

SARS-CoV-2 Spike Protein and Long COVID—Part 2: Understanding the Impact of Spike Protein and Cellular Receptor Interactions on the Pathophysiology of Long COVID Syndrome

 , , , , , and
, , , , , and
Abstract
1. Introduction
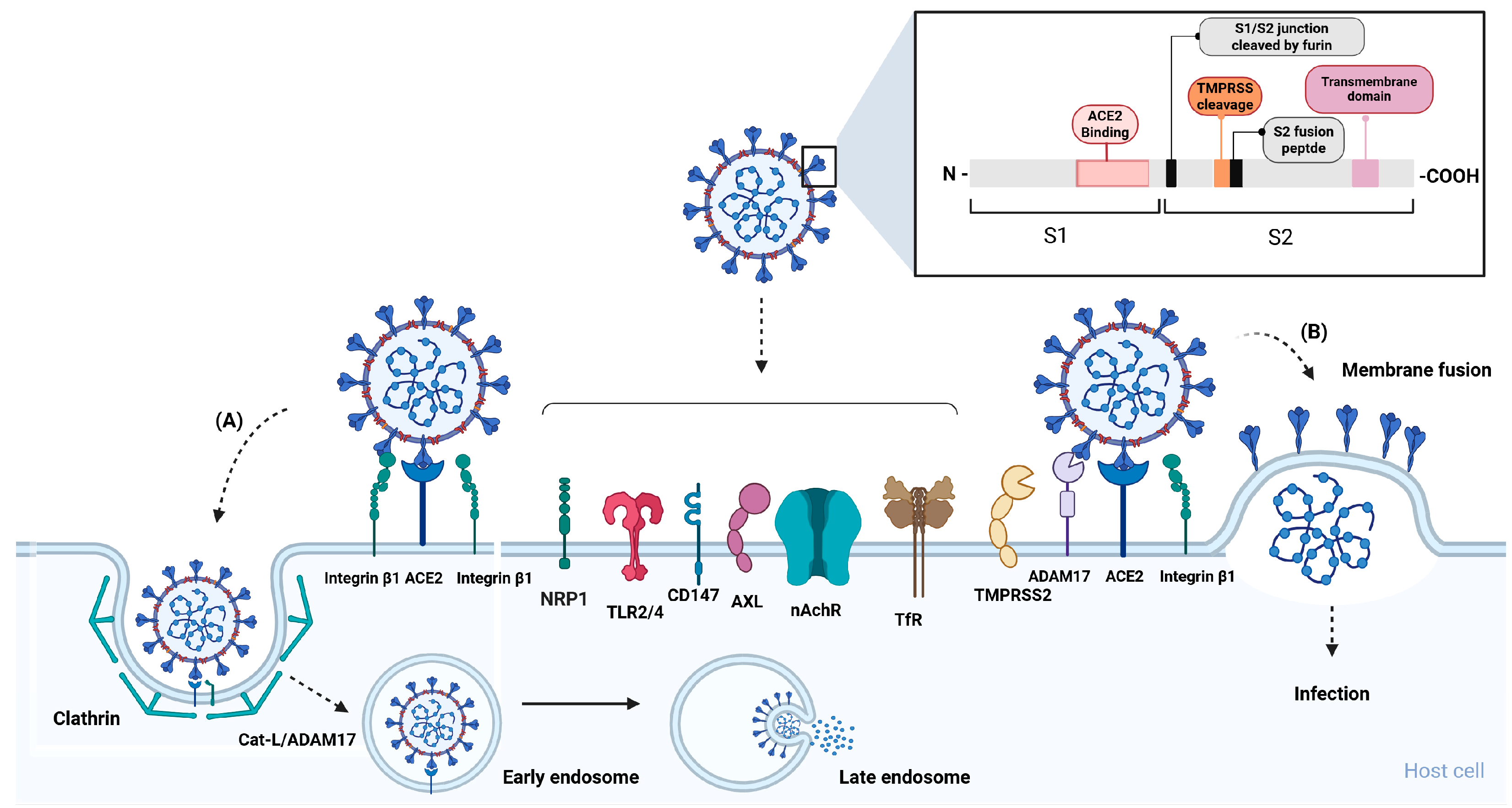
2. Cellular Receptors and Co-Receptors of SARS-CoV-2
Spike Protein
3. SARS-CoV-2 Spike Protein and Cellular Receptor Interplay Effect in Pathophysiology of Long COVID
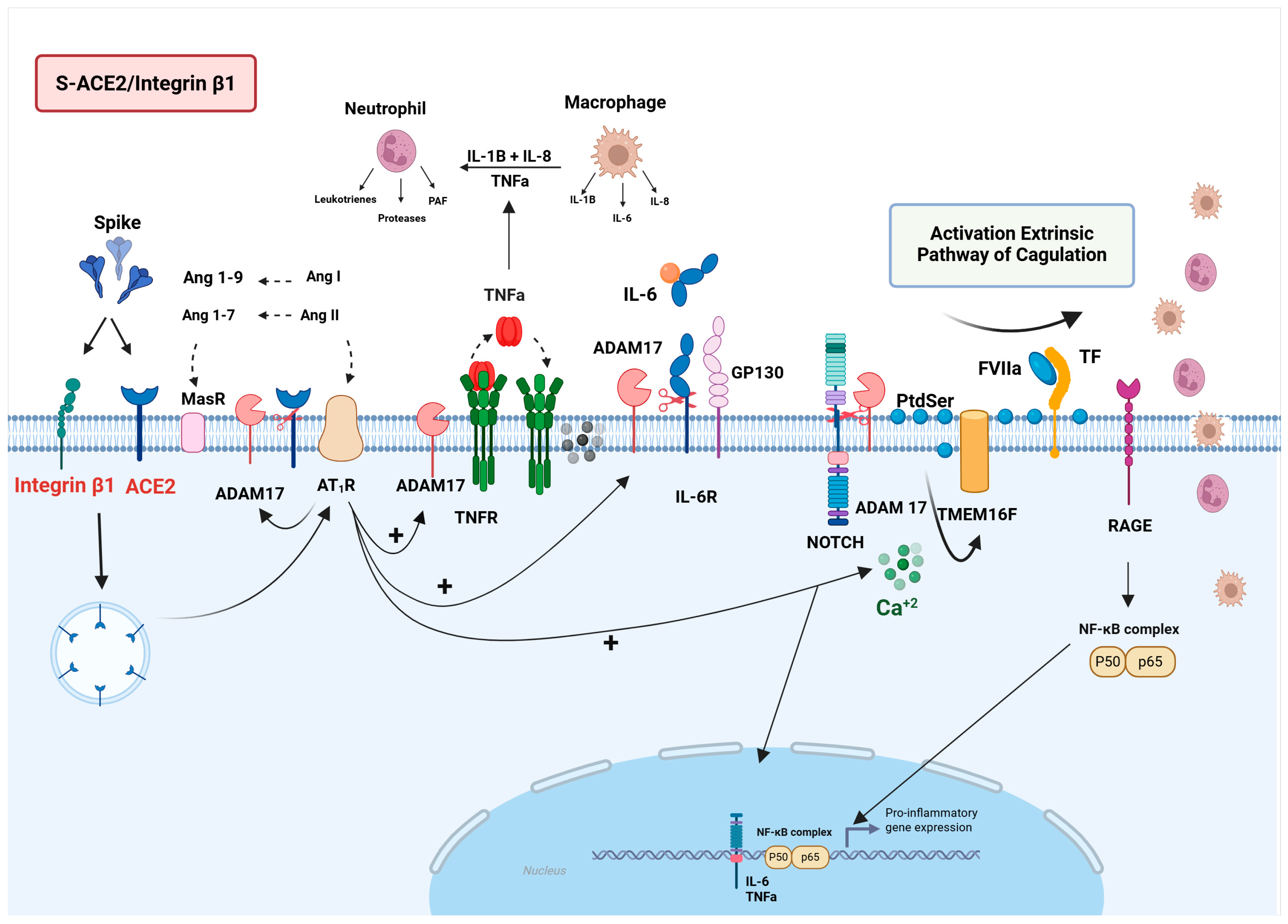
3.1. ACE2
3.1.1. S-ACE2 Interaction and Pathophysiology of SARS-CoV-2 Infection
S-ACE2/ADAM17/NLRP3 Signaling
S-ACE2/ADAM17/NOTCH/RAGE Signaling
3.1.2. Impact of the S-ACE2/Integrin β1 Interaction in Long COVID Pathophysiology
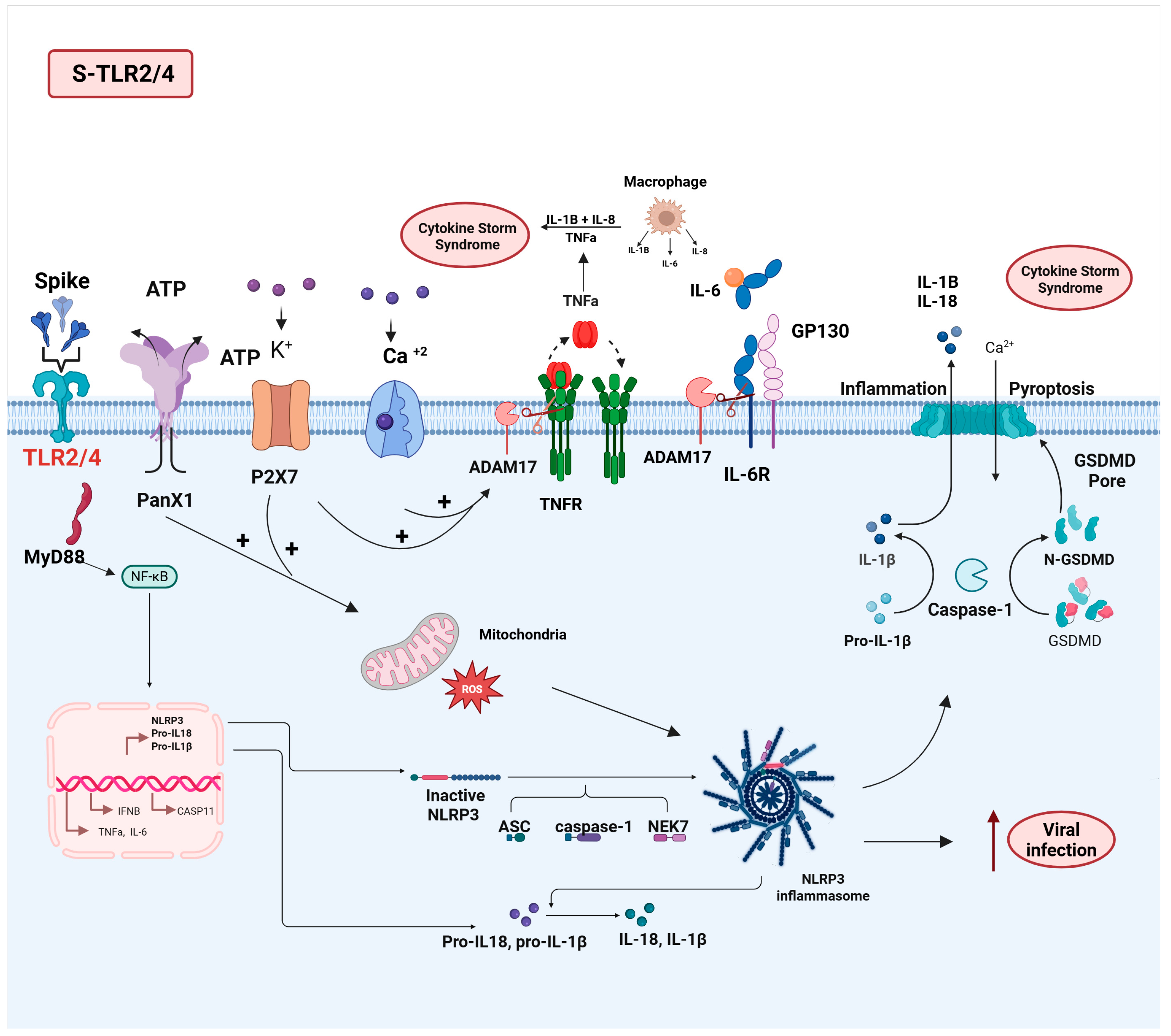
3.2. TLR2/4
3.2.1. S-TL2/4 Interaction and Pathophysiology of SARS-CoV-2 Infection
3.2.2. Impact of the S-TLR2/4 Interaction on Long COVID Pathophysiology
3.3. NRP1
3.3.1. S-NRP1 Interaction and Pathophysiology of SARS-CoV-2 Infection
3.3.2. Impact of the S-NRP1 Interaction in Long COVID Pathophysiology
3.4. DPP4
3.4.1. S-DPP4 Interaction in the Pathophysiology of SARS-CoV-2 Infection
3.4.2. Impact of S/DPP4 Interaction in the Long COVID Pathophysiology
3.5. NRP1/DPP4 Interplay in COVID-19 and Long COVID Pathophysiology
3.6. CD147
3.6.1. S-CD147 Interaction and SARS-CoV-2 Infection
3.6.2. CD147 and Pathophysiology of COVID-19
3.6.3. CD147/NRP1/DPP4/ACE2 Signaling
3.7. TfR
Impact of the S-TfR Interaction in the Pathophysiology of SARS-CoV-2 Infection and Long COVID
3.8. nAchRs
Impact of the S-nAchR Interplay in the Pathophysiology of SARS-CoV-2 Infection and Long COVID
4. Additional Potential Pathophysiological Effects Based on S Protein and Cellular Receptor Interactions
4.1. ERα
S-ERα Interplay and Physiopathology of SARS-CoV-2 Infection and Long COVID
5. Concluding Remarks and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE2 | Angiotensin-converting enzyme 2 |
| ADAM17 | A disintegrin and metalloprotease 17 |
