
Surveillance and Molecular Characterization of Marek’s Disease Virus (MDV) Strains Circulating in Tanzania

Abstract
1. Introduction
2. Materials and Methods
2.1. Study Sites
2.2. Study Design
2.3. Samples and Sampling Approach
2.4. Sample Processing and DNA Extraction
2.5. Molecular Screening for MDV Field Strain
2.6. PCR Amplification of MDV Oncogenic Genes
2.7. Sequencing of the Gene Amplicons and Sequence Analysis
2.8. Ethical Clearance
3. Results
3.1. Screening of MDV Genome by PCR
3.2. PCR Amplification and Sequence Analysis of Meq, pp38, and vIL-8 Genes
3.3. Pairwise Distance and Phylogenetic Analysis of meq Gene
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kleyn, F.J.; Ciacciariello, M. Future Demands of the Poultry Industry: Will We Meet Our Commitments Sustainably in Developed and Developing Economies ? World’s Poult. Sci. J. 2021, 77, 267–278. [Google Scholar] [CrossRef]
- Mramba, R.P.; Mwantambo, P.A. The Impact of Management Practices on the Disease and Mortality Rates of Broilers and Layers Kept by Small-Scale Farmers in Dodoma Urban District, Tanzania. Heliyon 2024, 10, e29624. [Google Scholar] [CrossRef]
- Komwihangilo, D. The Role of Chicken in the Tanzanian Economy and Opportunities for Development: An Overview. In Proceedings of the First ACGG Tanzania Innovation Platform Meeting, Dar Es Salaam, Tanzania, 13–14 July 2015; pp. 1–36. Available online: https://cgspace.cgiar.org/items/81bc61b9-e580-43ca-b849-92d8c44a3aab (accessed on 11 May 2025).
- Alemayehu, T.; Bruno, J.E.; Poole, E.J.; Getachew, F.; Goromela, E.; Mbaga, S.; Dessie, T. Household Baseline Data in Tanzania: Monitoring Delivery of Chicken Genetic Gains; ILRI Research Report 49; International Livestock Research Institute (ILRI): Nairobi, Kenya, 2018; pp. 1–23. Available online: https://cgspace.cgiar.org/server/api/core/bitstreams/a131ff7c-0a1d-4355-83b4-a814c4553dae/content (accessed on 11 May 2025).
- Bell, A.S.; Kennedy, D.A.; Jones, M.J.; Cairns, C.L.; Pandey, U.; Dunn, P.A.; Szpara, M.L.; Read, A.F. Molecular Epidemiology of Marek’s Disease Virus in Central Pennsylvania, USA. Virus Evol. 2019, 5, vey042. [Google Scholar] [CrossRef] [PubMed]
- OECD-FAO. Chapter 6: Meat. In OECD-FAO Agricultural Outlook 2021–2030; OECD Publishing: Paris, France, 2021; pp. 163–177. Available online: https://openknowledge.fao.org/server/api/core/bitstreams/313b0161-6176-4a76-b505-6f6d3836b9c7/content (accessed on 11 May 2025).
