
KRT6A Restricts Influenza A Virus Replication by Inhibiting the Nuclear Import and Assembly of Viral Ribonucleoprotein Complex

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Plasmids and Small Interfering RNAs
2.3. Antibodies
2.4. Transfection and Virus Titration
2.5. Co-Immunoprecipitation Assay
2.6. Indirect Immunofluorescence Assay
2.7. RNA Quantification
2.8. Nuclear and Cytoplasmic Fractionation
2.9. Western Blotting
2.10. Dual-Luciferase Reporter Assay
2.11. Cell Viability Assay
2.12. Statistical Analysis
3. Results
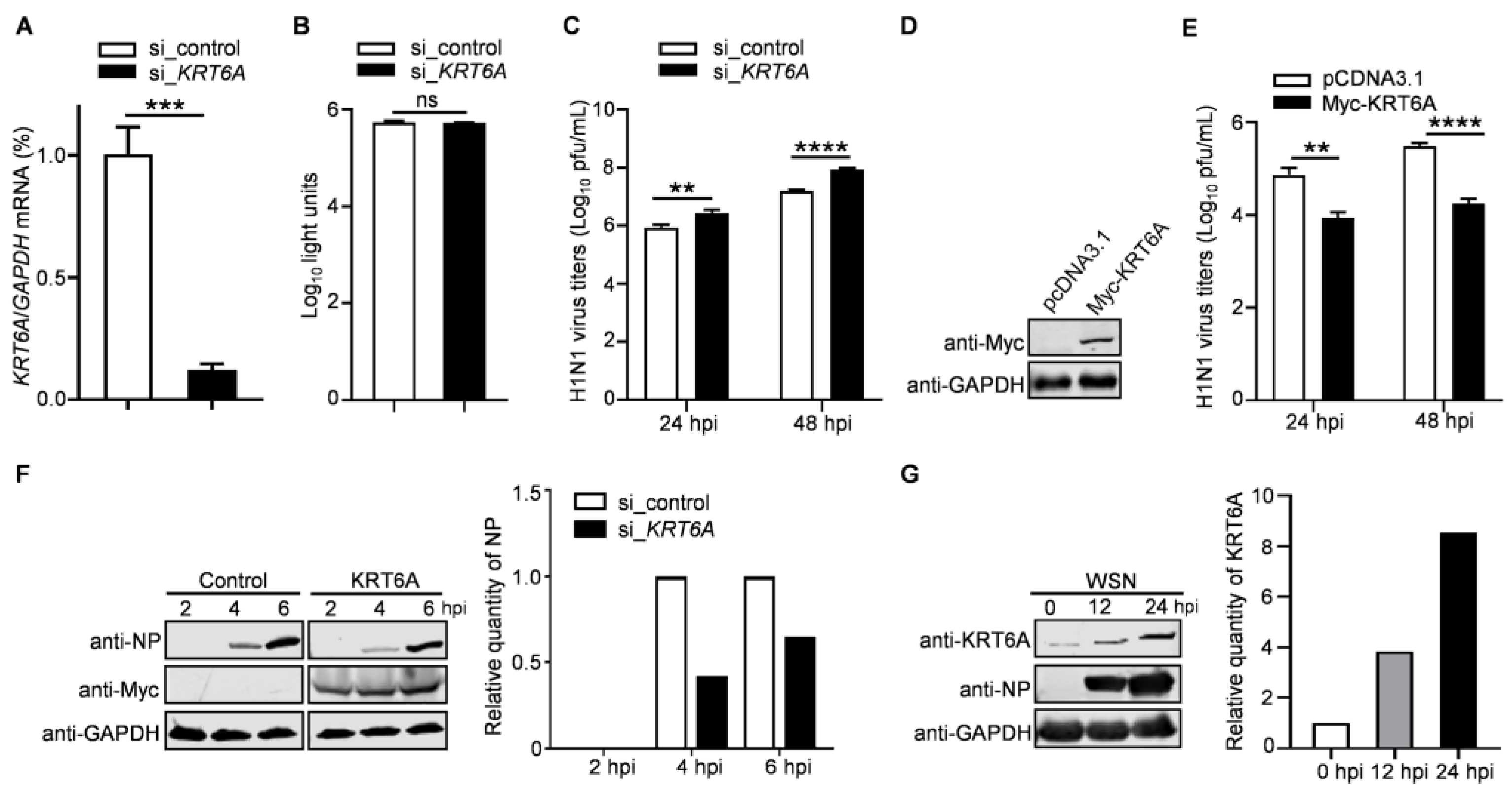
3.1. KRT6A Negatively Regulates the Replication of IAV
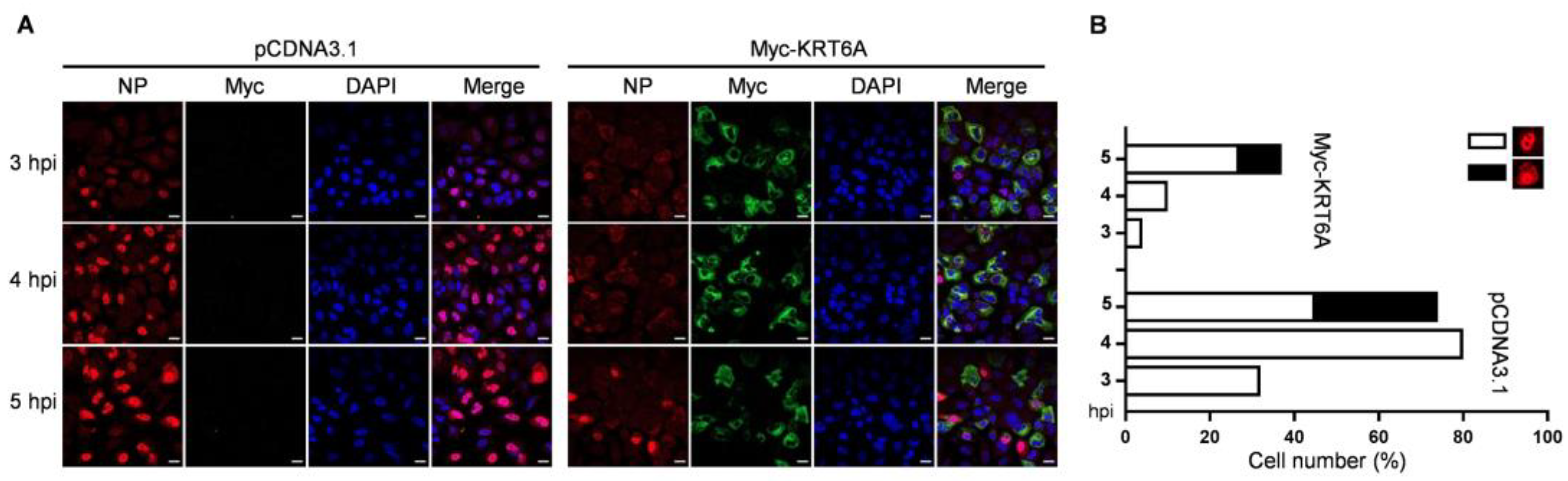
3.2. KRT6A Suppresses the Nuclear Import of Incoming vRNP Complex and Newly Synthesized NP
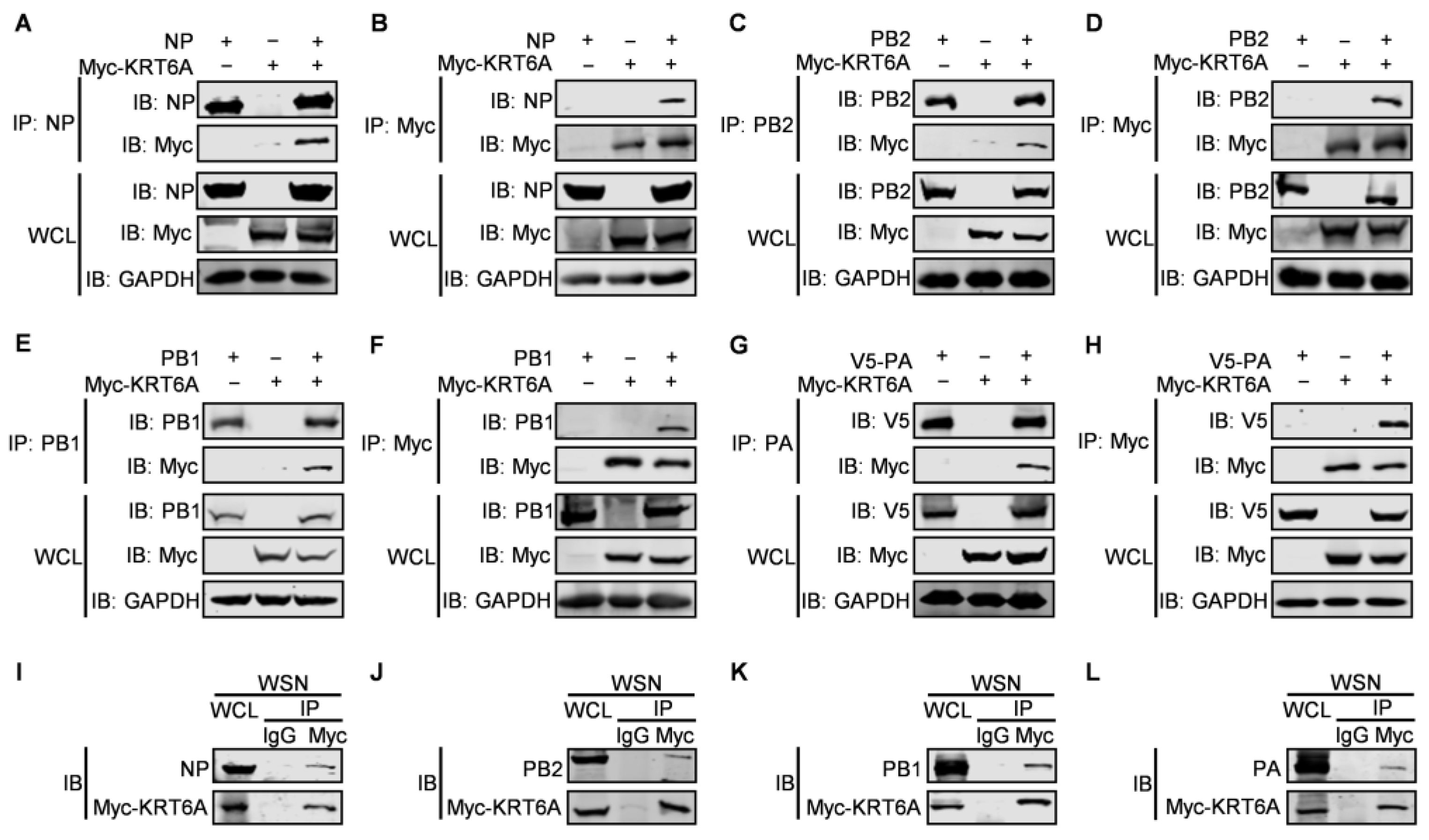
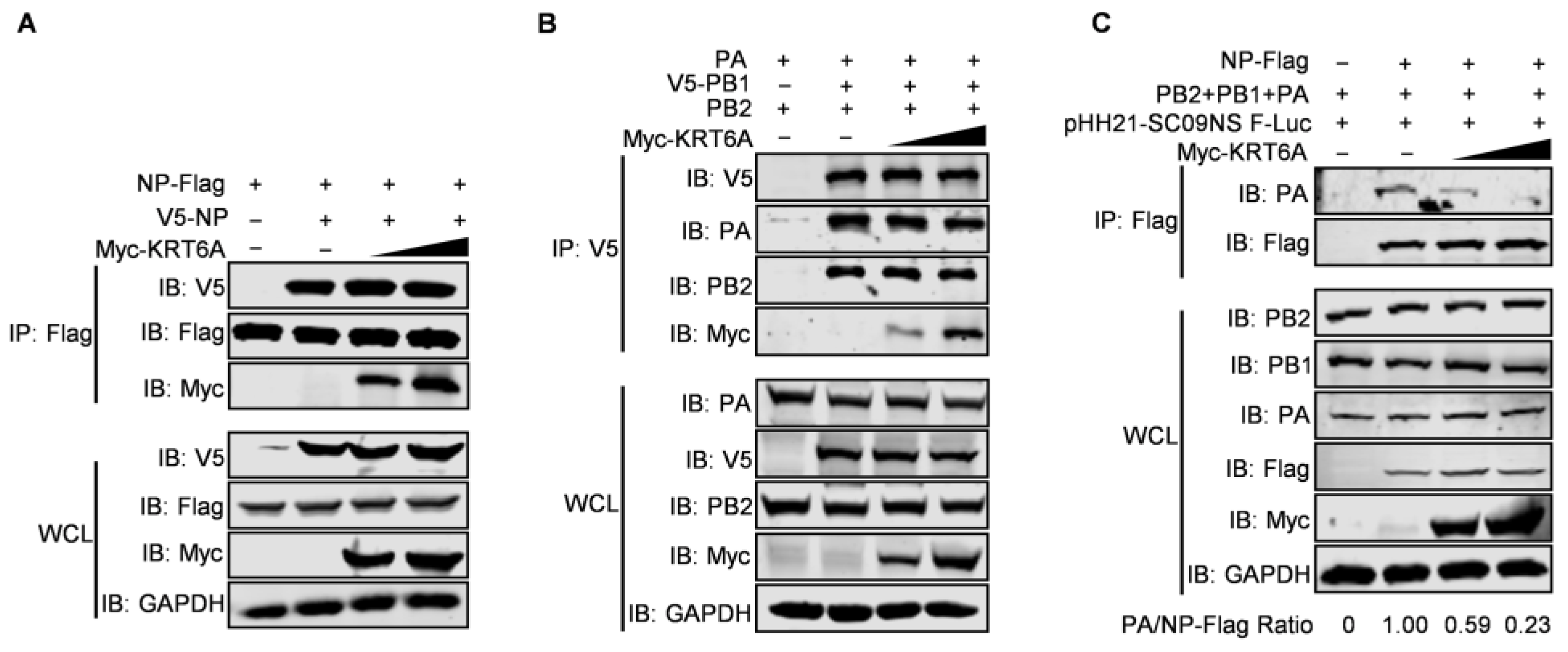
3.3. KRT6A Interacts with RNP Complex Proteins of IAV
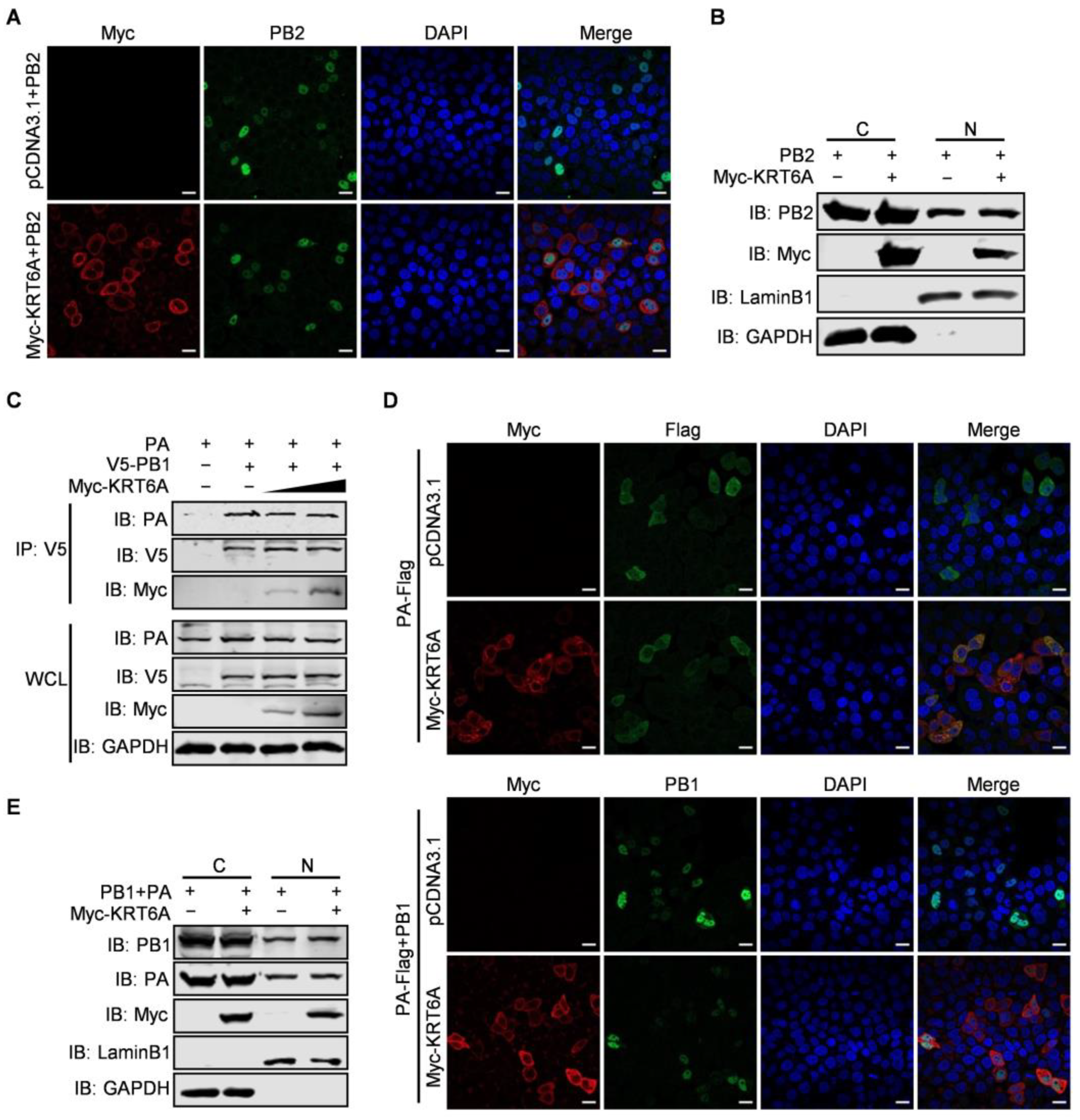
3.4. KRT6A Has No Effect on the Nuclear Import of PB2 or PB1-PA Heterodimer
3.5. KRT6A Weakens the Binding of NP with Importin α3
3.6. KRT6A Impairs vRNP Complex Assembly
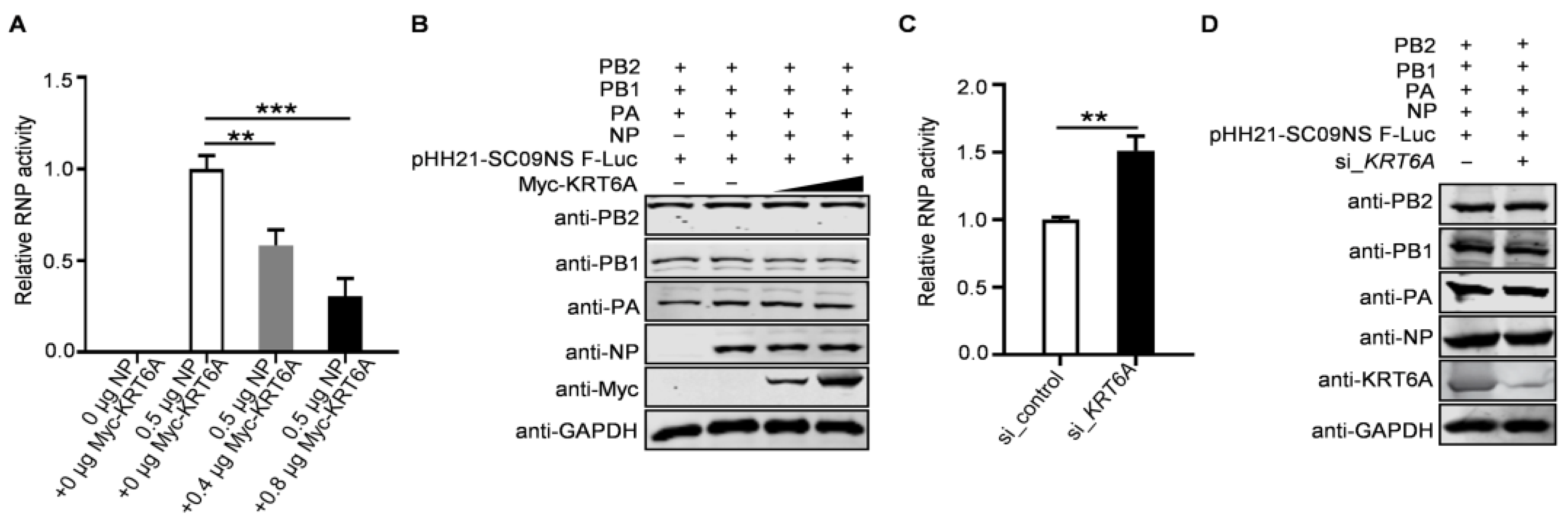
3.7. KRT6A Reduces the Polymerase Activity of IAV
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yamayoshi, S.; Watanabe, M.; Goto, H.; Kawaoka, Y. Identification of a Novel Viral Protein Expressed from the PB2 Segment of Influenza A Virus. J. Virol. 2016, 90, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef]
- Karakus, U.; Mena, I.; Kottur, J.; El Zahed, S.S.; Seoane, R.; Yildiz, S.; Chen, L.; Plancarte, M.; Lindsay, L.; Halpin, R.; et al. H19 influenza A virus exhibits species-specific MHC class II receptor usage. Cell Host Microbe 2024, 32, 1089–1102.e10. [Google Scholar] [CrossRef]
- Neumann, G.; Chen, H.; Gao, G.F.; Shu, Y.; Kawaoka, Y. H5N1 influenza viruses: Outbreaks and biological properties. Cell Res. 2010, 20, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Shi, J.; Cui, P.; Yan, C.; Zhang, Y.; Wang, C.; Zhang, Y.; Xing, X.; Zeng, X.; Liu, L.; et al. Novel H5N6 reassortants bearing the clade 2.3.4.4b HA gene of H5N8 virus have been detected in poultry and caused multiple human infections in China. Emerg. Microbes Infect. 2022, 11, 1174–1185. [Google Scholar] [CrossRef]