| TMPRSS2 | Transmembrane protease serine 2 |
| MAPK | Mitogen-activated protein kinase |
| NF-kB | Nuclear factor kappa B |
| IL-6 | Interleukin 6 |
| IL-8 | Interleukin 8 |
| IL-1β | Interleukin 1 beta |
| TNF-α | Tumor necrosis factor alpha |
| JAK | Janus kinase |
| STAT 1/2 | Signal transducer and activator of transcription 1/2 |
| IFN | Interferon |
| TNFR | Tumor necrosis factor receptor |
| TLR | Toll-like receptor |
| PtdSer | Phosphatidylserine |
| ICAM-1 | Intercellular adhesion molecule 1 |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| VEGF | Vascular endothelial growth factor |
References
- Cascella, M.; Rajnik, M.; Aleem, A.; Dulebohn, S.C.; Di Napoli, R. Features, Evaluation, and Treatment of Coronavirus (COVID-19). In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2024. [Google Scholar]
- Pizzato, M.; Baraldi, C.; Boscato Sopetto, G.; Finozzi, D.; Gentile, C.; Gentile, M.D.; Marconi, R.; Paladino, D.; Raoss, A.; Riedmiller, I.; et al. SARS-CoV-2 and the Host Cell: A Tale of Interactions. Front. Virol. 2022, 1, 815388. [Google Scholar] [CrossRef]
- Hikmet, F.; Mear, L.; Edvinsson, A.; Micke, P.; Uhlen, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef] [PubMed]
- Fontes-Dantas, F.L.; Fernandes, G.G.; Gutman, E.G.; De Lima, E.V.; Antonio, L.S.; Hammerle, M.B.; Mota-Araujo, H.P.; Colodeti, L.C.; Araujo, S.M.B.; Froz, G.M.; et al. SARS-CoV-2 Spike protein induces TLR4-mediated long-term cognitive dysfunction recapitulating post-COVID-19 syndrome in mice. Cell Rep. 2023, 42, 112189. [Google Scholar] [CrossRef]
- Kong, W.; Montano, M.; Corley, M.J.; Helmy, E.; Kobayashi, H.; Kinisu, M.; Suryawanshi, R.; Luo, X.; Royer, L.A.; Roan, N.R.; et al. Neuropilin-1 Mediates SARS-CoV-2 Infection of Astrocytes in Brain Organoids, Inducing Inflammation Leading to Dysfunction and Death of Neurons. mBio 2022, 13, e0230822. [Google Scholar] [CrossRef]
