
Progress in Pseudotyping Lentiviral Vectors Towards Cell-Specific Gene Delivery In Vivo

Abstract
1. Introduction
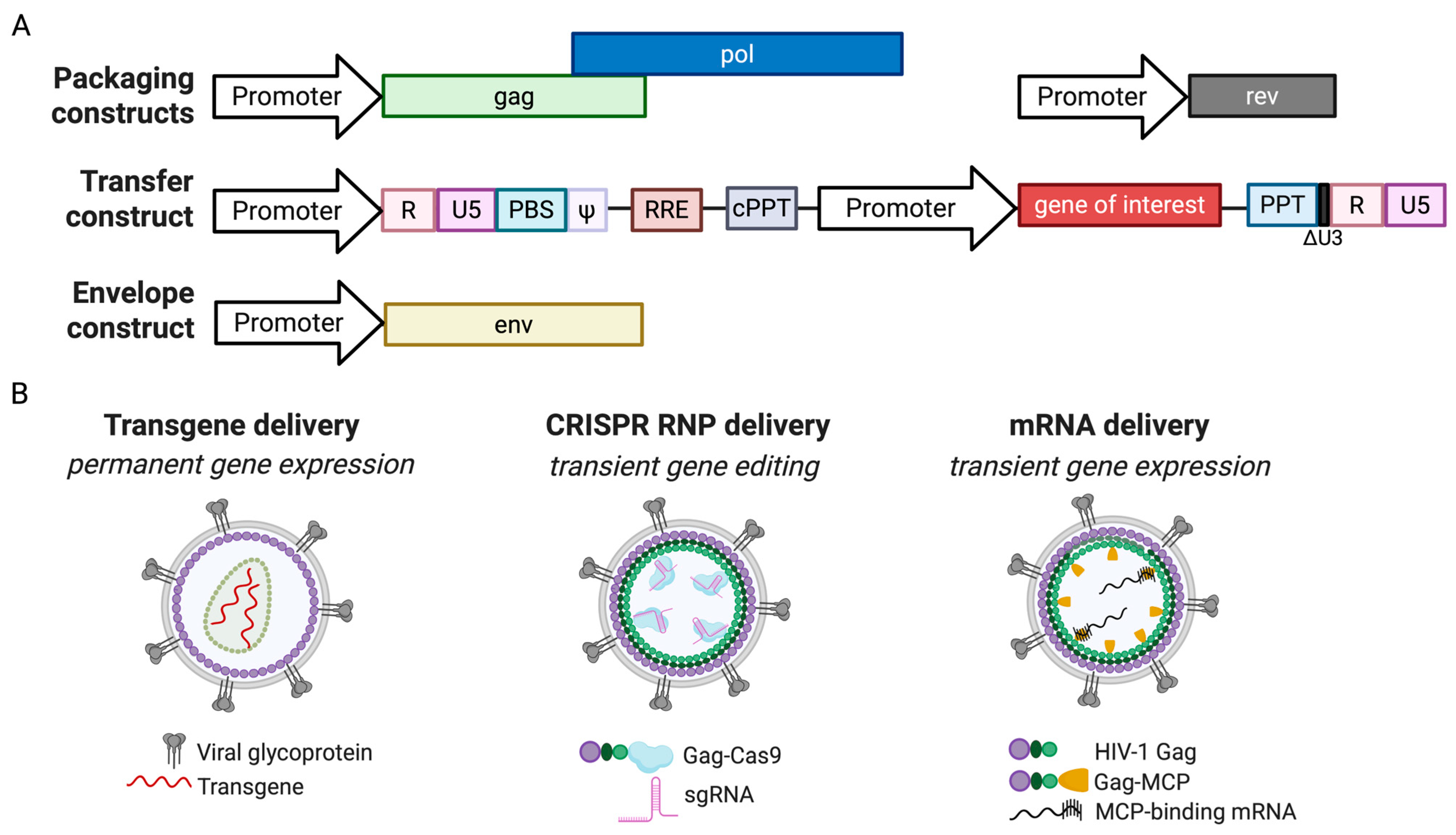
2. The Utility of Lentiviral Vectors in Gene Delivery
3. Pseudotyping: Acquiring the Right Coat
4. Engineering Viral Tropism to Achieve Targeted Delivery
4.1. Pseudotyping with Paramyxoviruses
4.2. Pseudotyping with Togaviruses
4.3. Pseudotyping with Rhabdoviruses

{kind=link}
{kind=link}
{kind=link}
| Targeting Ligand | Viral Glycoprotein | Examples | Target Cell Types | Potential Applications | References |
|---|---|---|---|---|---|
| Antibodies (mAb, bsAb) | SINV E2 * | membrane-bound anti-CD20 mAb | B lymphocytes | Cancer immunotherapy | Yang, L. et al. [113]; Ziegler, L. et al. [114]; Lei, Y. et al. [115] |
| SINV E, E2 displaying the ZZ domain * | anti-CD4 mAb | CD4+ T cells | Immunomodulation | Morizono, K. et al. [108]; Liang, M. et al. [109] | |
| anti-P-glycoprotein | Metastatic melanoma cells | Cancer immunotherapy | Morizono, K. et al. [110] | ||
| SINV E, E2 * | anti-E2xHER2 bsAb | Cancer cells (breast, ovarian, gastric) | Cancer immunotherapy | Parker, C.L. et al. [117] | |
| anti-E2xCD3 bsAb | T cells | Immunomodulation, CAR-T cell therapy | Huckaby, J.T. et al. [118] | ||
| scFvs | Measles (MV) H * | EGFR-specific scFv | Tumor cells | Cancer immunotherapy | Funke, S. et al. [100] |
| CD20-specific scFv | B lymphocytes | Cancer immunotherapy | Funke, S. et al. [100]; Anliker, B. et al. [101] | ||
| CD8-specific scFv | CD8+ T cells | CAR-T cell therapy | Zhou, Q. et al. [81] | ||
| CD105-specific scFv | Endothelial cells | Vascular targeting | Anliker, B. et al. [101] | ||
| GluA-specific scFv | Neurons | Neuromodulation | |||
| CD90-specific scFv | Hematopoietic stem cells (HSCs) | Regenerative medicine | Berckmueller, K. et al. [102] | ||
| Tupaia (TPMV) G | CD20-specific scFv | B lymphocytes | Cancer (lymphoma) immunotherapy | Enkirch, T. et al. [106] | |
| Nipah (NiV) G * | EpCAM-specific scFv | Tumor cells | Cancer immunotherapy | Bender, R.R. et al. [82] | |
| CD8-specific scFv | CD8+ T cells | CAR-T cell therapy | |||
| CD20-specific scFv | B Lymphocytes | Cancer (lymphoma) immunotherapy | |||
| VSV-G | anti-MHC-I scFv | Nucleated cells | Immunomodulation | Dreja, H. et al. [123] | |
| anti-CD30 scFv | Lymphocytes | Cancer (lymphoma) immunotherapy | Anastasov, N. et al. [124] | ||
| anti-CD34 scFv | Hematopoietic stem cells (HSCs) | Regenerative medicine | |||
| anti-EGFR scFv | Tumor cells | Cancer immunotherapy | Höfig, I. et al. [92] | ||
| VSV-G * (VSVGmut) | anti-CD19 scFv | B lymphocytes | Cancer (lymphoma) immunotherapy | Yu, B. et al. [84] | |
| anti-CD3 and CD4 scFv | T cells | CRISPR-Cas9 delivery CAR-T cell therapy | Hamilton, J.R. et al. [127] | ||
| DARPins | Measles (MV) H * | HER2/neu-specific DARPin | Cancer cells (breast, ovarian, gastric) | Cancer immunotherapy | Münch, R.C. et al. [87] |
| CD4-specific DARPin | CD4+ T cells | HIV entry inhibition | Zhou, Q. et al. [96] | ||
| CD8-specific DARPin | CD8+ T cells | CAR-T cell therapy | Michels, A. et al. [89] | ||
| SINV E2 * | HER2/neu-specific DARPin | Cancer cells (breast, ovarian, gastric) | Cancer immunotherapy | Kasaraneni, N. et al. [116] | |
| Cytokines | VSV-G | membrane-bound IL-7 | T cells | Immunotherapy CAR-T cell therapy | Verhoeyen, E. et al. [90] |
| VSV-G * (VSVGmut) | membrane-bound IL-13 | Immune cells | Cancer immunotherapy | Dobson, C.S. et al. [83] |
5. Overcoming Immune Barriers to LV Transduction
5.1. Host Adaptive Immunity as a Barrier to LV Transduction
5.2. Host Intrinsic Immunity as a Barrier to LV Transduction
6. Emerging Directions for LV Targeting
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aiuti, A.; Cattaneo, F.; Galimberti, S.; Benninghoff, U.; Cassani, B.; Callegaro, L.; Scaramuzza, S.; Andolfi, G.; Mirolo, M.; Brigida, I.; et al. Gene Therapy for Immunodeficiency Due to Adenosine Deaminase Deficiency. N. Engl. J. Med. 2009, 360, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Cicalese, M.P.; Ferrua, F.; Castagnaro, L.; Pajno, R.; Barzaghi, F.; Giannelli, S.; Dionisio, F.; Brigida, I.; Bonopane, M.; Casiraghi, M.; et al. Update on the Safety and Efficacy of Retroviral Gene Therapy for Immunodeficiency Due to Adenosine Deaminase Deficiency. Blood 2016, 128, 45–54. [Google Scholar] [CrossRef]
- Awasthi, R.; Maier, H.J.; Zhang, J.; Lim, S. Kymriah® (Tisagenlecleucel)—An Overview of the Clinical Development Journey of the First Approved CAR-T Therapy. Hum. Vaccines Immunother. 2023, 19, 2210046. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- U.S Food and Drug Administration. Approved Cellular and Gene Therapy Products. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products (accessed on 23 March 2025).