- Lai, S.; Qin, Y.; Cowling, B.J.; Ren, X.; Wardrop, N.A.; Gilbert, M.; Tsang, T.K.; Wu, P.; Feng, L.; Jiang, H.; et al. Global epidemiology of avian influenza A H5N1 virus infection in humans, 1997–2015: A systematic review of individual case data. Lancet Infect. Dis. 2016, 16, e108–e118. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Cao, B.; Hu, Y.; Feng, Z.; Wang, D.; Hu, W.; Chen, J.; Jie, Z.; Qiu, H.; Xu, K.; et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N. Engl. J. Med. 2013, 368, 1888–1897. [Google Scholar] [CrossRef]
- Shi, J.; Deng, G.; Kong, H.; Gu, C.; Ma, S.; Yin, X.; Zeng, X.; Cui, P.; Chen, Y.; Yang, H.; et al. H7N9 virulent mutants detected in chickens in China pose an increased threat to humans. Cell Res. 2017, 27, 1409–1421. [Google Scholar] [CrossRef]
- Li, C.; Chen, H. H7N9 Influenza Virus in China. Cold Spring Harb. Perspect. Med. 2021, 11, a038349. [Google Scholar] [CrossRef]
- Shi, J.Z.; Zeng, X.Y.; Cui, P.F.; Yan, C.; Chen, H.L. Alarming situation of emerging H5 and H7 avian influenza and effective control strategies. Emerg. Microbes Infect. 2023, 12, 2155072. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, J. H9N2 influenza virus in China: A cause of concern. Protein Cell 2015, 6, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Bao, P.; Liu, Y.; Zhang, X.; Fan, H.; Zhao, J.; Mu, M.; Li, H.; Wang, Y.; Ge, H.; Li, S.; et al. Human infection with a reassortment avian influenza A H3N8 virus: An epidemiological investigation study. Nat. Commun. 2022, 13, 6817. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yuan, H.; Gao, R.; Zhang, J.; Wang, D.; Xiong, Y.; Fan, G.; Yang, F.; Li, X.; Zhou, J.; et al. Clinical and epidemiological characteristics of a fatal case of avian influenza A H10N8 virus infection: A descriptive study. Lancet 2014, 383, 714–721. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, R.E.; Jaskunas, R.; Blobel, G.; Palese, P.; Moroianu, J. Nuclear import of influenza virus RNA can be mediated by viral nucleoprotein and transport factors required for protein import. J. Biol. Chem. 1995, 270, 22701–22704. [Google Scholar] [CrossRef]
- Martin, K.; Helenius, A. Transport of incoming influenza virus nucleocapsids into the nucleus. J. Virol. 1991, 65, 232–244. [Google Scholar] [CrossRef]
- Neumann, G.; Castrucci, M.R.; Kawaoka, Y. Nuclear import and export of influenza virus nucleoprotein. J. Virol. 1997, 71, 9690–9700. [Google Scholar] [CrossRef]