- Liao, Z.; Wang, C.; Tang, X.; Yang, M.; Duan, Z.; Liu, L.; Lu, S.; Ma, L.; Cheng, R.; Wang, G.; et al. Human transferrin receptor can mediate SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 2024, 121, e2317026121. [Google Scholar] [CrossRef] [PubMed]
- Gadanec, L.K.; McSweeney, K.R.; Qaradakhi, T.; Ali, B.; Zulli, A.; Apostolopoulos, V. Can SARS-CoV-2 Virus Use Multiple Receptors to Enter Host Cells? Int. J. Mol. Sci. 2021, 22, 992. [Google Scholar] [CrossRef]
- Katopodis, P.; Randeva, H.S.; Spandidos, D.A.; Saravi, S.; Kyrou, I.; Karteris, E. Host cell entry mediators implicated in the cellular tropism of SARS-CoV-2, the pathophysiology of COVID-19 and the identification of microRNAs that can modulate the expression of these mediators (Review). Int. J. Mol. Med. 2022, 49, 20. [Google Scholar] [CrossRef]
- Olajide, O.A.; Iwuanyanwu, V.U.; Adegbola, O.D.; Al-Hindawi, A.A. SARS-CoV-2 Spike Glycoprotein S1 Induces Neuroinflammation in BV-2 Microglia. Mol. Neurobiol. 2022, 59, 445–458. [Google Scholar] [CrossRef]
- Oka, N.; Shimada, K.; Ishii, A.; Kobayashi, N.; Kondo, K. SARS-CoV-2 S1 protein causes brain inflammation by reducing intracerebral acetylcholine production. iScience 2023, 26, 106954. [Google Scholar] [CrossRef]
- Montezano, A.C.; Camargo, L.L.; Mary, S.; Neves, K.B.; Rios, F.J.; Stein, R.; Lopes, R.A.; Beattie, W.; Thomson, J.; Herder, V.; et al. SARS-CoV-2 spike protein induces endothelial inflammation via ACE2 independently of viral replication. Sci. Rep. 2023, 13, 14086. [Google Scholar] [CrossRef]
- Tortorici, M.A.; Walls, A.C.; Lang, Y.; Wang, C.; Li, Z.; Koerhuis, D.; Boons, G.J.; Bosch, B.J.; Rey, F.A.; de Groot, R.J.; et al. Structural basis for human coronavirus attachment to sialic acid receptors. Nat. Struct. Mol. Biol. 2019, 26, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Pohlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784 e775. [Google Scholar] [CrossRef]