- Liu, A. Pfizer Discontinues Hemophilia Treatment Beqvez, Emptying Its Gene Therapy Portfolio. Available online: https://www.fiercepharma.com/pharma/pfizer-empties-gene-therapy-portfolio-discontinues-hemophilia-treatment-beqvez (accessed on 23 March 2025).
- Santhosh, C. Pfizer Stops Commercialization of Hemophilia Gene Therapy Beqvez. Available online: https://www.reuters.com/business/healthcare-pharmaceuticals/pfizer-says-it-will-end-global-development-gene-therapy-beqvez-nikkei-reports-2025-02-20/ (accessed on 27 March 2025).
- DiCarlo, J.E.; Mahajan, V.B.; Tsang, S.H. Gene Therapy and Genome Surgery in the Retina. J. Clin. Investig. 2018, 128, 2177–2188. [Google Scholar] [CrossRef] [PubMed]
- First Inhaled Lentiviral Gene Therapy Enters Cystic Fibrosis Trial. Nat. Biotechnol. 2025, 43, 288. [CrossRef]
- Davies, J.C.; Polineni, D.; Boyd, A.C.; Donaldson, S.; Gill, D.R.; Griesenbach, U.; Hyde, S.C.; Jain, R.; McLachlan, G.; Mall, M.A.; et al. Lentiviral Gene Therapy for Cystic Fibrosis: A Promising Approach and First-in-Human Trial. Am. J. Respir. Crit. Care Med. 2024, 210, 1398–1408. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, F.; Kay, M.A. The deLIVERed Promises of Gene Therapy: Past, Present, and Future of Liver-Directed Gene Therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2025, 33, 1966–1987. [Google Scholar] [CrossRef]
- Kliegman, M.; Zaghlula, M.; Abrahamson, S.; Esensten, J.H.; Wilson, R.C.; Urnov, F.D.; Doudna, J.A. A Roadmap for Affordable Genetic Medicines. Nature 2024, 634, 307–314. [Google Scholar] [CrossRef]
- Aiuti, A.; Slavin, S.; Aker, M.; Ficara, F.; Deola, S.; Mortellaro, A.; Morecki, S.; Andolfi, G.; Tabucchi, A.; Carlucci, F.; et al. Correction of ADA-SCID by Stem Cell Gene Therapy Combined with Nonmyeloablative Conditioning. Science 2002, 296, 2410–2413. [Google Scholar] [CrossRef]
- Czechowicz, A.; Kraft, D.; Weissman, I.L.; Bhattacharya, D. Efficient Transplantation via Antibody-Based Clearance of Hematopoietic Stem Cell Niches. Science 2007, 318, 1296–1299. [Google Scholar] [CrossRef]
- Mullard, A. In Vivo CAR T Cells Move into Clinical Trials. Nat. Rev. Drug Discov. 2024, 23, 727–730. [Google Scholar] [CrossRef]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and Problems with the Use of Viral Vectors for Gene Therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. Viral Vectors in Gene Therapy: Where Do We Stand in 2023? Viruses 2023, 15, 698. [Google Scholar] [CrossRef]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral Vector Platforms within the Gene Therapy Landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional Oncogenesis in 4 Patients after Retrovirus-Mediated Gene Therapy of SCID-X1. J. Clin. Invest. 2008, 118, 3132–3142. [Google Scholar] [CrossRef]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional Mutagenesis Combined with Acquired Somatic Mutations Causes Leukemogenesis Following Gene Therapy of SCID-X1 Patients. J. Clin. Invest. 2008, 118, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Bartolomae, C.C.; Cesana, D.; Cartier, N.; Aubourg, P.; Ranzani, M.; Cesani, M.; Benedicenti, F.; Plati, T.; Rubagotti, E.; et al. Lentiviral Vector Common Integration Sites in Preclinical Models and a Clinical Trial Reflect a Benign Integration Bias and Not Oncogenic Selection. Blood 2011, 117, 5332–5339. [Google Scholar] [CrossRef]
- Cohn, L.B.; Silva, I.T.; Oliveira, T.Y.; Rosales, R.A.; Parrish, E.H.; Learn, G.H.; Hahn, B.H.; Czartoski, J.L.; McElrath, M.J.; Lehmann, C.; et al. HIV-1 Integration Landscape during Latent and Active Infection. Cell 2015, 160, 420–432. [Google Scholar] [CrossRef]
- Wang, J.-H.; Gessler, D.J.; Zhan, W.; Gallagher, T.L.; Gao, G. Adeno-Associated Virus as a Delivery Vector for Gene Therapy of Human Diseases. Signal Transduct. Target. Ther. 2024, 9, 78. [Google Scholar] [CrossRef]
- Gutierrez-Guerrero, A.; Cosset, F.-L.; Verhoeyen, E. Lentiviral Vector Pseudotypes: Precious Tools to Improve Gene Modification of Hematopoietic Cells for Research and Gene Therapy. Viruses 2020, 12, 1016. [Google Scholar] [CrossRef] [PubMed]
- Mendes, B.B.; Conniot, J.; Avital, A.; Yao, D.; Jiang, X.; Zhou, X.; Sharf-Pauker, N.; Xiao, Y.; Adir, O.; Liang, H.; et al. Nanodelivery of Nucleic Acids. Nat. Rev. Methods Primer 2022, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Könnyű, B.; Sadiq, S.K.; Turányi, T.; Hírmondó, R.; Müller, B.; Kräusslich, H.-G.; Coveney, P.V.; Müller, V. Gag-Pol Processing during HIV-1 Virion Maturation: A Systems Biology Approach. PLoS Comput. Biol. 2013, 9, e1003103. [Google Scholar] [CrossRef]
- Freed, E.O. HIV-1 Replication. Somat. Cell Mol. Genet. 2001, 26, 13–33. [Google Scholar] [CrossRef]
- Solomon, M.; Liang, C. Pseudotyped Viruses for Retroviruses. Adv. Exp. Med. Biol. 2023, 1407, 61–84. [Google Scholar] [CrossRef]
- Kleinpeter, A.B.; Freed, E.O. HIV-1 Maturation: Lessons Learned from Inhibitors. Viruses 2020, 12, 940. [Google Scholar] [CrossRef]
- Hu, W.-S.; Hughes, S.H. HIV-1 Reverse Transcription. Cold Spring Harb. Perspect. Med. 2012, 2, a006882. [Google Scholar] [CrossRef] [PubMed]
- Charneau, P.; Clavel, F. A Single-Stranded Gap in Human Immunodeficiency Virus Unintegrated Linear DNA Defined by a Central Copy of the Polypurine Tract. J. Virol. 1991, 65, 2415–2421. [Google Scholar] [CrossRef]
- Cullen, B.R.; Malim, M.H. The HIV-1 Rev Protein: Prototype of a Novel Class of Eukaryotic Post-Transcriptional Regulators. Trends Biochem. Sci. 1991, 16, 346–350. [Google Scholar] [CrossRef]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-Inactivating Lentivirus Vector for Safe and Efficient in Vivo Gene Delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar] [CrossRef]
