
Current Evolutionary Dynamics of Porcine Epidemic Diarrhea Virus (PEDV) in the U.S. a Decade After Introduction

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. Sequence Alignment
2.3. Sequences Comparison
2.4. Recombination Analysis
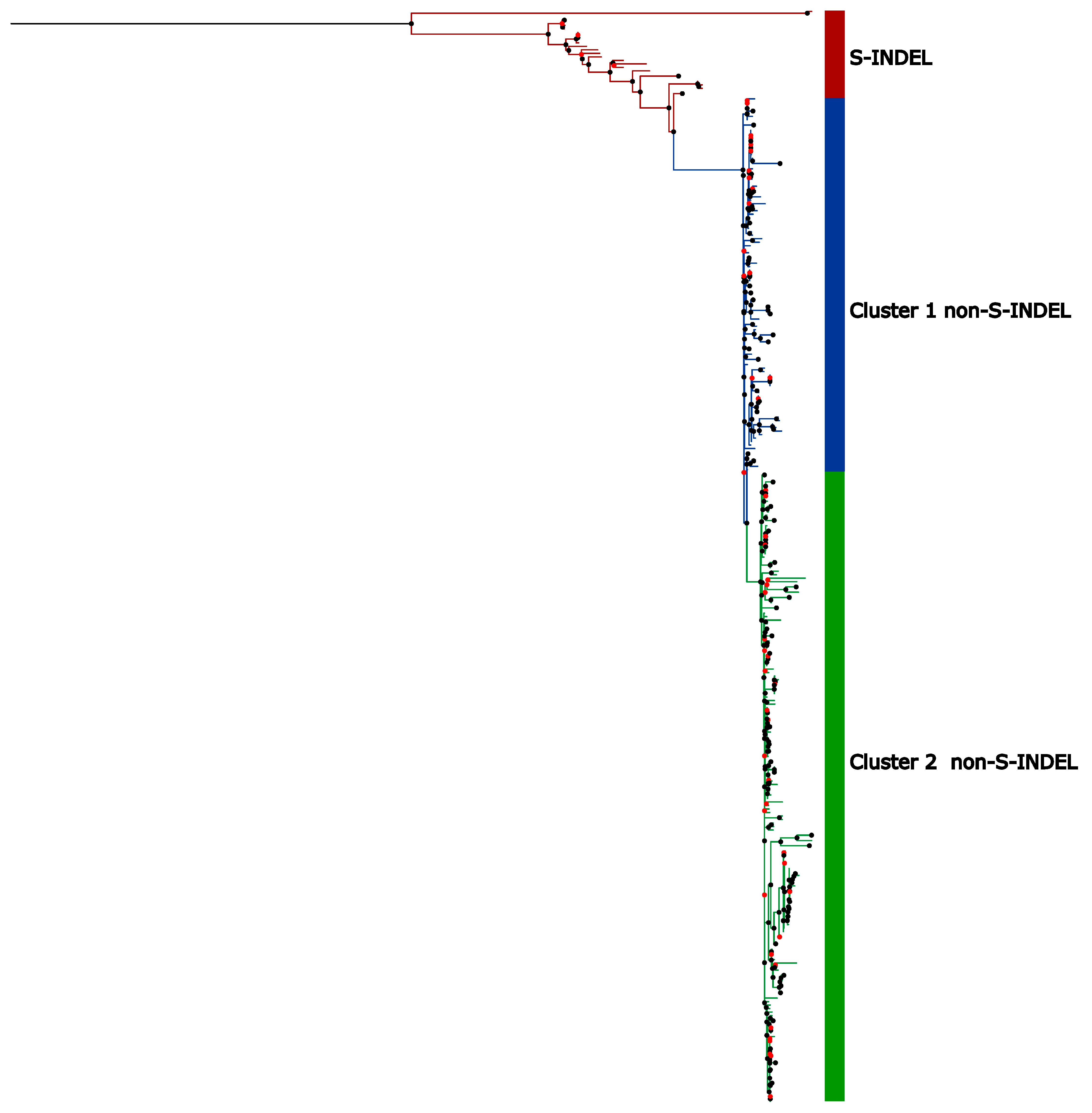
2.5. Phylogenetic Analysis
2.6. Genetic Diversity
2.7. Selections Analysis
3. Results
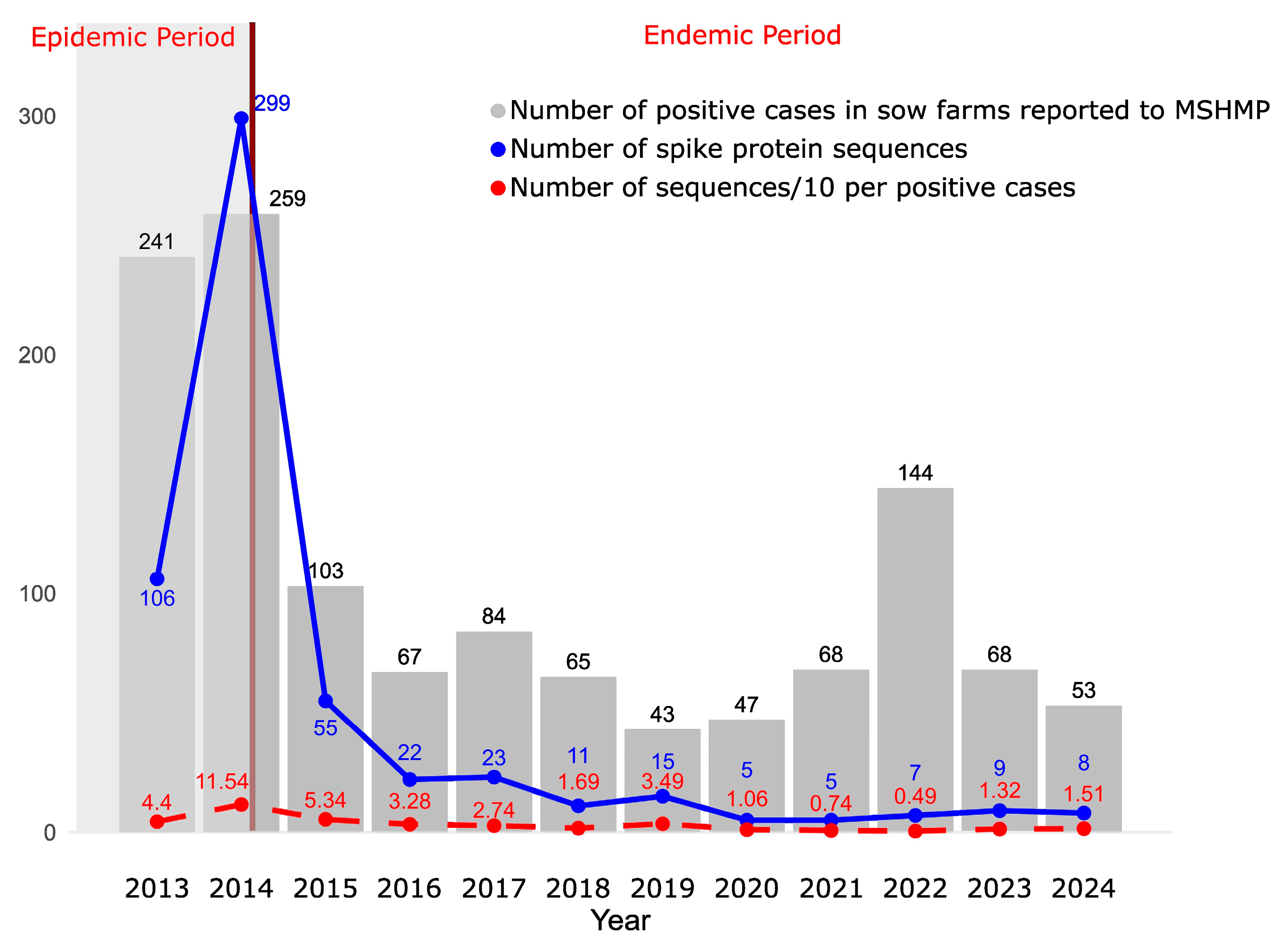
3.1. Stabilization of PEDV Case Numbers After the Epidemic Period
3.2. Evidence of Multiple Recombination Events
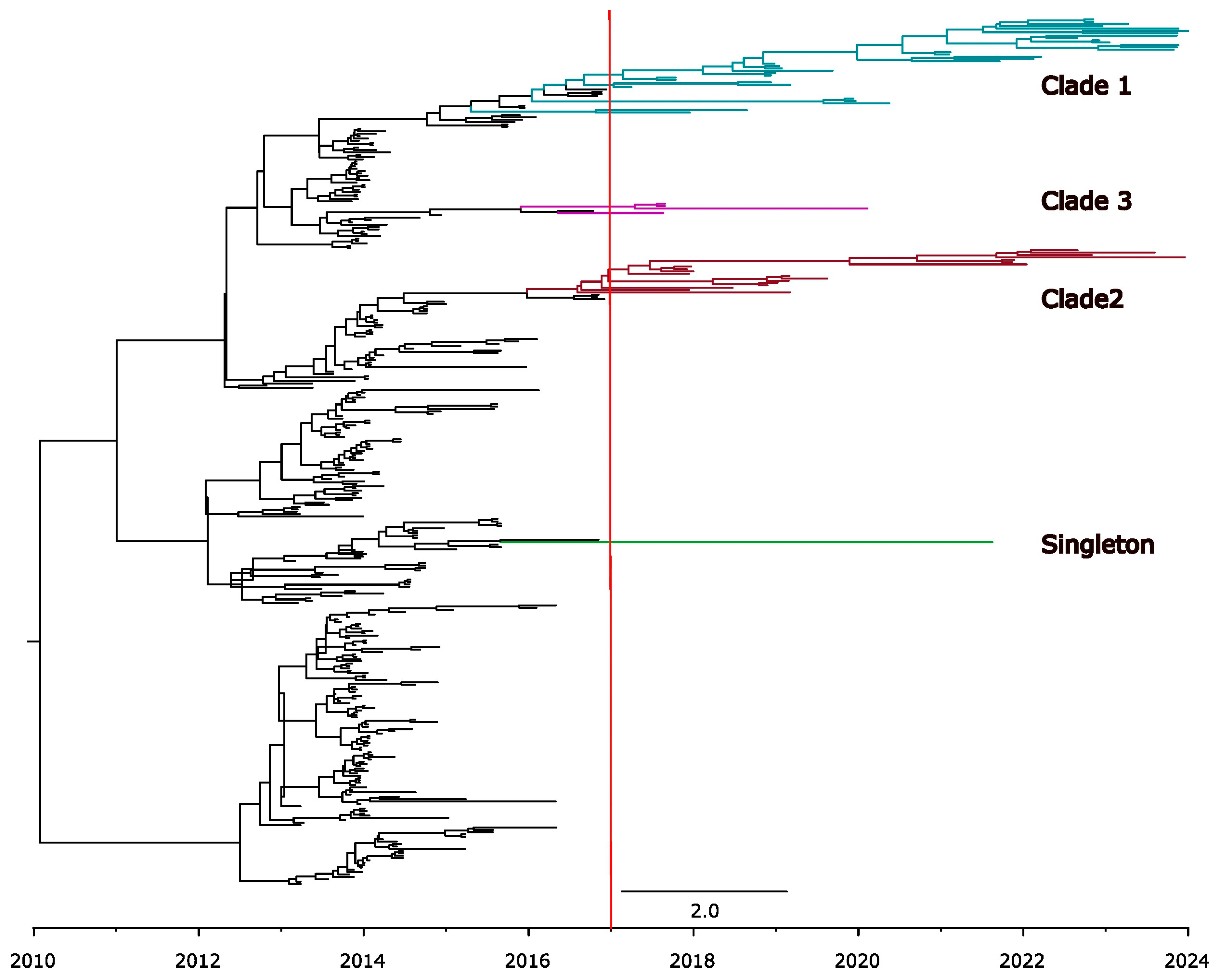
3.3. Temporal Signal and Nucleotide Substitution Rates
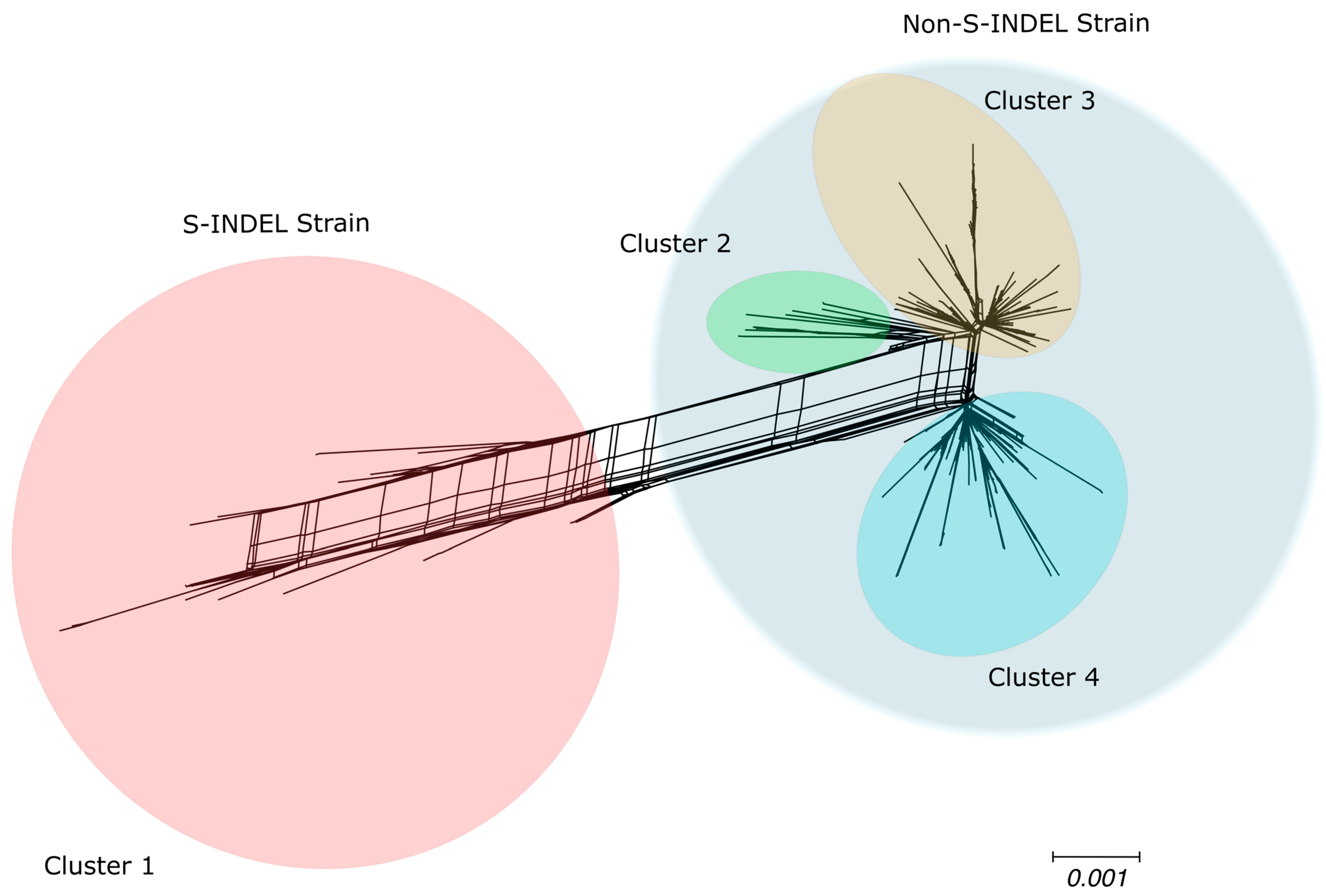
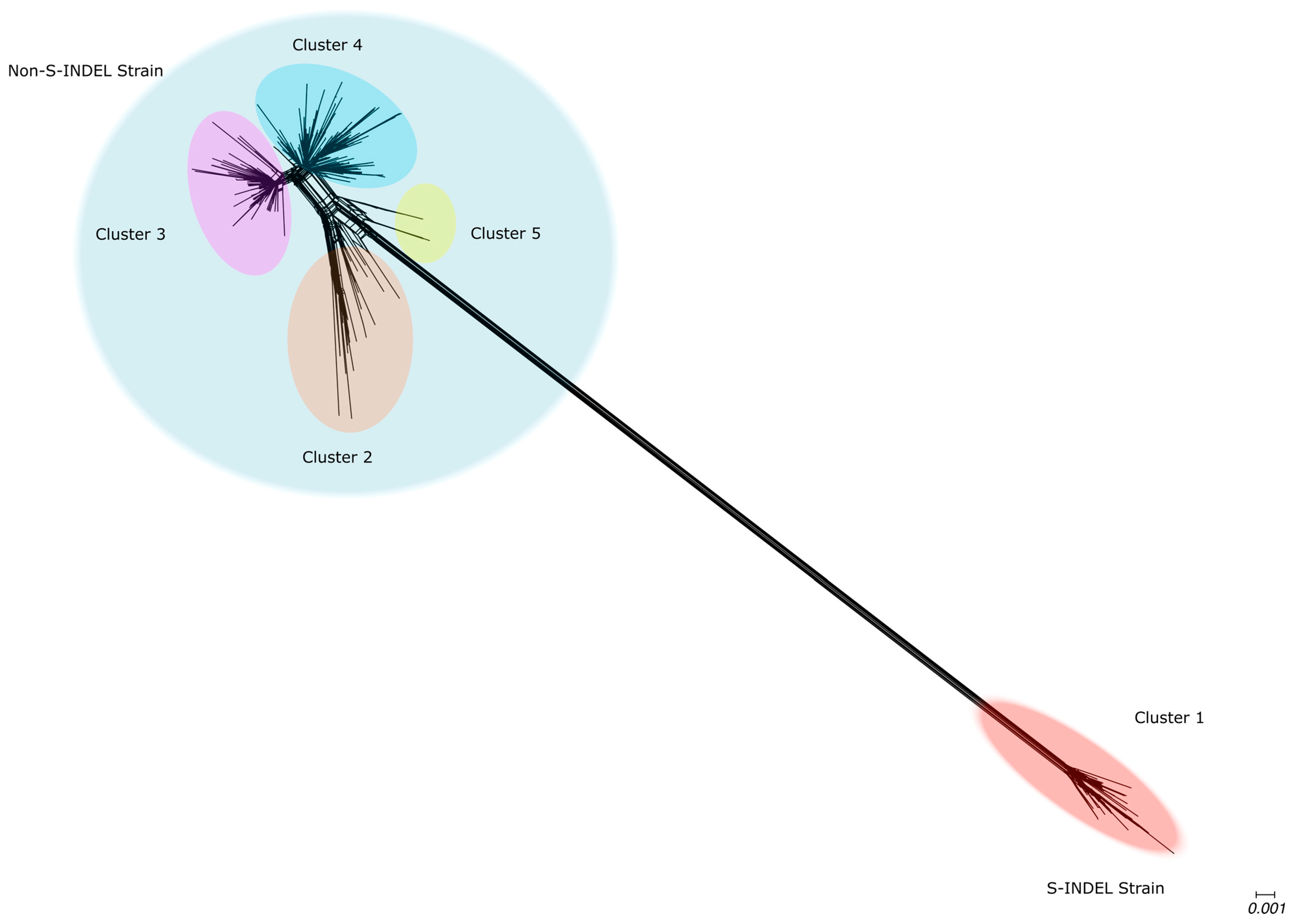
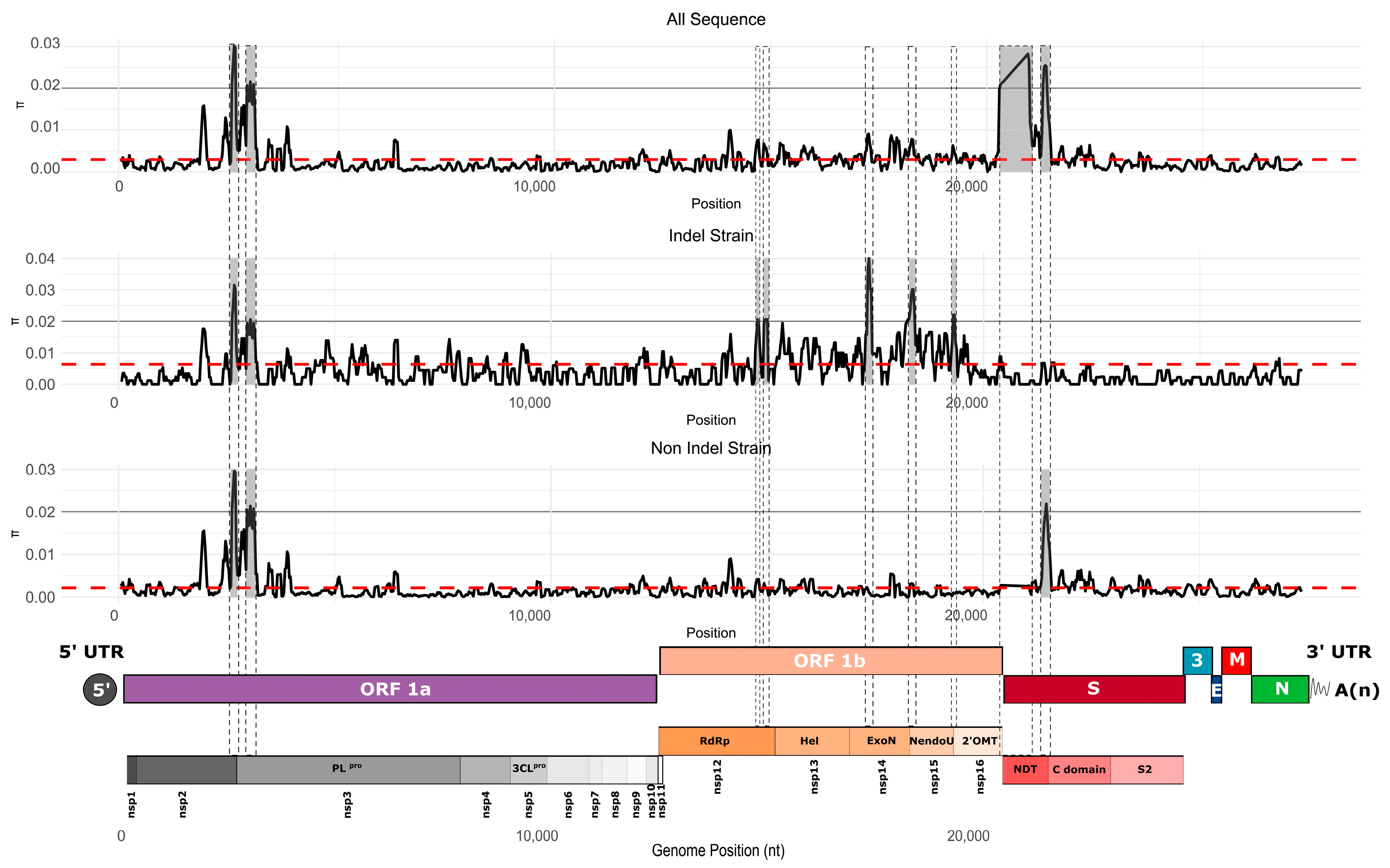
3.4. Genetic Variability in U.S. Population
3.5. High Sequence Similarity Between PEDV Strains and Among the Current Clades
3.6. Positive Selection Evidence in a Spike Protein Epitope
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.-m.; Niu, B.-b.; Yan, H.; Gao, D.-s.; Yang, X.; Chen, L.; Chang, H.-t.; Zhao, J.; Wang, C.-q. Genetic properties of endemic Chinese porcine epidemic diarrhea virus strains isolated since 2010. Arch. Virol. 2013, 158, 2487–2494. [Google Scholar] [PubMed]
- Stadler, J.; Zoels, S.; Fux, R.; Hanke, D.; Pohlmann, A.; Blome, S.; Weissenböck, H.; Weissenbacher-Lang, C.; Ritzmann, M.; Ladinig, A. Emergence of porcine epidemic diarrhea virus in southern Germany. BMC Vet. Res. 2015, 11, 142. [Google Scholar] [CrossRef]
- Stevenson, G.W.; Hoang, H.; Schwartz, K.J.; Burrough, E.R.; Sun, D.; Madson, D.; Cooper, V.L.; Pillatzki, A.; Gauger, P.; Schmitt, B.J. Emergence of Porcine epidemic diarrhea virus in the United States: Clinical signs, lesions, and viral genomic sequences. J. Vet. Diagn. Investig. 2013, 25, 649–654. [Google Scholar]