- Biggs, P.M. The History and Biology of Marek’s Disease Virus. In Marek’s Disease. Current Topics in Microbiology and Immunology; Hirai, K., Ed.; Springer: Berlin/Heidelberg, Germany, 2000; Volume 255, pp. 1–24. [Google Scholar] [CrossRef]
- Calnek, B.W. Pathogenesis of Marek’s Disease Virus Infection Introduction. In Marek’s Disease. Current Topics in Microbiology and Immunology; Hirai, K., Ed.; Springer: Berlin/Heidelberg, Germany, 2000; Volume 255, pp. 25–55. [Google Scholar] [CrossRef]
- Bertzbach, L.D.; Kheimar, A.; Ali, F.A.Z.; Kaufer, B.B. Viral Factors Involved in Marek’s Disease Virus (MDV) Pathogenesis. Curr. Clin. Microbiol. Reports 2018, 5, 238–244. [Google Scholar] [CrossRef]
- McPherson, M.C.; Delany, M.E. Virus and Host Genomic, Molecular, and Cellular Interactions during Marek’s Disease Pathogenesis and Oncogenesis. Poult. Sci. 2016, 95, 412–429. [Google Scholar] [CrossRef]
- Nair, V. Latency and Tumorigenesis in Marek ’ s Disease. Avian Dis. 2013, 57, 360–365. [Google Scholar] [CrossRef]
- Witter, R.L.; Calnek, B.W.; Buscaglia, C.; Gimeno, I.M.; Schat, K.A. Classification of Marek’s Disease Viruses According to Pathotype: Philosophy and Methodology. Avian Pathol. 2005, 34, 75–90. [Google Scholar] [CrossRef]
- Emad, A.; El-Kenawy, A.A.; El-Tholoth, M. Molecular Characterization of Marek’s Disease Virus Reveals Reticuloendotheliosis Virus-Long Terminal Repeat Integration in the Genome of the Field Isolates in Egypt. Poult. Sci. 2024, 103, 103722. [Google Scholar] [CrossRef]
- Cui, X.; Lee, L.F.; Reed, W.M.; Kung, H.-J.; Reddy, S.M. Marek’s Disease Virus-Encoded VIL-8 Gene Is Involved in Early Cytolytic Infection but Dispensable for Establishment of Latency. J. Virol. 2004, 78, 4753–4760. [Google Scholar] [CrossRef]
- Stolz, M.L.; McCormick, C. The BZIP Proteins of Oncogenic Viruses. Viruses 2020, 12, 757. [Google Scholar] [CrossRef]
- Motai, Y.; Murata, S.; Sato, J.; Nishi, A.; Maekawa, N.; Okagawa, T.; Konnai, S.; Ohashi, K. Characterization of a Very Short Meq Protein Isoform in a Marek’s Disease Virus Strain in Japan. Vet. Sci. 2024, 11, 43. [Google Scholar] [CrossRef]
- Chacón, R.D.; Sánchez-Llatas, C.J.; Pajuelo, S.L.; Diaz Forero, A.J.; Jimenez-Vasquez, V.; Médico, J.A.; Soto-Ugaldi, L.F.; Astolfi-Ferreira, C.S.; Piantino Ferreira, A.J. Molecular Characterization of the Meq Oncogene of Marek’s Disease Virus in Vaccinated Brazilian Poultry Farms Reveals Selective Pressure on Prevalent Strains. Vet. Q. 2024, 44, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.S.; Ohashi, K.; Onuma, M. Diversity (Polymorphism) of the Meq Gene in the Attenuated Marek’s Disease Virus (MDV) Serotype 1 and MDV-Transformed Cell Lines. J. Vet. Med. Sci. 2002, 64, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Shamblin, C.E.; Greene, N.; Arumugaswami, V.; Dienglewicz, R.L.; Parcells, M.S. Comparative Analysis of Marek’s Disease Virus (MDV) Glycoprotein-, Lytic Antigen Pp38- and Transformation Antigen Meq-Encoding Genes: Association of Meq Mutations with MDVs of High Virulence. Vet. Microbiol. 