- Zufferey, R.; Nagy, D.; Mandel, R.J.; Naldini, L.; Trono, D. Multiply Attenuated Lentiviral Vector Achieves Efficient Gene Delivery in Vivo. Nat. Biotechnol. 1997, 15, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Barry, M.A.; Ikeda, Y. Lentiviral Vectors: Basic to Translational. Biochem. J. 2012, 443, 603–618. [Google Scholar] [CrossRef] [PubMed]
- Duvergé, A.; Negroni, M. Pseudotyping Lentiviral Vectors: When the Clothes Make the Virus. Viruses 2020, 12, 1311. [Google Scholar] [CrossRef]
- Milone, M.C.; O’Doherty, U. Clinical Use of Lentiviral Vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef]
- Rahmat, Z.S.; Ali, M.H.; Talha, M.; Hasibuzzaman, M.A. FDA Approval of Casgevy and Lyfgenia: A Dual Breakthrough in Gene Therapies for Sickle Cell Disease. Ann. Med. Surg. 2024, 86, 4966–4968. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved Vectors and Genome-Wide Libraries for CRISPR Screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Lyu, P.; Wang, L.; Lu, B. Virus-Like Particle Mediated CRISPR/Cas9 Delivery for Efficient and Safe Genome Editing. Life 2020, 10, 366. [Google Scholar] [CrossRef]
- Hamilton, J.R.; Tsuchida, C.A.; Nguyen, D.N.; Shy, B.R.; McGarrigle, E.R.; Sandoval Espinoza, C.R.; Carr, D.; Blaeschke, F.; Marson, A.; Doudna, J.A. Targeted Delivery of CRISPR-Cas9 and Transgenes Enables Complex Immune Cell Engineering. Cell Rep. 2021, 35, 109207. [Google Scholar] [CrossRef]
- Zhang, S.; Shen, J.; Li, D.; Cheng, Y. Strategies in the Delivery of Cas9 Ribonucleoprotein for CRISPR/Cas9 Genome Editing. Theranostics 2021, 11, 614–648. [Google Scholar] [CrossRef]
- Banskota, S.; Raguram, A.; Suh, S.; Du, S.W.; Davis, J.R.; Choi, E.H.; Wang, X.; Nielsen, S.C.; Newby, G.A.; Randolph, P.B.; et al. Engineered Virus-like Particles for Efficient in Vivo Delivery of Therapeutic Proteins. Cell 2022, 185, 250–265.e16. [Google Scholar] [CrossRef]
- Ling, S.; Zhang, X.; Dai, Y.; Jiang, Z.; Zhou, X.; Lu, S.; Qian, X.; Liu, J.; Selfjord, N.; Satir, T.M.; et al. Customizable Virus-like Particles Deliver CRISPR-Cas9 Ribonucleoprotein for Effective Ocular Neovascular and Huntington’s Disease Gene Therapy. Nat. Nanotechnol. 2025, 20, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Ngo, W.; Peukes, J.; Baldwin, A.; Xue, Z.W.; Hwang, S.; Stickels, R.R.; Lin, Z.; Satpathy, A.T.; Wells, J.A.; Schekman, R.; et al. Mechanism-Guided Engineering of a Minimal Biological Particle for Genome Editing. Proc. Natl. Acad. Sci. USA 2025, 122, e2413519121. [Google Scholar] [CrossRef] [PubMed]
- Prel, A.; Caval, V.; Gayon, R.; Ravassard, P.; Duthoit, C.; Payen, E.; Maouche-Chretien, L.; Creneguy, A.; Nguyen, T.H.; Martin, N.; et al. Highly Efficient in Vitro and in Vivo Delivery of Functional RNAs Using New Versatile MS2-Chimeric Retrovirus-like Particles. Mol. Ther. Methods Clin. Dev. 2015, 2, 15039. [Google Scholar] [CrossRef]
- Lu, B.; Javidi-Parsijani, P.; Makani, V.; Mehraein-Ghomi, F.; Sarhan, W.M.; Sun, D.; Yoo, K.W.; Atala, Z.P.; Lyu, P.; Atala, A. Delivering SaCas9 mRNA by Lentivirus-like Bionanoparticles for Transient Expression and Efficient Genome Editing. Nucleic Acids Res. 2019, 47, e44. [Google Scholar] [CrossRef]
- Moore, J.P.; Trkola, A.; Dragic, T. Co-Receptors for HIV-1 Entry. Curr. Opin. Immunol. 1997, 9, 551–562. [Google Scholar] [CrossRef]
- Chen, B. Molecular Mechanism of HIV-1 Entry. Trends Microbiol. 2019, 27, 878–891. [Google Scholar] [CrossRef]
- Burns, J.C.; Friedmann, T.; Driever, W.; Burrascano, M.; Yee, J.K. Vesicular Stomatitis Virus G Glycoprotein Pseudotyped Retroviral Vectors: Concentration to Very High Titer and Efficient Gene Transfer into Mammalian and Nonmammalian Cells. Proc. Natl. Acad. Sci. USA 1993, 90, 8033–8037. [Google Scholar] [CrossRef]
- Finkelshtein, D.; Werman, A.; Novick, D.; Barak, S.; Rubinstein, M. LDL Receptor and Its Family Members Serve as the Cellular Receptors for Vesicular Stomatitis Virus. Proc. Natl. Acad. Sci. USA 2013, 110, 7306–7311. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, J.; Belot, L.; Raux, H.; Legrand, P.; Gaudin, Y.; Albertini, A.A. Structural Basis for the Recognition of LDL-Receptor Family Members by VSV Glycoprotein. Nat. Commun. 2018, 9, 1029. [Google Scholar] [CrossRef]
- Akkina, R.K.; Walton, R.M.; Chen, M.L.; Li, Q.X.; Planelles, V.; Chen, I.S. High-Efficiency Gene Transfer into CD34+ Cells with a Human Immunodeficiency Virus Type 1-Based Retroviral Vector Pseudotyped with Vesicular Stomatitis Virus Envelope Glycoprotein G. J. Virol. 1996, 70, 2581–2585. [Google Scholar] [CrossRef]
- Hanawa, H.; Kelly, P.F.; Nathwani, A.C.; Persons, D.A.; Vandergriff, J.A.; Hargrove, P.; Vanin, E.F.; Nienhuis, A.W. Comparison of Various Envelope Proteins for Their Ability to Pseudotype Lentiviral Vectors and Transduce Primitive Hematopoietic Cells from Human Blood. Mol. Ther. J. Am. Soc. Gene Ther. 2002, 5, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In Vivo Gene Delivery and Stable Transduction of Nondividing Cells by a Lentiviral Vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef]
- Johnson, L.G.; Olsen, J.C.; Naldini, L.; Boucher, R.C. Pseudotyped Human Lentiviral Vector-Mediated Gene Transfer to Airway Epithelia in Vivo. Gene Ther. 2000, 7, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Borok, Z.; Harboe-Schmidt, J.E.; Brody, S.L.; You, Y.; Zhou, B.; Li, X.; Cannon, P.M.; Kim, K.J.; Crandall, E.D.; Kasahara, N. Vesicular Stomatitis Virus G-Pseudotyped Lentivirus Vectors Mediate Efficient Apical Transduction of Polarized Quiescent Primary Alveolar Epithelial Cells. J. Virol. 2001, 75, 11747–11754. [Google Scholar] [CrossRef]
