocol for high throughput analysis of SARS-CoV-2 serology based on the Roche Elecsys anti-N assay. EBioMedicine 2021, 70, 103502. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Chu, C.-H.; Sarioglu, A.F. Point-of-Care Toolkit for Multiplex Molecular Diagnosis of SARS-CoV-2 and Influenza A and B Viruses. ACS Sensors 2021, 6, 3204–3213. [Google Scholar] [CrossRef] [PubMed]
- Krambrich, J.; Bringeland, E.; Hesson, J.C.; Hoffman, T.; Lundkvist, Å.; Lindahl, J.F.; Ling, J. Usage of FTA® Classic Cards for Safe Storage, Shipment, and Detection of Arboviruses. Microorganisms 2022, 10, 1445. [Google Scholar] [CrossRef] [PubMed]
- Tully, D.C.; Power, K.A.; Sarette, J.; Stopka, T.J.; Friedmann, P.D.; Korthuis, P.T.; Cooper, H.; Young, A.M.; Seal, D.W.; Westergaard, R.P.; et al. Validation of Dried Blood Spots for Capturing Hepatitis C Virus Diversity for Genomic Surveillance. medRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Lago, B.V.D.; Bezerra, C.S.; Moreira, D.A.; Parente, T.E.; Portilho, M.M.; Pessôa, R.; Sanabani, S.S.; Villar, L.M. Genetic diversity of hepatitis B virus quasispecies in different biological compartments reveals distinct genotypes. Sci. Rep. 2023, 13, 17023. [Google Scholar] [CrossRef] [PubMed]
- Kanta, P.; Chhikara, K.; Mohan, L.; Ghosh, A.; Goyal, K.; Singh, M.P. Alternate Approach in Storing and Shipment of SARS-CoV-2 RNA Samples with the Use of FTA Cards. Curr. Microbiol. 2022, 79, 396. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wang, J.; Li, W.; Zhang, Y.; Xiu, L.; Hu, Q.; Ruan, Z.; Chen, P.; Yin, K. Visual and Rapid Detection of Escherichia coli O157: H7 in Stool Samples by FTA Card-based Loop-mediated Isothermal Amplification. Zoonoses 2023, 3, 963. [Google Scholar] [CrossRef]
- Veiga, I.B.; Mühldorfer, K.; Hafez, H.M.; Lüschow, D. Whatman® FTA® Cards Performance for Ornithobacterium rhinotracheale DNA Amplification. Avian Dis. 2020, 64, 496–498. [Google Scholar] [CrossRef] [PubMed]
- Melanson, V.R.; Jochim, R.; Yarnell, M.; Ferlez, K.B.; Shashikumar, S.; Richardson, J.H. Improving vector-borne pathogen surveillance: A laboratory-based study exploring the potential to detect dengue virus and malaria parasites in mosquito saliva. J. Vector Borne Dis. 2017, 54, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Kariithi, H.M.; Christy, N.; Decanini, E.L.; Lemiere, S.; Volkening, J.D.; Afonso, C.L.; Suarez, D.L. Detection and Genome Sequence Analysis of Avian Metapneumovirus Subtype A Viruses Circulating in Commercial Chicken Flocks in Mexico. Veter- Sci. 2022, 9, 579. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Zhang, J.; Rogers, J.; Campbell, A.; Zhao, J.; Harding, D.; Sahr, F.; Liu, Y.; Wurie, I. The dynamic change of SARS-CoV-2 variants in Sierra Leone. Infect. Genet. Evol. 2022, 98, 105208. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lu, X.; Tie, A.; Ngegba, E.; Wang, L.; Sun, L.; Liang, Y.; Abdulai, M.K.; Bah, S.; Wang, G.; et al. Molecular diagnostics and next-generation sequencing reveal real etiological characteristics of invasive Salmonella infection in febrile illness in Freetown, Sierra Leone. Emerg. Microbes Infect. 2022, 11, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Morel, A.; Mougin, C.; Averous, G.; Legrain, M.; Fender, M.; Risch, S.; Fafi-Kremer, S.; Velten, M.; Oudet, P.; et al. Long-term storage and safe retrieval of human papillomavirus DNA using FTA elute cards. J. Virol. Methods 2016, 229, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Taesuji, M.; Rattanamas, K.; Yim, P.B.; Ruenphet, S. Stability and Detection Limit of Avian Influenza, Newcastle Disease Virus, and African Horse Sickness Virus on Flinders Technology Associates Card by Conventional Polymerase Chain Reaction. Animals 2024, 14, 1242. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Ishii, A.; Segawa, T.; Takagi, Y.; Kobayashi, Y.; Itou, T. Establishing conditions for the storage and elution of rabies virus RNA using FTA® cards. J. Vet. Med. Sci. 2015, 77, 461–465. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Lineage | Number of Seqes | Ancestral Lineage | WHO Clade | Collection Date |
|---|---|---|---|---|
| B.1.617.2 | 43 | Delta | 2021/12/25~2022/10/1 | |
| B.1.1.529 | 3 | Omicron | 2022/1/1 | |
| BE.1.1 | 2 | BA.5.3 | Omicron | 2022/1/1, 2022/10/1 |
| BQ.1 | 2 | BA.5.3 | Omicron | 2022/7/5, 2022/7/26 |
| BQ.1.1 | 4 | BA.5.3 | Omicron | 2021/12/24~2022/1/1 |
| BQ.1.1.30 | 1 | BA.5.3 | Omicron | 2022/1/1 |
| BQ.1.5 | 1 | BA.5.3 | Omicron | 2021/12/25 |
| FC.1 | 1 | BA.5.3 | Omicron | 2022/10/1 |
| B.1 | 2 | 2021/12/25, 2021/12/30 | ||
| B.1.1 | 10 | 2021/12/26~2022/4/1 | ||
| B.1.1.318 | 3 | B.1.1 | 2022/2/1 | |
| R.1 | 14 | B.1.1 | 2021/12/25~2022/1/1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Yin, Q.; Tia, A.B.; Tian, F.; Gao, L.; Nie, K.; Xiao, K.; Ma, X.; Dong, X.; Harding, D.; et al. Facilitating Cross-border Viral Sequencing Through Nucleic Acid Sample Transport Using Dry Cards. Viruses 2025, 17, 804. https://doi.org/10.3390/v17060804
Wang L, Yin Q, Tia AB, Tian F, Gao L, Nie K, Xiao K, Ma X, Dong X, Harding D, et al. Facilitating Cross-border Viral Sequencing Through Nucleic Acid Sample Transport Using Dry Cards. Viruses. 2025; 17(6):804. https://doi.org/10.3390/v17060804
Chicago/Turabian StyleWang, Lili, Qikai Yin, Alie Brima Tia, Fengyu Tian, Liping Gao, Kai Nie, Kang Xiao, Xuejun Ma, Xiaoping Dong, Doris Harding, and et al. 2025. "Facilitating Cross-border Viral Sequencing Through Nucleic Acid Sample Transport Using Dry Cards" Viruses 17, no. 6: 804. https://doi.org/10.3390/v17060804
APA StyleWang, L., Yin, Q., Tia, A. B., Tian, F., Gao, L., Nie, K., Xiao, K., Ma, X., Dong, X., Harding, D., He, X., & Gao, G. F. (2025). Facilitating Cross-border Viral Sequencing Through Nucleic Acid Sample Transport Using Dry Cards. Viruses, 17(6), 804. https://doi.org/10.3390/v17060804