2004, 102, 147–167. [Google Scholar] [CrossRef]
- Oluwayinka, E.B.; Otesile, E.B.; Oni, O.O.; Ajayi, O.L.; Dunn, J.R. Molecular Characterization and Phylogenetic Analysis of Marek’s Disease Virus in Chickens from Ogun State, Nigeria. Avian Pathol. 2023, 52, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Lupiani, B.; Lee, L.F.; Cui, X.; Gimeno, I.; Anderson, A.; Morgan, R.W.; Silva, R.F.; Witter, R.L.; Kung, H.J.; Reddy, S.M. Marek’s Disease Virus-Encoded Meq Gene Is Involved in Transformation of Lymphocytes but Is Dispensable for Replication. Proc. Natl. Acad. Sci. USA 2004, 101, 11815–11820. [Google Scholar] [CrossRef]
- Reddy, S.M.; Lupiani, B.; Gimeno, I.M.; Silva, R.F.; Lee, L.F.; Witter, R.L. Rescue of a Pathogenic Marek’s Disease Virus with Overlapping Cosmid DNAs: Use of a Pp38 Mutant to Validate the Technology for the Study of Gene Function. Proc. Natl. Acad. Sci. USA 2002, 99, 7054–7059. [Google Scholar] [CrossRef]
- You, Y.; Hagag, I.T.; Kheimar, A.; Bertzbach, L.D.; Kaufer, B.B. Characterization of a Novel Viral Interleukin 8 (VIL-8) Splice Variant Encoded by Marek’s Disease Virus. Microorganisms 2021, 9, 1457. [Google Scholar] [CrossRef]
- Engel, A.T.; Selvaraj, R.K.; Kamil, J.P.; Osterrieder, N.; Kaufer, B.B. Marek’s Disease Viral Interleukin-8 Promotes Lymphoma Formation through Targeted Recruitment of B Cells and CD4 + CD25 + T Cells. J. Virol. 2012, 86, 8536–8545. [Google Scholar] [CrossRef]
- Parcells, M.S.; Lin, S.; Dienglewicz, R.L.; Majerciak, V.; Robinson, D.A.N.R.; Chen, H.; Wu, Z.; Dubyak, G.R.; Brunovskis, P.; Hunt, H.D.; et al. Marek’ s Disease Virus (MDV) Encodes an Interleukin-8 Homolog (VIL-8): Characterization of the VIL-8 Protein and a VIL-8 Deletion Mutant MDV†. J Virol. 2001, 75, 5159–5173. [Google Scholar] [CrossRef]
- Gong, Z.; Zhang, L.; Wang, J.; Chen, L.; Shan, H.; Wang, Z.; Ma, H. Isolation and Analysis of a Very Virulent Marek’s Disease Virus Strain in China. Virol. J. 2013, 10, 155. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Deka, D. Ramneek Sequence Analysis of Meq Oncogene among Indian Isolates of Marek’s Disease Herpesvirus. Meta Gene 2016, 9, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Lachheb, J.; Mastour, H.; Nsiri, J.; Kaboudi, K.; Choura, I.; Ammouna, F.; Amara, A.; Ghram, A. Newly Detected Mutations in the Meq Oncogene and Molecular Pathotyping of Very Virulent Marek’s Disease Herpesvirus in Tunisia. Arch. Virol. 2020, 165, 2589–2597. [Google Scholar] [CrossRef]