- Petersen, G.F.; Hilbert, B.; Trope, G.; Kalle, W.; Strappe, P. Efficient Transduction of Equine Adipose-Derived Mesenchymal Stem Cells by VSV-G Pseudotyped Lentiviral Vectors. Res. Vet. Sci. 2014, 97, 616–622. [Google Scholar] [CrossRef]
- Park, F. Correction of Bleeding Diathesis without Liver Toxicity Using Arenaviral-Pseudotyped HIV-1-Based Vectors in Hemophilia A Mice. Hum. Gene Ther. 2003, 14, 1489–1494. [Google Scholar] [CrossRef]
- Beyer, W.R.; Westphal, M.; Ostertag, W.; von Laer, D. Oncoretrovirus and Lentivirus Vectors Pseudotyped with Lymphocytic Choriomeningitis Virus Glycoprotein: Generation, Concentration, and Broad Host Range. J. Virol. 2002, 76, 1488–1495. [Google Scholar] [CrossRef]
- Miletic, H.; Fischer, Y.H.; Neumann, H.; Hans, V.; Stenzel, W.; Giroglou, T.; Hermann, M.; Deckert, M.; Von Laer, D. Selective Transduction of Malignant Glioma by Lentiviral Vectors Pseudotyped with Lymphocytic Choriomeningitis Virus Glycoproteins. Hum. Gene Ther. 2004, 15, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Miletic, H.; Fischer, Y.H.; Giroglou, T.; Rueger, M.A.; Winkeler, A.; Li, H.; Himmelreich, U.; Stenzel, W.; Jacobs, A.H.; von Laer, D. Normal Brain Cells Contribute to the Bystander Effect in Suicide Gene Therapy of Malignant Glioma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 6761–6768. [Google Scholar] [CrossRef]
- Huszthy, P.C.; Giroglou, T.; Tsinkalovsky, O.; Euskirchen, P.; Skaftnesmo, K.O.; Bjerkvig, R.; von Laer, D.; Miletic, H. Remission of Invasive, Cancer Stem-like Glioblastoma Xenografts Using Lentiviral Vector-Mediated Suicide Gene Therapy. PLoS ONE 2009, 4, e6314. [Google Scholar] [CrossRef]
- Funke, S.; Schneider, I.C.; Glaser, S.; Mühlebach, M.D.; Moritz, T.; Cattaneo, R.; Cichutek, K.; Buchholz, C.J. Pseudotyping Lentiviral Vectors with the Wild-Type Measles Virus Glycoproteins Improves Titer and Selectivity. Gene Ther. 2009, 16, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Frecha, C.; Costa, C.; Nègre, D.; Gauthier, E.; Russell, S.J.; Cosset, F.-L.; Verhoeyen, E. Stable Transduction of Quiescent T Cells without Induction of Cycle Progression by a Novel Lentiviral Vector Pseudotyped with Measles Virus Glycoproteins. Blood 2008, 112, 4843–4852. [Google Scholar] [CrossRef] [PubMed]
- Frecha, C.; Costa, C.; Lévy, C.; Nègre, D.; Russell, S.J.; Maisner, A.; Salles, G.; Peng, K.-W.; Cosset, F.-L.; Verhoeyen, E. Efficient and Stable Transduction of Resting B Lymphocytes and Primary Chronic Lymphocyte Leukemia Cells Using Measles Virus Gp Displaying Lentiviral Vectors. Blood 2009, 114, 3173–3180. [Google Scholar] [CrossRef] [PubMed]
- Humbert, J.-M.; Frecha, C.; Amirache Bouafia, F.; N’Guyen, T.H.; Boni, S.; Cosset, F.-L.; Verhoeyen, E.; Halary, F. Measles Virus Glycoprotein-Pseudotyped Lentiviral Vectors Are Highly Superior to Vesicular Stomatitis Virus G Pseudotypes for Genetic Modification of Monocyte-Derived Dendritic Cells. J. Virol. 2012, 86, 5192–5203. [Google Scholar] [CrossRef]
- Laubach, J.; Richardson, P.; Anderson, K. Multiple Myeloma. Annu. Rev. Med. 2011, 62, 249–264. [Google Scholar] [CrossRef]
- Schoenhals, M.; Frecha, C.; Bruyer, A.; Caraux, A.; Veyrune, J.L.; Jourdan, M.; Moreaux, J.; Cosset, F.-L.; Verhoeyen, E.; Klein, B. Efficient Transduction of Healthy and Malignant Plasma Cells by Lentiviral Vectors Pseudotyped with Measles Virus Glycoproteins. Leukemia 2012, 26, 1663–1670. [Google Scholar] [CrossRef]
- Bartosch, B.; Dubuisson, J.; Cosset, F.-L. Infectious Hepatitis C Virus Pseudo-Particles Containing Functional E1-E2 Envelope Protein Complexes. J. Exp. Med. 2003, 197, 633–642. [Google Scholar] [CrossRef]
- Lee, S.; Kim, Y.-Y.; Ahn, H.J. Systemic Delivery of CRISPR/Cas9 to Hepatic Tumors for Cancer Treatment Using Altered Tropism of Lentiviral Vector. Biomaterials 2021, 272, 120793. [Google Scholar] [CrossRef]
- Frank, A.M.; Buchholz, C.J. Surface-Engineered Lentiviral Vectors for Selective Gene Transfer into Subtypes of Lymphocytes. Mol. Ther. Methods Clin. Dev. 2019, 12, 19–31. [Google Scholar] [CrossRef]
- He, B.; Wilson, B.; Chen, S.-H.; Sharma, K.; Scappini, E.; Cook, M.; Petrovich, R.; Martin, N.P. Molecular Engineering of Virus Tropism. Int. J. Mol. Sci. 2024, 25, 11094. [Google Scholar] [CrossRef]
- Deng, L.; Liang, P.; Cui, H. Pseudotyped Lentiviral Vectors: Ready for Translation into Targeted Cancer Gene Therapy? Genes Dis. 2023, 10, 1937–1955. [Google Scholar] [CrossRef] [PubMed]
- Marsh, M.; Helenius, A. Virus Entry: Open Sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Marcink, T.C.; Zipursky, G.; Sobolik, E.B.; Golub, K.; Herman, E.; Stearns, K.; Greninger, A.L.; Porotto, M.; Moscona, A. How a Paramyxovirus Fusion/Entry Complex Adapts to Escape a Neutralizing Antibody. Nat. Commun. 2024, 15, 8831. [Google Scholar] [CrossRef]
- Sun, X.; Yau, V.K.; Briggs, B.J.; Whittaker, G.R. Role of Clathrin-Mediated Endocytosis during Vesicular Stomatitis Virus Entry into Host Cells. Virology 2005, 338, 53–60. [Google Scholar] [CrossRef]
- DeTulleo, L. The Clathrin Endocytic Pathway in Viral Infection. EMBO J. 1998, 17, 4585–4593. [Google Scholar] [CrossRef]
- Aganovic, A. pH-Dependent Endocytosis Mechanisms for Influenza A and SARS-Coronavirus. Front. Microbiol. 2023, 14, 1190463. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Z.A.; Yeap, S.K.; Ali, A.M.; Ho, W.Y.; Alitheen, N.B.M.; Hamid, M. scFv Antibody: Principles and Clinical Application. Clin. Dev. Immunol. 2012, 2012, 980250. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Schneider, I.C.; Edes, I.; Honegger, A.; Bach, P.; Schönfeld, K.; Schambach, A.; Wels, W.S.; Kneissl, S.; Uckert, W.; et al. T-Cell Receptor Gene Transfer Exclusively to Human CD8+ Cells Enhances Tumor Cell Killing. Blood 2012, 120, 4334–4342. [Google Scholar] [CrossRef]