- Bashir, A.; Li, S.; Ye, Y.; Zheng, Q.; Knanghat, R.; Bashir, F.; Shah, N.N.; Yang, D.; Xue, M.; Wang, H.; et al. SARS-CoV-2 S protein harbors furin cleavage site located in a short loop between antiparallel beta-strand. Int. J. Biol. Macromol. 2024, 281, 136020. [Google Scholar] [CrossRef]
- Jocher, G.; Grass, V.; Tschirner, S.K.; Riepler, L.; Breimann, S.; Kaya, T.; Oelsner, M.; Hamad, M.S.; Hofmann, L.I.; Blobel, C.P.; et al. ADAM10 and ADAM17 promote SARS-CoV-2 cell entry and spike protein-mediated lung cell fusion. EMBO Rep. 2022, 23, e54305. [Google Scholar] [CrossRef] [PubMed]
- Puelles, V.G.; Lutgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S.; et al. Multiorgan and Renal Tropism of SARS-CoV-2. N. Engl. J. Med. 2020, 383, 590–592. [Google Scholar] [CrossRef]
- Sungnak, W.; Huang, N.; Becavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.; Lopez, J.; Wyr, M.; Ramirez, P.W. Defining diverse spike-receptor interactions involved in SARS-CoV-2 entry: Mechanisms and therapeutic opportunities. Virology 2025, 607, 110507. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediators Inflamm. 2021, 2021, 8874339. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.X.; Gong, L.; et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020, 5, 283. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Qiu, Z.; Hou, Y.; Deng, X.; Xu, W.; Zheng, T.; Wu, P.; Xie, S.; Bian, W.; Zhang, C.; et al. AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. 2021, 31, 126–140. [Google Scholar] [CrossRef]
- Ha, D.P.; Shin, W.J.; Hernandez, J.C.; Neamati, N.; Dubeau, L.; Machida, K.; Lee, A.S. GRP78 Inhibitor YUM70 Suppresses SARS-CoV-2 Viral Entry, Spike Protein Production and Ameliorates Lung Damage. Viruses 2023, 15, 1118. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.; Yang, L.; Lian, X.; Xie, Y.; Li, S.; Xin, S.; Cao, P.; Lu, J. The MERS-CoV Receptor DPP4 as a Candidate Binding Target of the SARS-CoV-2 Spike. iScience 2020, 23, 101160. [Google Scholar] [CrossRef]