- Tian, M.; Zhao, Y.; Lin, Y.; Zou, N.; Liu, C.; Liu, P.; Cao, S.; Wen, X.; Huang, Y. Comparative Analysis of Oncogenic Genes Revealed Unique Evolutionary Features of Field Marek’s Disease Virus Prevalent in Recent Years in China. Virol. J. 2011, 8, 121. [Google Scholar] [CrossRef] [PubMed]
- Sailen, A.M.; Marisa, J.M.; Masanja, P.; Mchonde, S.P.; Anderson, M.M. Trends in Diagnosis of Marek’s Disease (MD) in Poultry at Central Veterinary Laboratory in Dar Es Salaam, Tanzania. Proc. Tanzan. Vet. J. 2001, 35, 54–57. [Google Scholar]
- Kimbita, E.; Maeda, A. A Survey of Marek’s Disease Outbreak in Morogoro Township. Tanzania Vet. Bull. 1998, 8, 14–17. Available online: https://tvj.sua.ac.tz/vet2/index.php/TVJ/article/view/323 (accessed on 11 May 2025).
- Witter, R.L. Control Strategies for Marek’s Disease: A Perspective for the Future. Poult. Sci. 1998, 77, 1197–1203. [Google Scholar] [CrossRef]
- Ahmed, S.K. How to Choose a Sampling Technique and Determine Sample Size for Research: A Simplified Guide for Researchers. Oral Oncol. Rep. 2024, 12, 100662. [Google Scholar] [CrossRef]
- Cao, W.; Mays, J.; Dunn, J.; Fulton, R.; Silva, R.; Fadly, A. Use of Polymerase Chain Reaction in Detection of Marek’s Disease and Reticuloendotheliosis Viruses in Formalin-Fixed, Paraffin-Embedded Tumorous Tissues. Avian Dis. 2013, 57, 785–789. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Sanderford, M.; Sharma, S.; Tamura, K. MEGA12: Molecular Evolutionary Genetic Analysis Version 12 for Adaptive and Green Computing. Mol. Biol. Evol. 2024, 41, msae263. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The Rapid Generation of Mutation Data Matrices from Protein Sequences. Bioinformatics 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Sato, J.; Murata, S.; Yang, Z.; Kaufer, B.B.; Fujisawa, S.; Seo, H.; Maekawa, N.; Okagawa, T.; Konnai, S.; Osterrieder, N.; et al. Effect of Insertion and Deletion in the Meq Protein Encoded by Highly Oncogenic Marek’s Disease Virus on Transactivation Activity and Virulence. Viruses 2022, 14, 382. [Google Scholar] [CrossRef]
- Lee, S.I.; Takagi, M.; Ohashi, K.; Sugimoto, C.; Onuma, M. Difference in the Meq Gene between Oncogenic and Attenuated Strains of Marek’s Disease Virus Serotype 1. J. Vet. Med. Sci. 2000, 62, 287–292. [Google Scholar] [CrossRef]
- Song, B.; Zeb, J.; Hussain, S.; Aziz, M.U.; Circella, E.; Casalino, G.; Camarda, A.; Yang, G.; Buchon, N.; Sparagano, O. A Review on the Marek’s Disease Outbreak and Its Virulence-Related Meq Genovariation in Asia between 2011 and 2021. Animals 2022, 12, 540. [Google Scholar] [CrossRef]