- Bender, R.R.; Muth, A.; Schneider, I.C.; Friedel, T.; Hartmann, J.; Plückthun, A.; Maisner, A.; Buchholz, C.J. Receptor-Targeted Nipah Virus Glycoproteins Improve Cell-Type Selective Gene Delivery and Reveal a Preference for Membrane-Proximal Cell Attachment. PLoS Pathog. 2016, 12, e1005641. [Google Scholar] [CrossRef]
- Dobson, C.S.; Reich, A.N.; Gaglione, S.; Smith, B.E.; Kim, E.J.; Dong, J.; Ronsard, L.; Okonkwo, V.; Lingwood, D.; Dougan, M.; et al. Antigen Identification and High-Throughput Interaction Mapping by Reprogramming Viral Entry. Nat. Methods 2022, 19, 449–460. [Google Scholar] [CrossRef]
- Yu, B.; Shi, Q.; Belk, J.A.; Yost, K.E.; Parker, K.R.; Li, R.; Liu, B.B.; Huang, H.; Lingwood, D.; Greenleaf, W.J.; et al. Engineered Cell Entry Links Receptor Biology with Single-Cell Genomics. Cell 2022, 185, 4904–4920.e22. [Google Scholar] [CrossRef] [PubMed]
- Stumpp, M.T.; Binz, H.K.; Amstutz, P. DARPins: A New Generation of Protein Therapeutics. Drug Discov. Today 2008, 13, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Plückthun, A. Designed Ankyrin Repeat Proteins (DARPins): Binding Proteins for Research, Diagnostics, and Therapy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 489–511. [Google Scholar] [CrossRef]
- Münch, R.C.; Mühlebach, M.D.; Schaser, T.; Kneissl, S.; Jost, C.; Plückthun, A.; Cichutek, K.; Buchholz, C.J. DARPins: An Efficient Targeting Domain for Lentiviral Vectors. Mol. Ther. 2011, 19, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Walser, M.; Mayor, J.; Rothenberger, S. Designed Ankyrin Repeat Proteins: A New Class of Viral Entry Inhibitors. Viruses 2022, 14, 2242. [Google Scholar] [CrossRef]
- Michels, A.; Frank, A.M.; Günther, D.M.; Mataei, M.; Börner, K.; Grimm, D.; Hartmann, J.; Buchholz, C.J. Lentiviral and Adeno-Associated Vectors Efficiently Transduce Mouse T Lymphocytes When Targeted to Murine CD8. Mol. Ther.-Methods Clin. Dev. 2021, 23, 334–347. [Google Scholar] [CrossRef]
- Verhoeyen, E.; Dardalhon, V.; Ducrey-Rundquist, O.; Trono, D.; Taylor, N.; Cosset, F.-L. IL-7 Surface-Engineered Lentiviral Vectors Promote Survival and Efficient Gene Transfer in Resting Primary T Lymphocytes. Blood 2003, 101, 2167–2174. [Google Scholar] [CrossRef]
- Joglekar, A.V.; Sandoval, S. Pseudotyped Lentiviral Vectors: One Vector, Many Guises. Hum. Gene Ther. Methods 2017, 28, 291–301. [Google Scholar] [CrossRef]
- Höfig, I.; Barth, S.; Salomon, M.; Jagusch, V.; Atkinson, M.J.; Anastasov, N.; Thirion, C. Systematic Improvement of Lentivirus Transduction Protocols by Antibody Fragments Fused to VSV-G as Envelope Glycoprotein. Biomaterials 2014, 35, 4204–4212. [Google Scholar] [CrossRef]
- Aguilar, H.C.; Henderson, B.A.; Zamora, J.L.; Johnston, G.P. Paramyxovirus Glycoproteins and the Membrane Fusion Process. Curr. Clin. Microbiol. Rep. 2016, 3, 142–154. [Google Scholar] [CrossRef]
- Kim, A.S.; Diamond, M.S. A Molecular Understanding of Alphavirus Entry and Antibody Protection. Nat. Rev. Microbiol. 2023, 21, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, O.; Holmes, A.C.; Kafai, N.M.; Adams, L.J.; Diamond, M.S. Entry Receptors—The Gateway to Alphavirus Infection. J. Clin. Invest. 2023, 133, e165307. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Uhlig, K.M.; Muth, A.; Kimpel, J.; Lévy, C.; Münch, R.C.; Seifried, J.; Pfeiffer, A.; Trkola, A.; Coulibaly, C.; et al. Exclusive Transduction of Human CD4+ T Cells upon Systemic Delivery of CD4-Targeted Lentiviral Vectors. J. Immunol. 2015, 195, 2493–2501. [Google Scholar] [CrossRef]
- Yanagi, Y.; Takeda, M.; Ohno, S.; Hashiguchi, T. Measles Virus Receptors. In Measles; Griffin, D.E., Oldstone, M.B.A., Eds.; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2009; Volume 329, pp. 13–30. ISBN 978-3-540-70522-2. [Google Scholar]
- Mühlebach, M.D.; Mateo, M.; Sinn, P.L.; Prüfer, S.; Uhlig, K.M.; Leonard, V.H.J.; Navaratnarajah, C.K.; Frenzke, M.; Wong, X.X.; Sawatsky, B.; et al. Adherens Junction Protein Nectin-4 Is the Epithelial Receptor for Measles Virus. Nature 2011, 480, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Peng, K.-W.; Harvey, M.; Greiner, S.; Lorimer, I.A.J.; James, C.D.; Russell, S.J. Rescue and Propagation of Fully Retargeted Oncolytic Measles Viruses. Nat. Biotechnol. 2005, 23, 209–214. [Google Scholar] [CrossRef]
- Funke, S.; Maisner, A.; Mühlebach, M.D.; Koehl, U.; Grez, M.; Cattaneo, R.; Cichutek, K.; Buchholz, C.J. Targeted Cell Entry of Lentiviral Vectors. Mol. Ther. 2008, 16, 1427–1436. [Google Scholar] [CrossRef]
- Anliker, B.; Abel, T.; Kneissl, S.; Hlavaty, J.; Caputi, A.; Brynza, J.; Schneider, I.C.; Münch, R.C.; Petznek, H.; Kontermann, R.E.; et al. Specific Gene Transfer to Neurons, Endothelial Cells and Hematopoietic Progenitors with Lentiviral Vectors. Nat. Methods 2010, 7, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Berckmueller, K.; Thomas, J.; Taha, E.A.; Choo, S.; Madhu, R.; Kanestrom, G.; Rupert, P.B.; Strong, R.; Kiem, H.-P.; Radtke, S. CD90-Targeted Lentiviral Vectors for HSC Gene Therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2023, 31, 2901–2913. [Google Scholar] [CrossRef]
- Marino, M.P.; Panigaj, M.; Ou, W.; Manirarora, J.; Wei, C.-H.; Reiser, J. A Scalable Method to Concentrate Lentiviral Vectors Pseudotyped with Measles Virus Glycoproteins. Gene Ther. 2015, 22, 280–285. [Google Scholar] [CrossRef]
- Kneissl, S.; Abel, T.; Rasbach, A.; Brynza, J.; Schneider-Schaulies, J.; Buchholz, C.J. Measles Virus Glycoprotein-Based Lentiviral Targeting Vectors That Avoid Neutralizing Antibodies. PLoS ONE 2012, 7, e46667. [Google Scholar] [CrossRef]