- Angioni, R.; Bonfanti, M.; Caporale, N.; Sanchez-Rodriguez, R.; Munari, F.; Savino, A.; Pasqualato, S.; Buratto, D.; Pagani, I.; Bertoldi, N.; et al. RAGE engagement by SARS-CoV-2 enables monocyte infection and underlies COVID-19 severity. Cell Rep. Med 2023, 4, 101266. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.N.; Richards, S.J.; Guy, C.S.; Congdon, T.R.; Hasan, M.; Zwetsloot, A.J.; Gallo, A.; Lewandowski, J.R.; Stansfeld, P.J.; Straube, A.; et al. The SARS-CoV-2 Spike Protein Binds Sialic Acids and Enables Rapid Detection in a Lateral Flow Point of Care Diagnostic Device. ACS Cent. Sci. 2020, 6, 2046–2052. [Google Scholar] [CrossRef]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057.e15. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, N.S.; Davanzo, G.G.; de Moraes, D.; Ferrari, A.J.R.; Souza, G.F.; Muraro, S.P.; Knittel, T.L.; Boldrini, V.O.; Monteiro, L.B.; Virgilio-da-Silva, J.V.; et al. SARS-CoV-2 uses CD4 to infect T helper lymphocytes. Elife 2023, 12, e84790. [Google Scholar] [CrossRef]
- Qian, J.; Zhang, S.; Wang, F.; Li, J.; Zhang, J. What makes SARS-CoV-2 unique? Focusing on the spike protein. Cell Biol. Int. 2024, 48, 404–430. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Su, C.; Xue, J.; Ye, C.; Chen, A. Role of the central renin-angiotensin system in hypertension (Review). Int. J. Mol. Med. 2021, 47, 95. [Google Scholar] [CrossRef] [PubMed]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef]
- Patel, V.B.; Zhong, J.C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/Angiotensin 1-7 Axis of the Renin-Angiotensin System in Heart Failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [PubMed]
- El-Arif, G.; Khazaal, S.; Farhat, A.; Harb, J.; Annweiler, C.; Wu, Y.; Cao, Z.; Kovacic, H.; Abi Khattar, Z.; Fajloun, Z.; et al. Angiotensin II Type I Receptor (AT1R): The Gate towards COVID-19-Associated Diseases. Molecules 2022, 27, 2048. [Google Scholar] [CrossRef]