- Wang, P.; Palese, P.; O’Neill, R.E. The NPI-1/NPI-3 (karyopherin alpha) binding site on the influenza a virus nucleoprotein NP is a nonconventional nuclear localization signal. J. Virol. 1997, 71, 1850–1856. [Google Scholar] [CrossRef]
- Weber, F.; Kochs, G.; Gruber, S.; Haller, O. A classical bipartite nuclear localization signal on Thogoto and influenza A virus nucleoproteins. Virology 1998, 250, 9–18. [Google Scholar] [CrossRef]
- Ye, Q.; Krug, R.M.; Tao, Y.J. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 2006, 444, 1078–1082. [Google Scholar] [CrossRef]
- Zhu, W.; Feng, Z.; Chen, Y.; Yang, L.; Liu, J.; Li, X.; Liu, S.; Zhou, L.; Wei, H.; Gao, R.; et al. Mammalian-adaptive mutation NP-Q357K in Eurasian H1N1 Swine Influenza viruses determines the virulence phenotype in mice. Emerg. Microbes Infect. 2019, 8, 989–999. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, B.; Shi, J.; Yin, X.; Wang, G.; Cui, P.; Liu, L.; Deng, G.; Jiang, Y.; Li, C.; et al. Amino Acid Mutations A286V and T437M in the Nucleoprotein Attenuate H7N9 Viruses in Mice. J. Virol. 2020, 94, e01530-19. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, L.; Wang, G.; Shi, W.; Hu, Y.; Wang, B.; Zeng, X.; Tian, G.; Deng, G.; Shi, J.; et al. Influenza A virus use of BinCARD1 to facilitate the binding of viral NP to importin alpha7 is counteracted by TBK1-p62 axis-mediated autophagy. Cell. Mol. Immunol. 2022, 19, 1168–1184. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Kiyasu, Y.; Sugiyama, K.; Kimura, A.; Nakano, R.; Matsukage, A.; Nagata, K. An influenza virus replicon system in yeast identified Tat-SF1 as a stimulatory host factor for viral RNA synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18235–18240. [Google Scholar] [CrossRef]
- Momose, F.; Basler, C.F.; O’Neill, R.E.; Iwamatsu, A.; Palese, P.; Nagata, K. Cellular splicing factor RAF-2p48/NPI-5/BAT1/UAP56 interacts with the influenza virus nucleoprotein and enhances viral RNA synthesis. J. Virol. 2001, 75, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Moisy, D.; Avilov, S.V.; Jacob, Y.; Laoide, B.M.; Ge, X.Y.; Baudin, F.; Naffakh, N.; Jestin, J.L. HMGB1 Protein Binds to Influenza Virus Nucleoprotein and Promotes Viral Replication. J. Virol. 2012, 86, 9122–9133. [Google Scholar] [CrossRef]