- Panigaj, M.; Marino, M.P.; Reiser, J. Tagging and Capturing of Lentiviral Vectors Using Short RNAs. Int. J. Mol. Sci. 2021, 22, 10263. [Google Scholar] [CrossRef] [PubMed]
- Enkirch, T.; Kneissl, S.; Hoyler, B.; Ungerechts, G.; Stremmel, W.; Buchholz, C.J.; Springfeld, C. Targeted Lentiviral Vectors Pseudotyped with the Tupaia Paramyxovirus Glycoproteins. Gene Ther. 2013, 20, 16–23. [Google Scholar] [CrossRef]
- Ohno, K.; Sawai, K.; Lijima, Y.; Levin, B.; Meruelo, D. Cell-Specific Targeting of Sindbis Virus Vectors Displaying IgG-Binding Domains of Protein A. Nat. Biotechnol. 1997, 15, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Morizono, K.; Bristol, G.; Xie, Y.; Kung, S.K.-P.; Chen, I.S.Y. Antibody-Directed Targeting of Retroviral Vectors via Cell Surface Antigens. J. Virol. 2001, 75, 8016–8020. [Google Scholar] [CrossRef]
- Liang, M.; Morizono, K.; Pariente, N.; Kamata, M.; Lee, B.; Chen, I.S.Y. Targeted Transduction via CD4 by a Lentiviral Vector Uses a Clathrin-Mediated Entry Pathway. J. Virol. 2009, 83, 13026–13031. [Google Scholar] [CrossRef]
- Morizono, K.; Xie, Y.; Ringpis, G.-E.; Johnson, M.; Nassanian, H.; Lee, B.; Wu, L.; Chen, I.S.Y. Lentiviral Vector Retargeting to P-Glycoprotein on Metastatic Melanoma through Intravenous Injection. Nat. Med. 2005, 11, 346–352. [Google Scholar] [CrossRef]
- Morizono, K.; Pariente, N.; Xie, Y.; Chen, I.S.Y. Redirecting Lentiviral Vectors by Insertion of Integrin-tageting Peptides into Envelope Proteins. J. Gene Med. 2009, 11, 549–558. [Google Scholar] [CrossRef]
- Morizono, K.; Xie, Y.; Helguera, G.; Daniels, T.R.; Lane, T.F.; Penichet, M.L.; Chen, I.S.Y. A Versatile Targeting System with Lentiviral Vectors Bearing the Biotin-adaptor Peptide. J. Gene Med. 2009, 11, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Bailey, L.; Baltimore, D.; Wang, P. Targeting Lentiviral Vectors to Specific Cell Types in Vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 11479–11484. [Google Scholar] [CrossRef]
- Ziegler, L.; Yang, L.; Joo, K.I.; Yang, H.; Baltimore, D.; Wang, P. Targeting Lentiviral Vectors to Antigen-Specific Immunoglobulins. Hum. Gene Ther. 2008, 19, 861–872. [Google Scholar] [CrossRef]
- Lei, Y.; Joo, K.-I.; Wang, P. Engineering Fusogenic Molecules to Achieve Targeted Transduction of Enveloped Lentiviral Vectors. J. Biol. Eng. 2009, 3, 8. [Google Scholar] [CrossRef]
- Kasaraneni, N.; Chamoun-Emanuelli, A.M.; Wright, G.A.; Chen, Z. A Simple Strategy for Retargeting Lentiviral Vectors to Desired Cell Types via a Disulfide-Bond-Forming Protein-Peptide Pair. Sci. Rep. 2018, 8, 10990. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.L.; Jacobs, T.M.; Huckaby, J.T.; Harit, D.; Lai, S.K. Efficient and Highly Specific Gene Transfer Using Mutated Lentiviral Vectors Redirected with Bispecific Antibodies. mBio 2020, 11, e02990-19. [Google Scholar] [CrossRef]
- Huckaby, J.T.; Landoni, E.; Jacobs, T.M.; Savoldo, B.; Dotti, G.; Lai, S.K. Bispecific Binder Redirected Lentiviral Vector Enables in Vivo Engineering of CAR-T Cells. J. Immunother. Cancer 2021, 9, e002737. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Inoue, K.; Tanabe, S.; Kato, S.; Takada, M.; Kobayashi, K. Pseudotyped Lentiviral Vectors for Retrograde Gene Delivery into Target Brain Regions. Front. Neuroanat. 2017, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Kobayashi, K. Pseudotyped Lentiviral Vectors for Tract-Targeting and Application for the Functional Control of Selective Neural Circuits. J. Neurosci. Methods 2020, 344, 108854. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Wu, R.A.; Hansen, M.; Ironside, C.; Watts, K.L.; Olsen, P.; Beard, B.C.; Kiem, H.-P. Cocal-Pseudotyped Lentiviral Vectors Resist Inactivation by Human Serum and Efficiently Transduce Primate Hematopoietic Repopulating Cells. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 725–733. [Google Scholar] [CrossRef]
- Girard-Gagnepain, A.; Amirache, F.; Costa, C.; Lévy, C.; Frecha, C.; Fusil, F.; Nègre, D.; Lavillette, D.; Cosset, F.-L.; Verhoeyen, E. Baboon Envelope Pseudotyped LVs Outperform VSV-G-LVs for Gene Transfer into Early-Cytokine-Stimulated and Resting HSCs. Blood 2014, 124, 1221–1231. [Google Scholar] [CrossRef]
- Dreja, H.; Piechaczyk, M. The Effects of N-Terminal Insertion into VSV-G of an scFv Peptide. Virol. J. 2006, 3, 69. [Google Scholar] [CrossRef]
- Anastasov, N.; Höfig, I.; Mall, S.; Krackhardt, A.M.; Thirion, C. Optimized Lentiviral Transduction Protocols by Use of a Poloxamer Enhancer, Spinoculation, and scFv-Antibody Fusions to VSV-G. In Lentiviral Vectors and Exosomes as Gene and Protein Delivery Tools; Federico, M., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; Volume 1448, pp. 49–61. ISBN 978-1-4939-3751-6. [Google Scholar]
- Cordes, N.; Kolbe, C.; Lock, D.; Holzer, T.; Althoff, D.; Schäfer, D.; Blaeschke, F.; Kotter, B.; Karitzky, S.; Rossig, C.; et al. Anti-CD19 CARs Displayed at the Surface of Lentiviral Vector Particles Promote Transduction of Target-Expressing Cells. Mol. Ther. Methods Clin. Dev. 2021, 21, 42–53. [Google Scholar] [CrossRef]
- Strebinger, D.; Frangieh, C.J.; Friedrich, M.J.; Faure, G.; Macrae, R.K.; Zhang, F. Cell Type-Specific Delivery by Modular Envelope Design. Nat. Commun. 2023, 14, 5141. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.R.; Chen, E.; Perez, B.S.; Sandoval Espinoza, C.R.; Kang, M.H.; Trinidad, M.; Ngo, W.; Doudna, J.A. In Vivo Human T Cell Engineering with Enveloped Delivery Vehicles. Nat. Biotechnol. 2024, 42, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- EsoBiotec EsoBiotec Begins Clinical Trial of In Vivo BCMA CAR-T Candidate ESO-T01 for Multiple Myeloma. Available online: https://www.esobiotec.com/press-release-investigator-initiated-trial/ (accessed on 25 March 2025).