- Skidgel, R.A.; Erdos, E.G. Angiotensin converting enzyme (ACE) and neprilysin hydrolyze neuropeptides: A brief history, the beginning and follow-ups to early studies. Peptides 2004, 25, 521–525. [Google Scholar] [CrossRef]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B., Jr.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg(9) bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef]
- Zhong, J.; Basu, R.; Guo, D.; Chow, F.L.; Byrns, S.; Schuster, M.; Loibner, H.; Wang, X.H.; Penninger, J.M.; Kassiri, Z.; et al. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation 2010, 122, 717–728. [Google Scholar] [CrossRef]
- Sun, J.; Edsfeldt, A.; Svensson, J.; Ruge, T.; Goncalves, I.; Sward, P. ADAM-17 Activity and Its Relation to ACE2: Implications for Severe COVID-19. Int. J. Mol. Sci. 2024, 25, 5911. [Google Scholar] [CrossRef] [PubMed]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pohlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef]
- Arganaraz, G.A.; Palmeira, J.D.F.; Arganaraz, E.R. Phosphatidylserine inside out: A possible underlying mechanism in the inflammation and coagulation abnormalities of COVID-19. Cell Commun. Signal. 2020, 18, 190. [Google Scholar] [CrossRef]
- Senchenkova, E.Y.; Russell, J.; Esmon, C.T.; Granger, D.N. Roles of Coagulation and fibrinolysis in angiotensin II-enhanced microvascular thrombosis. Microcirculation 2014, 21, 401–407. [Google Scholar] [CrossRef] [PubMed]
- van de Veerdonk, F.L.; Netea, M.G.; van Deuren, M.; van der Meer, J.W.; de Mast, Q.; Bruggemann, R.J.; van der Hoeven, H. Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. Elife 2020, 9, e57555. [Google Scholar] [CrossRef]
- Patel, V.B.; Clarke, N.; Wang, Z.; Fan, D.; Parajuli, N.; Basu, R.; Putko, B.; Kassiri, Z.; Turner, A.J.; Oudit, G.Y. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: A positive feedback mechanism in the RAS. J. Mol. Cell. Cardiol. 2014, 66, 167–176. [Google Scholar] [CrossRef]
- Sommer, A.; Kordowski, F.; Buch, J.; Maretzky, T.; Evers, A.; Andra, J.; Dusterhoft, S.; Michalek, M.; Lorenzen, I.; Somasundaram, P.; et al. Phosphatidylserine exposure is required for ADAM17 sheddase function. Nat. Commun. 2016, 7, 11523. [Google Scholar] [CrossRef] [PubMed]