- Zhang, H.; Zou, C.; Peng, O.; Ashraf, U.; Xu, Q.; Gong, L.; Fan, B.; Zhang, Y.; Xu, Z.; Xue, C. Global dynamics of porcine enteric coronavirus PEDV epidemiology, evolution, and transmission. Mol. Biol. Evol. 2023, 40, msad052. [Google Scholar] [CrossRef]
- Ojkic, D.; Hazlett, M.; Fairles, J.; Marom, A.; Slavic, D.; Maxie, G.; Alexandersen, S.; Pasick, J.; Alsop, J.; Burlatschenko, S. The first case of porcine epidemic diarrhea in Canada. The first case of porcine epidemic diarrhea in Canada. Can. Vet. J. 2015, 56, 149. [Google Scholar]
- Vlasova, A.N.; Marthaler, D.; Wang, Q.; Culhane, M.R.; Rossow, K.D.; Rovira, A.; Collins, J.; Saif, L.J. Distinct characteristics and complex evolution of PEDV strains, North America, May 2013–February 2014. Emerg. Infect. Dis. 2014, 20, 1620. [Google Scholar] [CrossRef]
- Song, D.; Park, B. Porcine epidemic diarrhoea virus: A comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus Genes 2012, 44, 167–175. [Google Scholar] [PubMed]
- Wang, L.; Byrum, B.; Zhang, Y. New variant of porcine epidemic diarrhea virus, United States, 2014. Emerg. Infect. Dis. 2014, 20, 917. [Google Scholar] [CrossRef]
- Makau, D.N.; Pamornchainavakul, N.; VanderWaal, K.; Kikuti, M.; Picasso-Risso, C.; Geary, E.; Corzo, C.A. Postepidemic Epidemiology of Porcine Epidemic Diarrhea Virus in the United States. Transbound. Emerg. Dis. 2024, 2024, 5531899. [Google Scholar] [CrossRef]
- MSHMP. PED Aggregate Prevalence of Sow Herd Status (Last Updated April 2024). 2024. Available online: https://mshmp.umn.edu/reports#Charts (accessed on 1 April 2024).
- Jung, K.; Saif, L.J. Porcine epidemic diarrhea virus infection: Etiology, epidemiology, pathogenesis and immunoprophylaxis. Vet. J. 2015, 204, 134–143. [Google Scholar] [CrossRef]
- O’Dea, E.B.; Snelson, H.; Bansal, S. Using heterogeneity in the population structure of US swine farms to compare transmission models for porcine epidemic diarrhoea. Sci. Rep. 2016, 6, 22248. [Google Scholar] [CrossRef]