- Deng, Q.; Shi, M.; Li, Q.; Wang, P.; Li, M.; Wang, W.; Gao, Y.; Li, H.; Lin, L.; Huang, T.; et al. Analysis of the Evolution and Transmission Dynamics of the Field MDV in China during the Years 1995–2020, Indicating the Emergence of a Unique Cluster with the Molecular Characteristics of Vv+ MDV That Has Become Endemic in Southern China. Transbound. Emerg. Dis. 2021, 68, 3574–3587. [Google Scholar] [CrossRef] [PubMed]
- Buscaglia, C.; Nervi, P.; Risso, M. Characterization of Four Very Virulent Argentinian Strains of Marek’s Disease Virus and the Influence of One of Those Isolates on Synergism between Marek’s Disease Vaccine Viruses. Avian Pathol. 2004, 33, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, I.M.; Witter, R.L.; Hunt, H.D.; Reddy, S.M.; Lee, L.F.; Silva, R.F. The Pp38 Gene of Marek’s Disease Virus (MDV) Is Necessary for Cytolytic Infection of B Cells and Maintenance of the Transformed State but Not for Cytolytic Infection of the Feather Follicle Epithelium and Horizontal Spread of MDV. J. Virol. 2005, 79, 4545–4549. [Google Scholar] [CrossRef]
- Abd-Ellatieff, H.A.; Abou Rawash, A.A.; Ellakany, H.F.; Goda, W.M.; Suzuki, T.; Yanai, T. Molecular Characterization and Phylogenetic Analysis of a Virulent Marek’s Disease Virus Field Strain in Broiler Chickens in Japan. Avian Pathol. 2018, 47, 47–57. [Google Scholar] [CrossRef]
- Schat, K.; Nair, V. Neoplastic Diseases: Marek’s Disease. In Diseases of Poultry; Swayne, D.E., Glisson, J.R., McDougald, L.R., Nolan, L.K., Suarez, D.L., Nair, V.L., Eds.; WyleyBlackwell: Hoboken, NJ, USA, 2013; pp. 515–552. [Google Scholar]
- Tulman, E.R.; Afonso, C.L.; Lu, Z.; Zsak, L.; Rock, D.L.; Kutish, G.F. The Genome of a Very Virulent Marek’s Disease Virus. J. Virol. 2000, 74, 7980–7988. [Google Scholar] [CrossRef]
- Spatz, S.J.; Petherbridge, L.; Zhao, Y.; Nair, V. Comparative Full-Length Sequence Analysis of Oncogenic and Vaccine (Rispens) Strains of Marek’s Disease Virus. J. Gen. Virol. 2007, 88, 1080–1096. [Google Scholar] [CrossRef] [PubMed]
- Renz, K.G.; Cheetham, B.F.; Walkden-Brown, S.W. Differentiation between Pathogenic Serotype 1 Isolates of Marek’s Disease Virus and the Rispens CVI988 Vaccine in Australia Using Real-Time PCR and High Resolution Melt Curve Analysis. J. Virol. Methods 2013, 187, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Atkins, K.E.; Read, A.F.; Savill, N.J.; Renz, K.G.; Walkden-Brown, S.W.; Woolhouse, M.E.J. Modelling Marek’s Disease Virus (MDV) Infection: Parameter Estimates for Mortality Rate and Infectiousness. BMC Vet. Res. 2011, 7, 70. [Google Scholar] [CrossRef]