- Herzog, R.W. Complexity of Immune Responses to AAV Transgene Products—Example of Factor IX. Cell. Immunol. 2019, 342, 103658. [Google Scholar] [CrossRef]
- Follenzi, A.; Santambrogio, L.; Annoni, A. Immune Responses to Lentiviral Vectors. Curr. Gene Ther. 2007, 7, 306–315. [Google Scholar] [CrossRef]
- Annoni, A.; Gregori, S.; Naldini, L.; Cantore, A. Modulation of Immune Responses in Lentiviral Vector-Mediated Gene Transfer. Cell. Immunol. 2019, 342, 103802. [Google Scholar] [CrossRef]
- Ertl, O.T.; Wenz, D.C.; Bouche, F.B.; Berbers, G.A.M.; Muller, C.P. Immunodominant Domains of the Measles Virus Hemagglutinin Protein Eliciting a Neutralizing Human B Cell Response. Arch. Virol. 2003, 148, 2195–2206. [Google Scholar] [CrossRef] [PubMed]
- Lévy, C.; Amirache, F.; Costa, C.; Frecha, C.; Muller, C.P.; Kweder, H.; Buckland, R.; Cosset, F.-L.; Verhoeyen, E. Lentiviral Vectors Displaying Modified Measles Virus Gp Overcome Pre-Existing Immunity in in Vivo-like Transduction of Human T and B Cells. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 1699–1712. [Google Scholar] [CrossRef]
- Maguire, C.A.; Ramirez, S.H.; Merkel, S.F.; Sena-Esteves, M.; Breakefield, X.O. Gene Therapy for the Nervous System: Challenges and New Strategies. Neurother. J. Am. Soc. Exp. Neurother. 2014, 11, 817–839. [Google Scholar] [CrossRef]
- Abordo-Adesida, E.; Follenzi, A.; Barcia, C.; Sciascia, S.; Castro, M.G.; Naldini, L.; Lowenstein, P.R. Stability of Lentiviral Vector-Mediated Transgene Expression in the Brain in the Presence of Systemic Antivector Immune Responses. Hum. Gene Ther. 2005, 16, 741–751. [Google Scholar] [CrossRef]
- Rust, B.J.; Becker, P.S.; Chandrasekaran, D.; Kubek, S.P.; Peterson, C.W.; Adair, J.E.; Kiem, H.-P. Envelope-Specific Adaptive Immunity Following Transplantation of Hematopoietic Stem Cells Modified with VSV-G Lentivirus. Mol. Ther. Methods Clin. Dev. 2020, 19, 438–446. [Google Scholar] [CrossRef]
- Munis, A.M.; Mattiuzzo, G.; Bentley, E.M.; Collins, M.K.; Eyles, J.E.; Takeuchi, Y. Use of Heterologous Vesiculovirus G Proteins Circumvents the Humoral Anti-Envelope Immunity in Lentivector-Based In Vivo Gene Delivery. Mol. Ther. Nucleic Acids 2019, 17, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef]
- Piras, F.; Kajaste-Rudnitski, A. Antiviral Immunity and Nucleic Acid Sensing in Haematopoietic Stem Cell Gene Engineering. Gene Ther. 2021, 28, 16–28. [Google Scholar] [CrossRef]
- Kajaste-Rudnitski, A.; Naldini, L. Cellular Innate Immunity and Restriction of Viral Infection: Implications for Lentiviral Gene Therapy in Human Hematopoietic Cells. Hum. Gene Ther. 2015, 26, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Bieniasz, P.D. HIV Restriction Factors and Mechanisms of Evasion. Cold Spring Harb. Perspect. Med. 2012, 2, a006940. [Google Scholar] [CrossRef]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad Antiretroviral Defence by Human APOBEC3G through Lethal Editing of Nascent Reverse Transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA Deamination Mediates Innate Immunity to Retroviral Infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a Human Gene That Inhibits HIV-1 Infection and Is Suppressed by the Viral Vif Protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The Cytoplasmic Body Component TRIM5alpha Restricts HIV-1 Infection in Old World Monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific Recognition and Accelerated Uncoating of Retroviral Capsids by the TRIM5alpha Restriction Factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin Inhibits Retrovirus Release and Is Antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Baldauf, H.-M.; Pan, X.; Erikson, E.; Schmidt, S.; Daddacha, W.; Burggraf, M.; Schenkova, K.; Ambiel, I.; Wabnitz, G.; Gramberg, T.; et al. SAMHD1 Restricts HIV-1 Infection in Resting CD4(+) T Cells. Nat. Med. 2012, 18, 1682–1687. [Google Scholar] [CrossRef] [PubMed]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. SAMHD1 Restricts the Replication of Human Immunodeficiency Virus Type 1 by Depleting the Intracellular Pool of Deoxynucleoside Triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Pan, Q.; Ding, S.; Qian, J.; Xu, F.; Zhou, J.; Cen, S.; Guo, F.; Liang, C. The Interferon-Inducible MxB Protein Inhibits HIV-1 Infection. Cell Host Microbe 2013, 14, 398–410. [Google Scholar] [CrossRef]
- Lu, J.; Pan, Q.; Rong, L.; He, W.; Liu, S.-L.; Liang, C. The IFITM Proteins Inhibit HIV-1 Infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 Restrict HIV-1 Infectivity and Are Counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.J.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T.; et al. MX2 Is an Interferon-Induced Inhibitor of HIV-1 Infection. Nature 2013, 502, 563–566. [Google Scholar] [CrossRef]
- Goujon, C.; Moncorgé, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hué, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human MX2 Is an Interferon-Induced Post-Entry Inhibitor of HIV-1 Infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx Relieves Inhibition of HIV-1 Infection of Macrophages Mediated by the SAMHD1 Protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Ségéral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 Is the Dendritic- and Myeloid-Cell-Specific HIV-1 Restriction Factor Counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- Kim, K.; Dauphin, A.; Komurlu, S.; McCauley, S.M.; Yurkovetskiy, L.; Carbone, C.; Diehl, W.E.; Strambio-De-Castillia, C.; Campbell, E.M.; Luban, J. Cyclophilin A Protects HIV-1 from Restriction by Human TRIM5α. Nat. Microbiol. 2019, 4, 2044–2051. [Google Scholar] [CrossRef] [PubMed]
- Franzolin, E.; Pontarin, G.; Rampazzo, C.; Miazzi, C.; Ferraro, P.; Palumbo, E.; Reichard, P.; Bianchi, V. The Deoxynucleotide Triphosphohydrolase SAMHD1 Is a Major Regulator of DNA Precursor Pools in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 14272–14277. [Google Scholar] [CrossRef]
- Brass, A.L.; Huang, I.-C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM Proteins Mediate Cellular Resistance to Influenza A H1N1 Virus, West Nile Virus, and Dengue Virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.-C.; Bailey, C.C.; Weyer, J.L.; Radoshitzky, S.R.; Becker, M.M.; Chiang, J.J.; Brass, A.L.; Ahmed, A.A.; Chi, X.; Dong, L.; et al. Distinct Patterns of IFITM-Mediated Restriction of Filoviruses, SARS Coronavirus, and Influenza A Virus. PLoS Pathog. 2011, 7, e1001258. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.C.; Zhong, G.; Huang, I.-C.; Farzan, M. IFITM-Family Proteins: The Cell’s First Line of Antiviral Defense. Annu. Rev. Virol. 2014, 1, 261–283. [Google Scholar] [CrossRef]
- Zhao, X.; Li, J.; Winkler, C.A.; An, P.; Guo, J.-T. IFITM Genes, Variants, and Their Roles in the Control and Pathogenesis of Viral Infections. Front. Microbiol. 2018, 9, 3228. [Google Scholar] [CrossRef]
- Li, K.; Markosyan, R.M.; Zheng, Y.-M.; Golfetto, O.; Bungart, B.; Li, M.; Ding, S.; He, Y.; Liang, C.; Lee, J.C.; et al. IFITM Proteins Restrict Viral Membrane Hemifusion. PLoS Pathog. 2013, 9, e1003124. [Google Scholar] [CrossRef]