- Zhang, X.X.; Pu, J.; Sun, Y.P.; Bi, Y.H.; Jiang, Z.M.; Xu, G.L.; Zhang, H.Y.; Cao, J.; Chang, K.C.; Liu, J.H.; et al. Neurovirulence of Avian Influenza Virus Is Dependent on the Interaction of Viral NP Protein with FMRP in the Murine Brain. J. Virol. 2021, 95, e01272-20. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Zhang, J.; Liang, L.; Wang, G.; Li, Q.; Zhu, P.; Zhou, Y.; Li, J.; Zhao, Y.; Sun, N.; et al. Phospholipid scramblase 1 interacts with influenza A virus NP, impairing its nuclear import and thereby suppressing virus replication. PLoS Pathog. 2018, 14, e1006851. [Google Scholar] [CrossRef]
- Zhang, J.S.; Huang, F.; Tan, L.K.; Bai, C.; Chen, B.; Liu, J.; Liang, J.R.; Liu, C.; Zhang, S.Y.; Lu, G.; et al. Host Protein Moloney Leukemia Virus 10 (MOV10) Acts as a Restriction Factor of Influenza A Virus by Inhibiting the Nuclear Import of the Viral Nucleoprotein. J. Virol. 2016, 90, 3966–3980. [Google Scholar] [CrossRef]
- Di Pietro, A.; Kajaste-Rudnitski, A.; Oteiza, A.; Nicora, L.; Towers, G.J.; Mechti, N.; Vicenzi, E. TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J. Virol. 2013, 87, 4523–4533. [Google Scholar] [CrossRef]
- Patil, G.; Zhao, M.; Song, K.; Hao, W.; Bouchereau, D.; Wang, L.; Li, S. TRIM41-Mediated Ubiquitination of Nucleoprotein Limits Influenza A Virus Infection. J. Virol. 2018, 92, e00905-18. [Google Scholar] [CrossRef]
- Wu, X.; Wang, J.; Wang, S.; Wu, F.; Chen, Z.; Li, C.; Cheng, G.; Qin, F.X. Inhibition of Influenza A Virus Replication by TRIM14 via Its Multifaceted Protein-Protein Interaction with NP. Front. Microbiol. 2019, 10, 344. [Google Scholar] [CrossRef]
- Shi, W.J.; Shan, Z.B.; Jiang, L.; Wang, G.W.; Wang, X.Y.; Chang, Y.; Hu, Y.Z.; Wang, B.; Li, Q.B.; Wang, Y.H.; et al. ABTB1 facilitates the replication of influenza A virus by counteracting TRIM4-mediated degradation of viral NP protein. Emerg. Microbes Infect. 2023, 12, 2270073. [Google Scholar] [CrossRef]
- Toivola, D.M.; Boor, P.; Alam, C.; Strnad, P. Keratins in health and disease. Curr. Opin. Cell Biol. 2015, 32, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.T.; Coulombe, P.A.; Kwan, R.; Omary, M.B. Types I and II Keratin Intermediate Filaments. Cold Spring Harb. Perspect. Biol. 2018, 10, a018275. [Google Scholar] [CrossRef] [PubMed]