- Witter, R.L. Increased Virulence of Marek’s Disease Virus Field Isolates. Avian Dis. 1997, 41, 149–163. [Google Scholar] [CrossRef]
- Nair, V. Evolution of Marek’s Disease—A Paradigm for Incessant Race between the Pathogen and the Host. Vet. J. 2005, 170, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Yehia, N.; El-Sayed, H.S.; Omar, S.E.; Erfan, A.; Amer, F. Genetic Evolution of Marek’s Disease Virus in Vaccinated Poultry Farms. Vet. World 2021, 14, 1342–1353. [Google Scholar] [CrossRef]
- Patria, J.N.; Jwander, L.; Mbachu, I.; Parcells, L.; Ladman, B.; Trimpert, J.; Kaufer, B.B.; Tavlarides-hontz, P.; Parcells, M.S. The Meq Genes of Nigerian Marek’s Disease Virus (MDV) Field Isolates Contain Mutations Common to Both European and US High Virulence Strains. Viruses 2025, 17, 56. [Google Scholar] [CrossRef]
- Trimpert, J.; Groenke, N.; Jenckel, M.; He, S.; Kunec, D.; Szpara, M.L.; Spatz, S.J.; Osterrieder, N.; McMahon, D.P. A Phylogenomic Analysis of Marek’s Disease Virus Reveals Independent Paths to Virulence in Eurasia and North America. Evol. Appl. 2017, 10, 1091–1101. [Google Scholar] [CrossRef]
- Haunshi, S.; Nishitha, Y.; Subbiah, M.; Krishna, S.V.; Kannaki, T.R.; Priyanka, E.; Nishitha, Y.; Krishna, S.V.; Haunshi, S.; Subbiah, M. Molecular Detection and Phylogenetic Analysis of Marek’s Disease Virus Virulence-Associated Genes from Vaccinated Flocks in Southern India Reveals Circulation of Virulent MDV Genotype. Transbound. Emerg. Dis. 2022, 69, e244–e253. [Google Scholar] [CrossRef]
- Liu, J.-L.; Teng, M.; Zheng, L.-P.; Zhu, F.-X.; Ma, S.-X.; Li, L.-Y.; Zhang, Z.-H.; Chai, S.-J.; Yao, Y.; Luo, J. Emerging Hypervirulent Marek’s Disease Virus Variants Significantly Overcome Protection Conferred by Commercial Vaccines. Viruses 2023, 15, 1434. [Google Scholar] [CrossRef]
- Bertzbach, L.D.; Conradie, A.M.; You, Y.; Kaufer, B.B. Latest Insights into Marek’s Disease Virus Pathogenesis and Tumorigenesis. Cancers 2020, 12, 647. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Zhao, X.; Zhu, X.; Kong, Z.; Liao, Y.; Teng, M.; Yao, Y.; Luo, J.; Nair, V.; Zhuang, G.; et al. Fully Attenuated Meq and Pp38 Double Gene Deletion Mutant Virus Confers Superior Immunological Protection against Highly Virulent Marek’s Disease Virus Infection. Microbiol. Spectr. 2022, 10, e02871-22. [Google Scholar] [CrossRef] [PubMed]
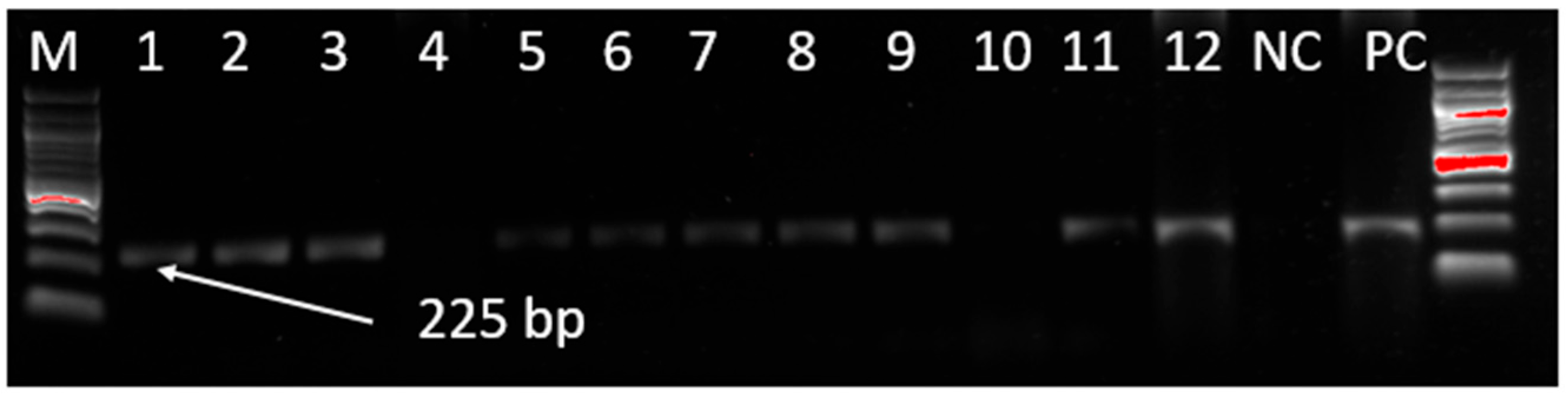
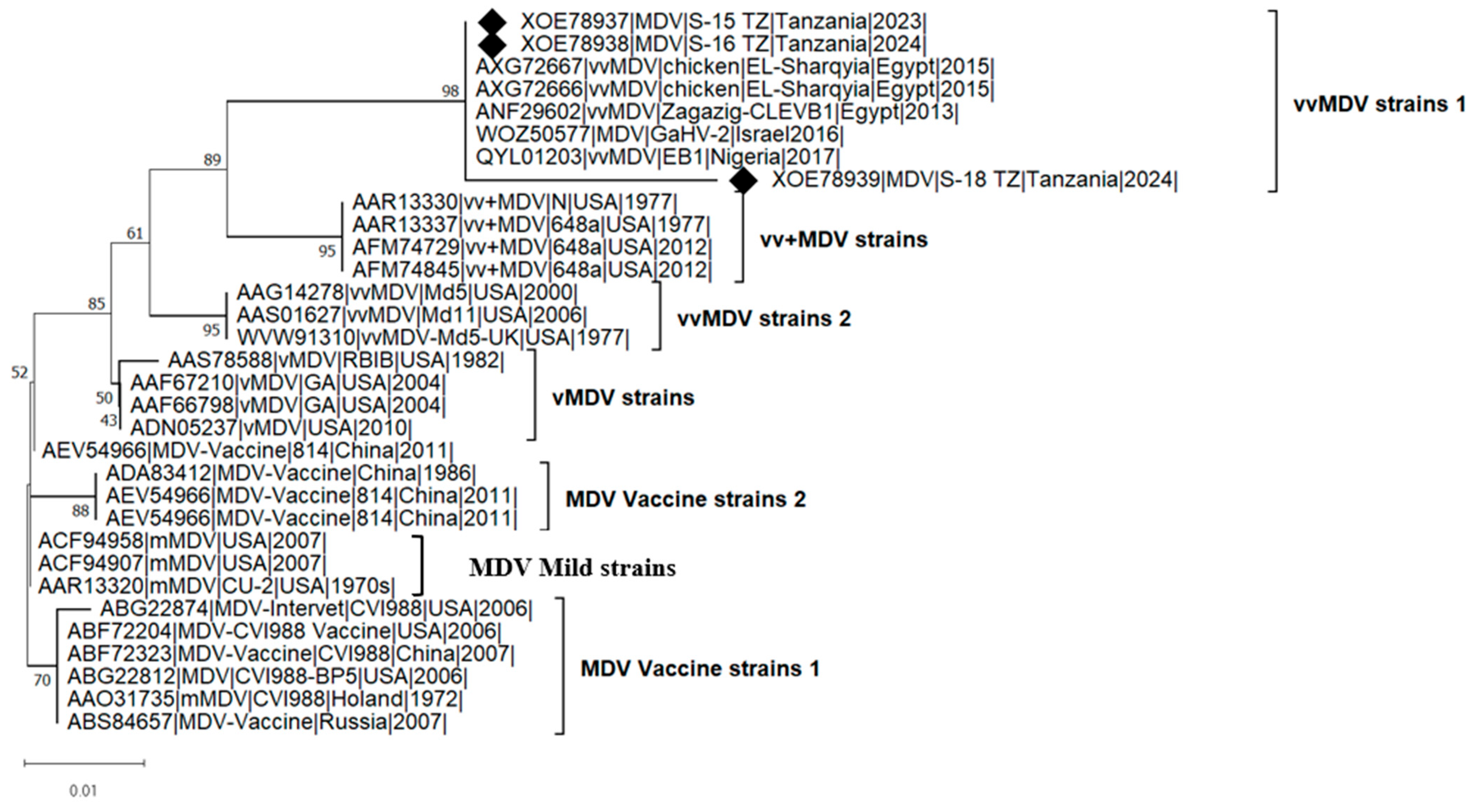

{kind=link}
{kind=link}
| Zone/Region | Ratio | Multiplier | Sample Size |
|---|---|---|---|
| Southern/Mtwara | 1 | 0.1 | 77 |
| Central/Dodoma | 1 | 0.1 | 77 |
| Southern Highland/Iringa | 1 | 0.1 | 77 |
| Northern/Arusha | 2 | 0.2 | 154 |