- Schumacher, L.L.; Huss, A.R.; Cochrane, R.A.; Stark, C.R.; Woodworth, J.C.; Bai, J.; Poulsen, E.G.; Chen, Q.; Main, R.G.; Zhang, J. Characterizing the rapid spread of porcine epidemic diarrhea virus (PEDV) through an animal food manufacturing facility. PLoS ONE 2017, 12, e0187309. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Yang, M.; Goyal, S.M.; Cheeran, M.C.; Torremorell, M. Evaluation of biosecurity measures to prevent indirect transmission of porcine epidemic diarrhea virus. BMC Vet. Res. 2017, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Dee, S.; Clement, T.; Schelkopf, A.; Nerem, J.; Knudsen, D.; Christopher-Hennings, J.; Nelson, E. An evaluation of contaminated complete feed as a vehicle for porcine epidemic diarrhea virus infection of naive pigs following consumption via natural feeding behavior: Proof of concept. BMC Vet. Res. 2014, 10, 176. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; de Groot, R.J.; Haagmans, B.; Lau, S.K.; Neuman, B.W.; Perlman, S.; Sola, I.; van der Hoek, L.; Wong, A.C.; Yeh, S.-H. ICTV Virus Taxonomy Profile: Coronaviridae 2023: This article is part of the ICTV Virus Taxonomy Profiles collection. J. Gen. Virol. 2023, 104, 001843. [Google Scholar] [CrossRef]
- Elena, S.F.; Sanjuán, R. Adaptive value of high mutation rates of RNA viruses: Separating causes from consequences. J. Virol. 2005, 79, 11555–11558. [Google Scholar] [CrossRef]
- Makau, D.N.; Lycett, S.; Michalska-Smith, M.; Paploski, I.A.D.; Cheeran, M.C.J.; Craft, M.E.; Kao, R.R.; Schroeder, D.C.; Doeschl-Wilson, A.; VanderWaal, K. Ecological and evolutionary dynamics of multi-strain RNA viruses. Nat. Ecol. Evol. 2022, 6, 1414–1422. [Google Scholar] [CrossRef]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Dolan, P.T.; Whitfield, Z.J.; Andino, R. Mechanisms and concepts in RNA virus population dynamics and evolution. Annu. Rev. Virol. 2018, 5, 69–92. [Google Scholar] [CrossRef]