- Hesse, M.; Zimek, A.; Weber, K.; Magin, T.M. Comprehensive analysis of keratin gene clusters in humans and rodents. Eur. J. Cell Biol. 2004, 83, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Chen, S.; Liu, H.B.; Parent, C.A.; Coulombe, P.A. Keratin 6 regulates collective keratinocyte migration by altering cell-cell and cell-matrix adhesion. J. Cell Biol. 2018, 217, 4314–4330. [Google Scholar] [CrossRef]
- Zhou, J.Z.; Jiang, G.Q.; Xu, E.W.; Zhou, J.X.; Liu, L.L.; Yang, Q.Y. Identification of SRXN1 and KRT6A as Key Genes in Smoking-Related Non-Small-Cell Lung Cancer Through Bioinformatics and Functional Analyses. Front. Oncol. 2022, 11, 810301. [Google Scholar] [CrossRef]
- Wang, H.J.; Liu, J.; Li, J.S.; Zang, D.; Wang, X.H.; Chen, Y.Y.; Gu, T.T.; Su, W.; Song, N. Identification of gene modules and hub genes in colon adenocarcinoma associated with pathological stage based on WGCNA analysis. Cancer Genet. 2020, 242, 1–7. [Google Scholar] [CrossRef]
- Chen, C.; Shan, H. Keratin 6A gene silencing suppresses cell invasion and metastasis of nasopharyngeal carcinoma via the beta-catenin cascade. Mol. Med. Rep. 2019, 19, 3477–3484. [Google Scholar]
- Yang, B.; Zhang, W.; Zhang, M.M.; Wang, X.H.; Peng, S.Z.; Zhang, R.S. KRT6A Promotes EMT and Cancer Stem Cell Transformation in Lung Adenocarcinoma. Technol. Cancer Res. Treat. 2020, 19, 1533033820921248. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, J.; Kong, F.; Li, Q.; Wang, J.; Ma, S.; Zhao, Y.; Liang, L.; Li, J.; Sun, N.; et al. Generation and application of replication-competent Venus-expressing H5N1, H7N9, and H9N2 influenza A viruses. Sci. Bull. 2018, 63, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wen, X.; Li, Q.; Jiang, L.; Wang, G.; Liang, L.; Wang, X.; Chen, H.; Li, C. Generation and application of two monoclonal antibodies targeting conserved linear epitopes in the NP protein of influenza A virus. J. Integr. Agric. 2021, 21, 2095–2105. [Google Scholar] [CrossRef]
- Zhu, P.; Liang, L.; Shao, X.; Luo, W.; Jiang, S.; Zhao, Q.; Sun, N.; Zhao, Y.; Li, J.; Wang, J.; et al. Host Cellular Protein TRAPPC6ADelta Interacts with Influenza A Virus M2 Protein and Regulates Viral Propagation by Modulating M2 Trafficking. J. Virol. 2017, 91, e01757-16. [Google Scholar] [CrossRef] [PubMed]
- Fodor, E.; Smith, M. The PA subunit is required for efficient nuclear accumulation of the PB1 subunit of the influenza A virus RNA polymerase complex. J. Virol. 2004, 78, 9144–9153. [Google Scholar] [CrossRef]