| Lake/Mwanza | 2 | 0.2 | 154 |
| Eastern/Dar | 3 | 0.3 | 230 |
| Total sample size (chickens) | 769 | ||
| Gene | Forward Primer | Reverse Primer | Amplicon Size |
|---|---|---|---|
| meq | 5′-GGCACGGTACAGGTGTAAAGAG-3′ | 5′-GCATAGACGATGTGCTGCTGAG-3′ | 1081 bp |
| pp38 | 5′-TCATCTTCAACCCACAGCCATCC-3′ | 5′-TCGCTTAATCTCCGCCTCCAAC-3′ | 1006 bp |
| vIL-8 | 5′-GAGACCCAATAACAGGGAAATC-3′ | 5′-TAGACCGTATCCCTGCTCCATC-3′ | 887 bp |
| Zone/Region | Total Number of Samples Collected | PCR Positive by Sample Type | PCR Positive Samples by Zone/Region (Prevalence) | |
|---|---|---|---|---|
| Feather Tips (Live Chickens) | Internal ORGANS (DISEASED) | |||
| Lake/Mwanza | 154 | 27 (n = 125) | 25 (n = 29) | 52 (33.77%) |
| Eastern/Dar es Salaam | 230 | 8 (n = 91) | 22 (n = 139) | 30 (13.04%) |
| Central/Dodoma | 77 | 0 (n = 31) | 4 (n = 46) | 4 (5.19) |
| Northern/Arusha | 154 | 7 (n = 129) | 3 (n = 25) | 10 (6.49%) |
| Southern Highland/Iringa | 77 | 10 (n = 67) | 3 (n = 10) | 13 (16.88%) |
| Southern/Mtwara | 77 | 15 (n = 55) | 8 (n = 22) | 23 (29.87%) |
| Total | 769 | 67 (n = 498) | 65 (n = 271) | 132 (18.08%) |
| Sample ID | Collection Date | Location | Target Gene | Nucleotide Length (nt) | Nucleotide Identity (%) | Amino Acids Length (aa) | Amino Acid Identity (%) | Accession Number |
|---|---|---|---|---|---|---|---|---|
| S-1 | 7 December 2023 | Mwanza | pp38 | 870 | 99.66–100 | 290 | 97.93–100 | PV008123 |
| S-2 | 12 December 2023 | Mwanza | pp38 | 870 | 99.66–100 | 290 | 97.93–100 | PV008124 |
| S-3 | 13 March 2024 | DSM | pp38 | 870 | 99.31–99.77 | 290 | 99.43–99.77 | PV008125 |
| S-4 | 15 March 2024 | DSM | pp38 | 870 | 99.66–99.89 | 290 | 98.28–100 | PV008126 |
| S-5 | 18 November 2024 | Dodoma | pp38 | 870 | 99.54–99.89 | 290 | 96.47–100 | PV008127 |
| S-6 | 10 September 2023 | Arusha | pp38 | 786 | 99.62–100 | 262 | 98.85–100 | PV008128 |
| S-7 | 18 November 2024 | Iringa | pp38 | 870 | 99.77–100 | 290 | 98.28–100 | PV008129 |
| S-8 | 2 November 2023 | Mtwara | pp38 | 870 | 99.77–100 | 290 | 91.44–100 | PV008130 |
| S-9 | 5 November 2023 | Mtwara | pp38 | 870 | 99.66–100 | 290 | 97.93–100 | PV008131 |
| S-10 | 7 December 2023 | Mtwara | vIL-8 | 537 | 99.26–100 | 102 | 95.10–100 | PV008132 |
| S-12 | 13 March 2024 | DSM | vIL-8 | 816 | 99.02–99.88 | 102 | 99.02–100 | PV008133 |
| S-13 | 18 November 2024 | Dodoma | vIL-8 | 832 | 98.80–100 | 102 | 97.06–100 | PV008134 |
| S-15 | 7 December 2023 | Mwanza | meq | 1026 | 98.83–99.42 | 330 | 98.18–100 | PV082624 |
| S-16 | 15 January 2024 | Arusha | meq | 1026 | 98.83–99.42 | 330 | 98.18–100 | PV082625 |
| S-18 | 18 November 2024 | Dodoma | meq | 654 | 97.40–98.16 | 212 | 92.40–95.75 | PV082626 |