- Minskaia, E.; Hertzig, T.; Gorbalenya, A.E.; Campanacci, V.; Cambillau, C.; Canard, B.; Ziebuhr, J. Discovery of an RNA virus 3′→ 5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 5108–5113. [Google Scholar] [CrossRef]
- Eckerle, L.D.; Lu, X.; Sperry, S.M.; Choi, L.; Denison, M.R. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 2007, 81, 12135–12144. [Google Scholar] [CrossRef]
- Brown, D.W. Threat to humans from virus infections of non-human primates. Rev. Med. Virol. 1997, 7, 239–246. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Weiller, G.F. Evidence that a plant virus switched hosts to infect a vertebrate and then recombined with a vertebrate-infecting virus. Proc. Natl. Acad. Sci. USA 1999, 96, 8022–8027. [Google Scholar] [CrossRef] [PubMed]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef]
- Wells, H.L.; Bonavita, C.M.; Navarrete-Macias, I.; Vilchez, B.; Rasmussen, A.L.; Anthony, S.J. The coronavirus recombination pathway. Cell Host Microbe 2023, 31, 874–889. [Google Scholar] [CrossRef]
- Müller, N.F.; Kistler, K.E.; Bedford, T. A Bayesian approach to infer recombination patterns in coronaviruses. Nat. Comm. 2022, 13, 4186. [Google Scholar] [CrossRef] [PubMed]
- Mei, X.; Guo, J.; Fang, P.; Ma, J.; Li, M.; Fang, L. The characterization and pathogenicity of a recombinant porcine epidemic diarrhea virus variant ECQ1. Viruses 2023, 15, 1492. [Google Scholar] [CrossRef]
- Jarvis, M.C.; Lam, H.C.; Zhang, Y.; Wang, L.; Hesse, R.A.; Hause, B.M.; Vlasova, A.; Wang, Q.; Zhang, J.; Nelson, M.I. Genomic and evolutionary inferences between American and global strains of porcine epidemic diarrhea virus. Prev. Vet. Med. 2016, 123, 175–184. [Google Scholar] [CrossRef]
- Lee, C. Porcine epidemic diarrhea virus: An emerging and re-emerging epizootic swine virus. Virol. J. 2015, 12, 193. [Google Scholar] [CrossRef]