- Gabriel, G.; Klingel, K.; Otte, A.; Thiele, S.; Hudjetz, B.; Arman-Kalcek, G.; Sauter, M.; Shmidt, T.; Rother, F.; Baumgarte, S.; et al. Differential use of importin-alpha isoforms governs cell tropism and host adaptation of influenza virus. Nat. Commun. 2011, 2, 156. [Google Scholar] [CrossRef] [PubMed]
- Elton, D.; Medcalf, L.; Bishop, K.; Harrison, D.; Digard, P. Identification of amino acid residues of influenza virus nucleoprotein essential for RNA binding. J. Virol. 1999, 73, 7357–7367. [Google Scholar] [CrossRef]
- Mondal, A.; Dawson, A.R.; Potts, G.K.; Freiberger, E.C.; Baker, S.F.; Moser, L.A.; Bernard, K.A.; Coon, J.J.; Mehle, A. Influenza virus recruits host protein kinase C to control assembly and activity of its replication machinery. Elife 2017, 6, e26910. [Google Scholar] [CrossRef]
- Rogers, G.N.; Pritchett, T.J.; Lane, J.L.; Paulson, J.C. Differential sensitivity of human, avian, and equine influenza A viruses to a glycoprotein inhibitor of infection: Selection of receptor specific variants. Virology 1983, 131, 394–408. [Google Scholar] [CrossRef]
- Rust, M.J.; Lakadamyali, M.; Zhang, F.; Zhuang, X. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat. Struct. Mol. Biol. 2004, 11, 567–573. [Google Scholar] [CrossRef]
- Ni, Z.X.; Wang, J.L.; Yu, X.F.; Wang, Y.F.; Wang, J.F.; He, X.J.; Li, C.J.; Deng, G.H.; Shi, J.Z.; Kong, H.H.; et al. Influenza virus uses mGluR2 as an endocytic receptor to enter cells. Nat. Microbiol. 2024, 9, 1764–1777. [Google Scholar] [CrossRef]
- Lakadamyali, M.; Rust, M.J.; Babcock, H.P.; Zhuang, X. Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. USA 2003, 100, 9280–9285. [Google Scholar] [CrossRef] [PubMed]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef]
- Hu, Y.Z.; Jiang, L.; Wang, G.W.; Song, Y.M.; Shan, Z.B.; Wang, X.Y.; Deng, G.H.; Shi, J.Z.; Tian, G.B.; Zeng, X.Y.; et al. M6PR interacts with the HA2 subunit of influenza A virus to facilitate the fusion of viral and endosomal membranes. Sci. China Life Sci. 2024, 67, 579–595. [Google Scholar] [CrossRef]
- Miyake, Y.; Keusch, J.J.; Decamps, L.; Ho-Xuan, H.; Iketani, S.; Gut, H.; Kutay, U.; Helenius, A.; Yamauchi, Y. Influenza virus uses transportin 1 for vRNP debundling during cell entry. Nat. Microbiol. 2019, 4, 578–586. [Google Scholar] [CrossRef]
- Larson, G.P.; Tran, V.; Yu, S.; Cai, Y.; Higgins, C.A.; Smith, D.M.; Baker, S.F.; Radoshitzky, S.R.; Kuhn, J.H.; Mehle, A. EPS8 Facilitates Uncoating of Influenza A Virus. Cell Rep. 2019, 29, 2175–2183.e4. [Google Scholar] [CrossRef]