- Guo, X.; Steinkühler, J.; Marin, M.; Li, X.; Lu, W.; Dimova, R.; Melikyan, G.B. Interferon-Induced Transmembrane Protein 3 Blocks Fusion of Diverse Enveloped Viruses by Altering Mechanical Properties of Cell Membranes. ACS Nano 2021, 15, 8155–8170. [Google Scholar] [CrossRef]
- Wang, J.; Luo, Y.; Katiyar, H.; Liang, C.; Liu, Q. The Antiviral Activity of Interferon-Induced Transmembrane Proteins and Virus Evasion Strategies. Viruses 2024, 16, 734. [Google Scholar] [CrossRef]
- Rahman, K.; Wilt, I.; Jolley, A.A.; Chowdhury, B.; Datta, S.A.K.; Compton, A.A. SNARE Mimicry by the CD225 Domain of IFITM3 Enables Regulation of Homotypic Late Endosome Fusion. EMBO J. 2025, 44, 534–562. [Google Scholar] [CrossRef]
- Roesch, F.; OhAinle, M.; Emerman, M. A CRISPR Screen for Factors Regulating SAMHD1 Degradation Identifies IFITMs as Potent Inhibitors of Lentiviral Particle Delivery. Retrovirology 2018, 15, 26. [Google Scholar] [CrossRef] [PubMed]
- Hornick, A.L.; Li, N.; Oakland, M.; McCray, P.B.; Sinn, P.L. Human, Pig, and Mouse Interferon-Induced Transmembrane Proteins Partially Restrict Pseudotyped Lentiviral Vectors. Hum. Gene Ther. 2016, 27, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.-X.; Carmichael, G.G. Innate Immunity in Pluripotent Human Cells: Attenuated Response to Interferon-β. J. Biol. Chem. 2013, 288, 16196–16205. [Google Scholar] [CrossRef]
- Wu, X.; Dao Thi, V.L.; Huang, Y.; Billerbeck, E.; Saha, D.; Hoffmann, H.-H.; Wang, Y.; Silva, L.A.V.; Sarbanes, S.; Sun, T.; et al. Intrinsic Immunity Shapes Viral Resistance of Stem Cells. Cell 2018, 172, 423–438.e25. [Google Scholar] [CrossRef]
- Shi, G.; Ozog, S.; Torbett, B.E.; Compton, A.A. mTOR Inhibitors Lower an Intrinsic Barrier to Virus Infection Mediated by IFITM3. Proc. Natl. Acad. Sci. USA 2018, 115, E10069–E10078. [Google Scholar] [CrossRef] [PubMed]
- Tajer, P.; Karakaslar, E.O.; Canté-Barrett, K.; Naber, B.A.E.; Vloemans, S.A.; Van Eggermond, M.C.J.A.; Van Der Hoorn, M.-L.; Van Den Akker, E.; Pike-Overzet, K.; Staal, F.J.T. Utilizing Epigenetic Regulators to Improve HSC-Based Lentiviral Gene Therapy. Blood Adv. 2024, 8, 4936–4947. [Google Scholar] [CrossRef]
- Adabi, E.; Charitidis, F.T.; Thalheimer, F.B.; Guaza-Lasheras, M.; Clarke, C.; Buchholz, C.J. Enhanced Conversion of T Cells into CAR T Cells by Modulation of the MAPK/ERK Pathway. Cell Rep. Med. 2025, 6, 101970. [Google Scholar] [CrossRef]
- Ozog, S.; Timberlake, N.D.; Hermann, K.; Garijo, O.; Haworth, K.G.; Shi, G.; Glinkerman, C.M.; Schefter, L.E.; D’Souza, S.; Simpson, E.; et al. Resveratrol Trimer Enhances Gene Delivery to Hematopoietic Stem Cells by Reducing Antiviral Restriction at Endosomes. Blood 2019, 134, 1298–1311. [Google Scholar] [CrossRef]
- Petrillo, C.; Thorne, L.G.; Unali, G.; Schiroli, G.; Giordano, A.M.S.; Piras, F.; Cuccovillo, I.; Petit, S.J.; Ahsan, F.; Noursadeghi, M.; et al. Cyclosporine H Overcomes Innate Immune Restrictions to Improve Lentiviral Transduction and Gene Editing in Human Hematopoietic Stem Cells. Cell Stem Cell 2018, 23, 820–832.e9. [Google Scholar] [CrossRef]
- Suddala, K.C.; Lee, C.C.; Meraner, P.; Marin, M.; Markosyan, R.M.; Desai, T.M.; Cohen, F.S.; Brass, A.L.; Melikyan, G.B. Interferon-Induced Transmembrane Protein 3 Blocks Fusion of Sensitive but Not Resistant Viruses by Partitioning into Virus-Carrying Endosomes. PLoS Pathog. 2019, 15, e1007532. [Google Scholar] [CrossRef]
- Meischel, T.; Fritzlar, S.; Villalon-Letelier, F.; Tessema, M.B.; Brooks, A.G.; Reading, P.C.; Londrigan, S.L. IFITM Proteins That Restrict the Early Stages of Respiratory Virus Infection Do Not Influence Late-Stage Replication. J. Virol. 2021, 95, e00837-e21. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Guo, F.; Liu, F.; Cuconati, A.; Chang, J.; Block, T.M.; Guo, J.-T. Interferon Induction of IFITM Proteins Promotes Infection by Human Coronavirus OC43. Proc. Natl. Acad. Sci. USA 2014, 111, 6756–6761. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.L.; Wilson, H.; Iyer, S.S.; Coss, K.; Doores, K.; Smith, S.; Kellam, P.; Finzi, A.; Borrow, P.; Hahn, B.H.; et al. Resistance of Transmitted Founder HIV-1 to IFITM-Mediated Restriction. Cell Host Microbe 2016, 20, 429–442. [Google Scholar] [CrossRef]
- Wang, Y.; Pan, Q.; Ding, S.; Wang, Z.; Yu, J.; Finzi, A.; Liu, S.-L.; Liang, C. The V3 Loop of HIV-1 Env Determines Viral Susceptibility to IFITM3 Impairment of Viral Infectivity. J. Virol. 2017, 91, e02441-16. [Google Scholar] [CrossRef]
- Ding, S.; Pan, Q.; Liu, S.-L.; Liang, C. HIV-1 Mutates to Evade IFITM1 Restriction. Virology 2014, 454–455, 11–24. [Google Scholar] [CrossRef]
- Jia, R.; Ding, S.; Pan, Q.; Liu, S.-L.; Qiao, W.; Liang, C. The C-Terminal Sequence of IFITM1 Regulates Its Anti-HIV-1 Activity. PLoS ONE 2015, 10, e0118794. [Google Scholar] [CrossRef]
- Lista, M.J.; Winstone, H.; Wilson, H.D.; Dyer, A.; Pickering, S.; Galao, R.P.; De Lorenzo, G.; Cowton, V.M.; Furnon, W.; Suarez, N.; et al. The P681H Mutation in the Spike Glycoprotein of the Alpha Variant of SARS-CoV-2 Escapes IFITM Restriction and Is Necessary for Type I Interferon Resistance. J. Virol. 2022, 96, e0125022. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.L.; Juergens, D.; Bennett, N.R.; Trippe, B.L.; Yim, J.; Eisenach, H.E.; Ahern, W.; Borst, A.J.; Ragotte, R.J.; Milles, L.F.; et al. De Novo Design of Protein Structure and Function with RFdiffusion. Nature 2023, 620, 1089–1100. [Google Scholar] [CrossRef]
- Vázquez Torres, S.; Benard Valle, M.; Mackessy, S.P.; Menzies, S.K.; Casewell, N.R.; Ahmadi, S.; Burlet, N.J.; Muratspahić, E.; Sappington, I.; Overath, M.D.; et al. De Novo Designed Proteins Neutralize Lethal Snake Venom Toxins. Nature 2025, 639, 225–231. [Google Scholar] [CrossRef]
- Lauko, A.; Pellock, S.J.; Sumida, K.H.; Anishchenko, I.; Juergens, D.; Ahern, W.; Jeung, J.; Shida, A.; Hunt, A.; Kalvet, I.; et al. Computational Design of Serine Hydrolases. Science 2025, 388, eadu2454. [Google Scholar] [CrossRef]
- Glögl, M.; Krishnakumar, A.; Ragotte, R.J.; Goreshnik, I.; Coventry, B.; Bera, A.K.; Kang, A.; Joyce, E.; Ahn, G.; Huang, B.; et al. Target-Conditioned Diffusion Generates Potent TNFR Superfamily Antagonists and Agonists. Science 2024, 386, 1154–1161. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arduini, A.; Katiyar, H.; Liang, C. Progress in Pseudotyping Lentiviral Vectors Towards Cell-Specific Gene Delivery In Vivo. Viruses 2025, 17, 802. https://doi.org/10.3390/v17060802
Arduini A, Katiyar H, Liang C. Progress in Pseudotyping Lentiviral Vectors Towards Cell-Specific Gene Delivery In Vivo. Viruses. 2025; 17(6):802. https://doi.org/10.3390/v17060802
Chicago/Turabian StyleArduini, Ariana, Harshita Katiyar, and Chen Liang. 2025. "Progress in Pseudotyping Lentiviral Vectors Towards Cell-Specific Gene Delivery In Vivo" Viruses 17, no. 6: 802. https://doi.org/10.3390/v17060802
APA StyleArduini, A., Katiyar, H., & Liang, C. (2025). Progress in Pseudotyping Lentiviral Vectors Towards Cell-Specific Gene Delivery In Vivo. Viruses, 17(6), 802. https://doi.org/10.3390/v17060802