| Country | Strain | Type | Code | 71 | 77 | 80 | 88 | 93 | 119 | 139 | 153 | 176 | 180 | 277 | 337 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| USA | CVI988-BP5 | Vaccine | ABG22812 | S | E | D | A | Q | C | T | P | P | T | P | L |
| USA | CVI988 | Vaccine | ABF72204 | S | E | D | A | Q | C | T | P | P | T | P | L |
| China | 814 | Vaccine | AEV54966 | S | E | D | A | Q | C | T | P | P | T | P | L |
| Holand | CVI988 | mMDV | AA031735 | S | E | D | A | Q | C | T | P | P | T | P | L |
| USA | CU-2 | mMDV | AAR13320 | S | E | D | A | Q | C | T | P | P | T | P | L |
| USA | RBIB | vMDV | AAS78588 | A | K | D | A | Q | C | T | P | P | T | P | L |
| USA | GA | vMDV | AAF67210 | A | K | D | A | Q | C | T | P | P | T | P | L |
| USA | Md11 | vvMDV | AAS01627 | A | K | D | A | Q | C | T | P | P | T | A | L |
| Nigeria | Nigeria | vvMDV | WYC13993 | A | E | Y | T | R | C | A | P | A | A | A | L |
| Nigeria | EB1 | vvMDV | QYL01203 | A | E | Y | T | R | C | A | P | A | A | A | L |
| Egypt | EL-Sharqyia | vvMDV | AXG72666 | A | E | Y | T | R | C | A | P | A | A | A | L |
| Egypt | EL-Sharqyia | vvMDV | AXG72667 | A | E | Y | T | R | C | A | P | A | A | A | L |
| Egypt | CLEVB1 | vvMDV | ANF29602 | A | E | Y | T | R | C | A | P | A | A | A | L |
| Tanzania | S-15_TZ | vvMDV | XOE78937 | A | E | Y | T | R | C | A | P | A | A | A | L |
| Tanzania | S-16_TZ | vvMDV | XOE78937 | A | E | Y | T | R | C | A | P | A | A | A | L |
| Tanzania | S-18_TZ | vvMDV | XOE78937 | A | E | Y | T | R | C | A | P | A | A | A | L |
| USA | 684a | vv+MDV | AFM74845 | A | K | D | A | Q | R | T | Q | A | A | A | L |
| USA | N | vv+MDV | AAR13330 | A | K | D | A | Q | R | T | Q | A | A | A | L |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chengula, A.A.; Mpete, H.; Makasali, R.J. Surveillance and Molecular Characterization of Marek’s Disease Virus (MDV) Strains Circulating in Tanzania. Viruses 2025, 17, 698. https://doi.org/10.3390/v17050698
Chengula AA, Mpete H, Makasali RJ. Surveillance and Molecular Characterization of Marek’s Disease Virus (MDV) Strains Circulating in Tanzania. Viruses. 2025; 17(5):698. https://doi.org/10.3390/v17050698
Chicago/Turabian StyleChengula, Augustino Alfred, Herbertha Mpete, and Ramadhani Juma Makasali. 2025. "Surveillance and Molecular Characterization of Marek’s Disease Virus (MDV) Strains Circulating in Tanzania" Viruses 17, no. 5: 698. https://doi.org/10.3390/v17050698
APA StyleChengula, A. A., Mpete, H., & Makasali, R. J. (2025). Surveillance and Molecular Characterization of Marek’s Disease Virus (MDV) Strains Circulating in Tanzania. Viruses, 17(5), 698. https://doi.org/10.3390/v17050698