- Deng, T.; Engelhardt, O.G.; Thomas, B.; Akoulitchev, A.V.; Brownlee, G.G.; Fodor, E. Role of ran binding protein 5 in nuclear import and assembly of the influenza virus RNA polymerase complex. J. Virol. 2006, 80, 11911–11919. [Google Scholar] [CrossRef]
- Tarendeau, F.; Boudet, J.; Guilligay, D.; Mas, P.J.; Bougault, C.M.; Boulo, S.; Baudin, F.; Ruigrok, R.W.; Daigle, N.; Ellenberg, J.; et al. Structure and nuclear import function of the C-terminal domain of influenza virus polymerase PB2 subunit. Nat. Struct. Mol. Biol. 2007, 14, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, G.; Herwig, A.; Klenk, H.D. Interaction of polymerase subunit PB2 and NP with importin alpha1 is a determinant of host range of influenza A virus. PLoS Pathog. 2008, 4, e11. [Google Scholar] [CrossRef] [PubMed]
- Melen, K.; Fagerlund, R.; Franke, J.; Kohler, M.; Kinnunen, L.; Julkunen, I. Importin alpha nuclear localization signal binding sites for STAT1, STAT2, and influenza A virus nucleoprotein. J. Biol. Chem. 2003, 278, 28193–28200. [Google Scholar] [CrossRef]
- Resa-Infante, P.; Jorba, N.; Zamarreno, N.; Fernandez, Y.; Juarez, S.; Ortin, J. The host-dependent interaction of alpha-importins with influenza PB2 polymerase subunit is required for virus RNA replication. PLoS ONE 2008, 3, e3904. [Google Scholar] [CrossRef]
- Batra, J.; Tripathi, S.; Kumar, A.; Katz, J.M.; Cox, N.J.; Lal, R.B.; Sambhara, S.; Lal, S.K. Human Heat shock protein 40 (Hsp40/DnaJB1) promotes influenza A virus replication by assisting nuclear import of viral ribonucleoproteins. Sci. Rep. 2016, 6, 19063. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Yang, C.; Ren, C.; Zhang, S.; Gao, X.; Jin, M.; Chen, H.; Ma, W.; Zhou, H. Eukaryotic Translation Elongation Factor 1 Delta Inhibits the Nuclear Import of the Nucleoprotein and PA-PB1 Heterodimer of Influenza A Virus. J. Virol. 2020, 95, e01391-20. [Google Scholar] [CrossRef]