- Wang, Q.; Vlasova, A.N.; Kenney, S.P.; Saif, L.J. Emerging and re-emerging coronaviruses in pigs. Curr. Opin. Virol. 2019, 34, 39–49. [Google Scholar] [CrossRef]
- Huang, Y.-W.; Dickerman, A.W.; Piñeyro, P.; Li, L.; Fang, L.; Kiehne, R.; Opriessnig, T.; Meng, X.-J. Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the United States. mBio 2013, 4, 00737-13. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Saif, L.J.; Wang, Q. Porcine epidemic diarrhea virus (PEDV): An update on etiology, transmission, pathogenesis, and prevention and control. Virus Res. 2020, 286, 198045. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, C. Outbreak-related porcine epidemic diarrhea virus strains similar to US strains, South Korea, 2013. Emerg. Infect. Dis. 2014, 20, 1223. [Google Scholar] [CrossRef]
- Lin, C.-N.; Chung, W.-B.; Chang, S.-W.; Wen, C.-C.; Liu, H.; Chien, C.-H.; Chiou, M.-T. US-like strain of porcine epidemic diarrhea virus outbreaks in Taiwan, 2013–2014. J. Vet. Med. Sci. 2014, 76, 1297–1299. [Google Scholar] [CrossRef]
- He, W.-T.; Bollen, N.; Xu, Y.; Zhao, J.; Dellicour, S.; Yan, Z.; Gong, W.; Zhang, C.; Zhang, L.; Lu, M. Phylogeography reveals association between swine trade and the spread of porcine epidemic diarrhea virus in China and across the world. Mol. Biol. Evol. 2022, 39, msab364. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Ostell, J.; Pruitt, K.D.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2019, 47, D94–D99. [Google Scholar] [CrossRef] [PubMed]
- Perez, A.M.; Linhares, D.C.; Arruda, A.G.; VanderWaal, K.; Machado, G.; Vilalta, C.; Sanhueza, J.M.; Torrison, J.; Torremorell, M.; Corzo, C.A. Individual or common good? Voluntary data sharing to inform disease surveillance systems in food animals. Front. Vet. Sci. 2019, 6, 194. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Martin, D.; Rybicki, E.P. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–224. [Google Scholar] [CrossRef]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retroviruses 2005, 21, 98–102. [Google Scholar] [PubMed]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [PubMed]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [PubMed]
- Lam, H.M.; Ratmann, O.; Boni, M.F. Improved algorithmic complexity for the 3SEQ recombination detection algorithm. Mol. Biol. Evol. 2018, 35, 247–251. [Google Scholar] [CrossRef]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Duchene, S.; Lemey, P.; Stadler, T.; Ho, S.Y.W.; Duchene, D.A.; Dhanasekaran, V.; Baele, G. Bayesian Evaluation of Temporal Signal in Measurably Evolving Populations. Mol. Biol. Evol. 2020, 37, 3363–3379. [Google Scholar] [CrossRef]
- Duchene, S.; Featherstone, L.; Haritopoulou-Sinanidou, M.; Rambaut, A.; Lemey, P.; Baele, G. Temporal signal and the phylodynamic threshold of SARS-CoV-2. Virus Evol. 2020, 6, veaa061. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Kass, R.E.; Raftery, A.E. Bayes factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar]
- Baele, G.; Lemey, P.; Suchard, M.A. Genealogical working distributions for Bayesian model testing with phylogenetic uncertainty. Syst. Biol. 2016, 65, 250–264. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Lima, A.T.M.; Silva, J.C.F.; Silva, F.N.; Castillo-Urquiza, G.P.; Silva, F.F.; Seah, Y.M.; Mizubuti, E.S.G.; Duffy, S.; Zerbini, F.M. The diversification of begomovirus populations is predominantly driven by mutational dynamics. The diversification of begomovirus populations is predominantly driven by mutational dynamics. Virus Evol. 2017, 3, vex005. [Google Scholar] [CrossRef]
- Peng, R.D. Simpleboot: Simple Bootstrap Routines. 2019. Available online: https://cran.r-project.org/web/packages/simpleboot/simpleboot.pdf (accessed on 27 April 2025).