- Bleibaum, F.; Sommer, A.; Veit, M.; Rabe, B.; Andra, J.; Kunzelmann, K.; Nehls, C.; Correa, W.; Gutsmann, T.; Grotzinger, J.; et al. ADAM10 sheddase activation is controlled by cell membrane asymmetry. J. Mol. Cell Biol. 2019, 11, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Liu, S.; Yan, H.; Lu, W.; Shan, X.; Chen, H.; Bao, C.; Feng, H.; Liao, J.; Liang, S.; et al. SARS-CoV-2 spike protein receptor-binding domain perturbates intracellular calcium homeostasis and impairs pulmonary vascular endothelial cells. Signal Transduct. Target. Ther. 2023, 8, 276. [Google Scholar] [CrossRef]
- Bohan, D.; Van Ert, H.; Ruggio, N.; Rogers, K.J.; Badreddine, M.; Aguilar Briseno, J.A.; Elliff, J.M.; Rojas Chavez, R.A.; Gao, B.; Stokowy, T.; et al. Phosphatidylserine receptors enhance SARS-CoV-2 infection. PLoS Pathog. 2021, 17, e1009743. [Google Scholar] [CrossRef]
- Braga, L.; Ali, H.; Secco, I.; Chiavacci, E.; Neves, G.; Goldhill, D.; Penn, R.; Jimenez-Guardeno, J.M.; Ortega-Prieto, A.M.; Bussani, R.; et al. Drugs that inhibit TMEM16 proteins block SARS-CoV-2 spike-induced syncytia. Nature 2021, 594, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Ardiana, M.; Suryawan, I.G.R.; Hermawan, H.O.; Harsoyo, P.M.; Sufiyah, I.M.; Muhammad, A.R.; Zaini, B.S.I. Perindopril and losartan attenuate pro-coagulation factors in human adipocytes exposed to SARS-CoV-2 spike protein. J. Physiol. Pharmacol. 2023, 74, 275–280. [Google Scholar] [CrossRef]
- Liang, S.; Bao, C.; Yang, Z.; Liu, S.; Sun, Y.; Cao, W.; Wang, T.; Schwantes-An, T.H.; Choy, J.S.; Naidu, S.; et al. SARS-CoV-2 spike protein induces IL-18-mediated cardiopulmonary inflammation via reduced mitophagy. Signal Transduct. Target. Ther. 2023, 8, 108. [Google Scholar] [CrossRef]