- Hsu, W.B.; Shih, J.L.; Shih, J.R.; Du, J.L.; Teng, S.C.; Huang, L.M.; Wang, W.B. Cellular protein HAX1 interacts with the influenza A virus PA polymerase subunit and impedes its nuclear translocation. J. Virol. 2013, 87, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Yamada, K.; Yoneda, Y. Importin alpha: A key molecule in nuclear transport and non-transport functions. J. Biochem. 2016, 160, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kawakami, E.; Shoemaker, J.E.; Lopes, T.J.; Matsuoka, Y.; Tomita, Y.; Kozuka-Hata, H.; Gorai, T.; Kuwahara, T.; Takeda, E.; et al. Influenza virus-host interactome screen as a platform for antiviral drug development. Cell Host Microbe 2014, 16, 795–805. [Google Scholar] [CrossRef]
- De Conto, F.; Conversano, F.; Razin, S.V.; Belletti, S.; Arcangeletti, M.C.; Chezzi, C.; Calderaro, A. Host-cell dependent role of phosphorylated keratin 8 during influenza A/NWS/33 virus (H1N1) infection in mammalian cells. Virus Res. 2021, 295, 198333. [Google Scholar] [CrossRef]
- Yu, G.; Liang, W.; Liu, J.; Meng, D.; Wei, L.; Chai, T.; Cai, Y. Proteomic Analysis of Differential Expression of Cellular Proteins in Response to Avian H9N2 Virus Infection of A549 Cells. Front. Microbiol. 2016, 7, 1962. [Google Scholar] [CrossRef]
- Liu, N.; Song, W.; Wang, P.; Lee, K.; Chan, W.; Chen, H.; Cai, Z. Proteomics analysis of differential expression of cellular proteins in response to avian H9N2 virus infection in human cells. Proteomics 2008, 8, 1851–1858. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, Y.; Shan, Z.; Shi, W.; Li, Q.; Wang, Y.; Wang, B.; Wang, G.; Chen, H.; Jiang, L.; Li, C. KRT6A Restricts Influenza A Virus Replication by Inhibiting the Nuclear Import and Assembly of Viral Ribonucleoprotein Complex. Viruses 2025, 17, 671. https://doi.org/10.3390/v17050671
Chang Y, Shan Z, Shi W, Li Q, Wang Y, Wang B, Wang G, Chen H, Jiang L, Li C. KRT6A Restricts Influenza A Virus Replication by Inhibiting the Nuclear Import and Assembly of Viral Ribonucleoprotein Complex. Viruses. 2025; 17(5):671. https://doi.org/10.3390/v17050671
Chicago/Turabian StyleChang, Yu, Zhibo Shan, Wenjun Shi, Qibing Li, Yihan Wang, Bo Wang, Guangwen Wang, Hualan Chen, Li Jiang, and Chengjun Li. 2025. "KRT6A Restricts Influenza A Virus Replication by Inhibiting the Nuclear Import and Assembly of Viral Ribonucleoprotein Complex" Viruses 17, no. 5: 671. https://doi.org/10.3390/v17050671
APA StyleChang, Y., Shan, Z., Shi, W., Li, Q., Wang, Y., Wang, B., Wang, G., Chen, H., Jiang, L., & Li, C. (2025). KRT6A Restricts Influenza A Virus Replication by Inhibiting the Nuclear Import and Assembly of Viral Ribonucleoprotein Complex. Viruses, 17(5), 671. https://doi.org/10.3390/v17050671