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar]
- Pond, S.L.K.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. GARD: A genetic algorithm for recombination detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef]
- Kosakovsky-Pond, S.L.; Frost, S.D.W. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, Y.; Yan, Z.; Li, G.; Luo, J.; Huang, S.; Guo, X. A comprehensive view on the protein functions of porcine epidemic diarrhea virus. Genes 2024, 15, 165. [Google Scholar] [CrossRef]
- Weng, L.; Weersink, A.; Poljak, Z.; de Lange, K.; von Massow, M. An economic evaluation of intervention strategies for Porcine Epidemic Diarrhea (PED). Prev. Vet. Med. 2016, 134, 58–68. [Google Scholar] [CrossRef] [PubMed]
- VanderWaal, K.; Perez, A.; Torremorrell, M.; Morrison, R.M.; Craft, M. Role of animal movement and indirect contact among farms in transmission of porcine epidemic diarrhea virus. Epidemics 2018, 24, 67–75. [Google Scholar] [CrossRef]
- Gallien, S.; Andraud, M.; Moro, A.; Lediguerher, G.; Morin, N.; Gauger, P.C.; Bigault, L.; Paboeuf, F.; Berri, M.; Rose, N. Better horizontal transmission of a US non-InDel strain compared with a French InDel strain of porcine epidemic diarrhoea virus. Transbound. Emerg. Dis. 2018, 65, 1720–1732. [Google Scholar] [CrossRef]
- Kikuti, M.; Drebes, D.; Robbins, R.; Dufresne, L.; Sanhueza, J.M.; Corzo, C.A. Growing pig incidence rate, control and prevention of porcine epidemic diarrhea virus in a large pig production system in the United States. Porc. Health Manag. 2022, 8, 23. [Google Scholar] [CrossRef]
- Lowe, J.; Gauger, P.; Harmon, K.; Zhang, J.; Connor, J.; Yeske, P.; Loula, T.; Levis, I.; Dufresne, L.; Main, R. Role of transportation in spread of porcine epidemic diarrhea virus infection, United States. Emerg. Infect. Dis. 2014, 20, 872. [Google Scholar] [CrossRef]
- Bowman, A.S.; Krogwold, R.A.; Price, T.; Davis, M.; Moeller, S.J. Investigating the introduction of porcine epidemic diarrhea virus into an Ohio swine operation. BMC Vet. Res. 2015, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Narechania, A.; Bobo, D.; Deitz, K.; DeSalle, R.; Planet, P.J.; Mathema, B. Rapid SARS-CoV-2 surveillance using clinical, pooled, or wastewater sequence as a sensor for population change. Genome Res. 2024, 34, 1651–1660. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B. Tracking changes in SARS-CoV-2 spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Cardenas, N.C.; Valencio, A.; Sanchez, F.; O’Hara, K.C.; Machado, G. Analyzing the intrastate and interstate swine movement network in the United States. Prev. Vet. Med. 2024, 230, 106264. [Google Scholar] [CrossRef]
- Niederwerder, M.; Hesse, R. Swine enteric coronavirus disease: A review of 4 years with porcine epidemic diarrhoea virus and porcine deltacoronavirus in the United States and Canada. Transbound. Emerg. Dis. 2018, 65, 660–675. [Google Scholar] [CrossRef]
- Sun, M.; Ma, J.; Wang, Y.; Wang, M.; Song, W.; Zhang, W.; Lu, C.; Yao, H. Genomic and epidemiological characteristics provide new insights into the phylogeographical and spatiotemporal spread of porcine epidemic diarrhea virus in Asia. J. Clin. Microbiol. 2015, 53, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Yu, Z.; Cheng, K.; Liu, Y.; Huang, J.; Xin, Y.; Li, Y.; Fan, S.; Wang, T.; Huang, G. Molecular characterization and phylogenetic analysis of new variants of the porcine epidemic diarrhea virus in Gansu, China in 2012. Viruses 2013, 5, 1991–2004. [Google Scholar] [CrossRef]
- Sun, R.; Leng, Z.; Zhai, S.-L.; Chen, D.; Song, C. Genetic variability and phylogeny of current Chinese porcine epidemic diarrhea virus strains based on spike, ORF3, and membrane genes. Sci. World J. 2014, 2014, 208439. [Google Scholar]
- Peng, Q.; Fan, B.; Song, X.; He, W.; Wang, C.; Zhao, Y.; Guo, W.; Zhang, X.; Liu, S.; Gao, J. Genetic signatures associated with the virulence of porcine epidemic diarrhea virus AH2012/12. J. Virol. 2023, 97, e01063-23. [Google Scholar] [CrossRef]
- Li, D.; Li, Y.; Liu, Y.; Chen, Y.; Jiao, W.; Feng, H.; Wei, Q.; Wang, J.; Zhang, Y.; Zhang, G. Isolation and identification of a recombinant porcine epidemic diarrhea virus with a novel insertion in S1 domain. Front. Microbiol. 2021, 12, 667084. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, L.; Shang, Y.; Tan, R.; Ji, M.; Yue, X.; Wang, N.; Liu, J.; Wang, C.; Li, Y. Emergence and evolution of highly pathogenic porcine epidemic diarrhea virus by natural recombination of a low pathogenic vaccine isolate and a highly pathogenic strain in the spike gene. Virus Evol. 2020, 6, veaa049. [Google Scholar] [CrossRef] [PubMed]
- Kong, N.; Meng, Q.; Jiao, Y.; Wu, Y.; Zuo, Y.; Wang, H.; Sun, D.; Dong, S.; Zhai, H.; Tong, W. Identification of a novel B-cell epitope in the spike protein of porcine epidemic diarrhea virus. Virol. J. 2020, 17, 46. [Google Scholar] [CrossRef] [PubMed]
- Servín-Blanco, R.; Zamora-Alvarado, R.; Gevorkian, G.; Manoutcharian, K. Antigenic variability: Obstacles on the road to vaccines against traditionally difficult targets. Human. Vaccines Immunother. 2016, 12, 2640–2648. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breakpoint Position | ||||||||
| A | ||||||||
| Full Genome | In Recombinant Sequence | |||||||
| Event | Found In | Begin | End | Recombination Sequencies | Minor Parental | Major Parental | Detection Methods # | p-Value * |
| 1 | 6 | 16,816 | 22,432 | KR265760 | KM975738 | KF452322 | RGBMCSP3 | 2.64 × 10−76 |
| 2 | 5 | 3910 | 20,375 | KR265761 | KR265763 | KM975738 | RGMCS3 | 6.01 × 10−75 |
| 3 | 4 | 13,244 | 21,566 | KM975740 | KM975738 | KF452322 | RGMS3 | 3.03 × 10−53 |
| 4 | 2 | 13,244 | 17,879 | KR265786 | KJ645704 | KF468753 | RGMC3 | 3.02 × 10−46 |
| 5 | 2 | 11,075 | 27,425 | KM975738 | KR265759 | Unknown | RGBMCS3 | 5.96 × 10−37 |
| 6 | 106 | 16,816 | 19,902 | KU558702 | KJ645704 | 248 | RGMC3 | 3.48 × 10−32 |
| 7 | 12 | 20,376 | 21,478 | KR265761 | Unknown | KJ645643 | RGBMS3 | 1.48 × 10−33 |
| 8 | 7 | 4545 | 16,815 | KR265759 | Unknown | 205 | RGBMS3 | 7.98 × 10−28 |
| 9 | 6 | 4377 | 16,815 | 264 | Unknown | KJ645641 | MCS3 | 1.16 × 10−22 |
| 10 | 116 | 9921 | 14,874 | KU893873 | KF452322 | KJ645635 | RGMC3 | 3.91 × 10−19 |
| 11 | 223 | 7680 | 13,243 | KJ645704 | KJ645635 | KR265844 | RGM3 | 1.23 × 10−14 |
| 12 | 152 | 19,912 | 21,587 | KJ645641 | MG837058 | Unknown | RGMC3 | 1.20 × 10−6 |
| B | ||||||||
| Spike Protein | Breakpoint Position | |||||||
| Event | Found In | Begin | End | Recombination Sequencies | Minor Parental | Major Parental | Detection Methods # | p-Value * |
| 1 | 3 | 8 | 915 | KU982979 | 265 | 340 | RMSP3 | 1.75 × 10−23 |
| 2 | 34 | 8 | 955 | 340 | Unknown | 296 | RBMSP3 | 4.98 × 10−24 |
| 3 | 24 | 8 | 1056 | 262 | Unknown | KU982968 | MP3 | 3.88 × 10−23 |
| Clade | Min | Max | Average |
|---|---|---|---|
| Spike Protein | |||
| Non-S-INDEL | 97.6% | 99.8% | 99.1% |
| Clade1non-S-INDEL | 98.2% | 99.8% | 99.3% |
| Clade1non-S-INDEL vs. older cladesnon-S-INDEL | 97.7% | 99.6% | 98.6% |
| Clade2non-S-INDEL | 99.3% | 99.9% | 99.2% |
| Clade2non-S-INDEL vs. older cladesnon-S-INDEL | 97.8% | 99.7% | 99.3% |
| Clade1non-S-INDEL vs. Clade 2non-S-INDEL | 97.9% | 99.0% | 98.6% |
| S-INDEL strain | 94.5% | 100.0% | 99.1% |
| S-INDEL vs. non-S-INDEL strains | 91.14% | 94.25% | 93.61% |
| Full Genome | |||
| Non-S-INDEL strain | 98.75% | 100% | 99.78% |
| INDEL strain | 99.21% | 100% | 99.55% |
| INDEL vs. Non-INDEL strains | 98.52% | 99.85% | 98.99% |
| Gene | dN/dS | SLAC | MEME | FUBAR | |||
|---|---|---|---|---|---|---|---|
| Diversifying | Purifying | Diversifying | Purifying | Diversifying | Purifying | ||
| S-INDEL strain | |||||||
| Spike | 0.331 | - | 368, 659 | 27, 83, 240, 351, 429, 500, 632 | - | 83, 196, 310, 351, 719 | 21, 41, 44, 48, 73, 76, 92, 93, 125, 141, 149, 199, 226, 237, 238, 269, 312 |
| Envelop | 0.491 | - | - | - | - | 66 | - |
| Matrix | 0.0967 | - | - | - | - | - | 27, 41, 71, 78, 116, 121, 122, 188, 201 |
| Nucleo capside | 0.240 | - | - | 27, 54, 415 | - | 27 | 28, 43, 51, 140, 147, 207, 211, 249, 267, 271, 298, 327, 360, 364, 414 |
| Non-S-INDEL | |||||||
| Spike | 0.887 | 144, 380, 488, 525, 526, 568, 610, 614, 724 |
80, 94, 100, 101, 114, 139, 154, 209, 276, 359, 374, 462, 558, 582, 588, 621, 654, 664, 697, 729 |
24, 58, 144, 277, 355, 380, 412, 417, 433, 488, 495, 525, 526, 568, 610, 614, 724 | - | 24, 55, 58, 70, 144, 157, 196, 277, 355, 380, 412, 433, 488, 494, 501, 525, 526, 568, 610, 614, 637, 653, 676, 695, 724 | 53, 94, 100, 101, 109, 139, 154, 244, 276, 359, 374, 394, 462, 468, 558, 582, 621, 625, 654, 697, 729 |
| Envelop | 0.743 | - | - | - | - | - | - |
| Matrix | 0.195 | - | - | - | 200 | - | - |
| Nucleo capside | 0.585 | - | 240 | - | - | 193 | 55, 87, 100, 190, 240, 244, 252, 386 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, J.P.H.d.; Pamornchainavakul, N.; Kikuti, M.; Yue, X.; Corzo, C.A.; VanderWaal, K. Current Evolutionary Dynamics of Porcine Epidemic Diarrhea Virus (PEDV) in the U.S. a Decade After Introduction. Viruses 2025, 17, 654. https://doi.org/10.3390/v17050654
Silva JPHd, Pamornchainavakul N, Kikuti M, Yue X, Corzo CA, VanderWaal K. Current Evolutionary Dynamics of Porcine Epidemic Diarrhea Virus (PEDV) in the U.S. a Decade After Introduction. Viruses. 2025; 17(5):654. https://doi.org/10.3390/v17050654
Chicago/Turabian StyleSilva, Joao P. Herrera da, Nakarin Pamornchainavakul, Mariana Kikuti, Xiaomei Yue, Cesar A. Corzo, and Kimberly VanderWaal. 2025. "Current Evolutionary Dynamics of Porcine Epidemic Diarrhea Virus (PEDV) in the U.S. a Decade After Introduction" Viruses 17, no. 5: 654. https://doi.org/10.3390/v17050654
APA StyleSilva, J. P. H. d., Pamornchainavakul, N., Kikuti, M., Yue, X., Corzo, C. A., & VanderWaal, K. (2025). Current Evolutionary Dynamics of Porcine Epidemic Diarrhea Virus (PEDV) in the U.S. a Decade After Introduction. Viruses, 17(5), 654. https://doi.org/10.3390/v17050654