- Sun, N.N.; Yu, C.H.; Pan, M.X.; Zhang, Y.; Zheng, B.J.; Yang, Q.J.; Zheng, Z.M.; Meng, Y. Mir-21 Mediates the Inhibitory Effect of Ang (1-7) on AngII-induced NLRP3 Inflammasome Activation by Targeting Spry1 in lung fibroblasts. Sci. Rep. 2017, 7, 14369. [Google Scholar] [CrossRef]
- Zhao, M.; Bai, M.; Ding, G.; Zhang, Y.; Huang, S.; Jia, Z.; Zhang, A. Angiotensin II Stimulates the NLRP3 Inflammasome to Induce Podocyte Injury and Mitochondrial Dysfunction. Kidney Dis. 2018, 4, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Villacampa, A.; Alfaro, E.; Morales, C.; Diaz-Garcia, E.; Lopez-Fernandez, C.; Bartha, J.L.; Lopez-Sanchez, F.; Lorenzo, O.; Moncada, S.; Sanchez-Ferrer, C.F.; et al. SARS-CoV-2 S protein activates NLRP3 inflammasome and deregulates coagulation factors in endothelial and immune cells. Cell Commun. Signal. 2024, 22, 38. [Google Scholar] [CrossRef] [PubMed]
- Breikaa, R.M.; Lilly, B. The Notch Pathway: A Link Between COVID-19 Pathophysiology and Its Cardiovascular Complications. Front. Cardiovasc. Med. 2021, 8, 681948. [Google Scholar] [CrossRef] [PubMed]
- Bozkulak, E.C.; Weinmaster, G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol. Cell. Biol. 2009, 29, 5679–5695. [Google Scholar] [CrossRef]
- Doberstein, K.; Steinmeyer, N.; Hartmetz, A.K.; Eberhardt, W.; Mittelbronn, M.; Harter, P.N.; Juengel, E.; Blaheta, R.; Pfeilschifter, J.; Gutwein, P. MicroRNA-145 targets the metalloprotease ADAM17 and is suppressed in renal cell carcinoma patients. Neoplasia 2013, 15, 218–230. [Google Scholar] [CrossRef]
- Hamldar, S.; Kiani, S.J.; Khoshmirsafa, M.; Nahand, J.S.; Mirzaei, H.; Khatami, A.; Kahyesh-Esfandiary, R.; Khanaliha, K.; Tavakoli, A.; Babakhaniyan, K.; et al. Expression profiling of inflammation-related genes including IFI-16, NOTCH2, CXCL8, THBS1 in COVID-19 patients. Biologicals 2022, 80, 27–34. [Google Scholar] [CrossRef]
- Pickering, R.J.; Tikellis, C.; Rosado, C.J.; Tsorotes, D.; Dimitropoulos, A.; Smith, M.; Huet, O.; Seeber, R.M.; Abhayawardana, R.; Johnstone, E.K.; et al. Transactivation of RAGE mediates angiotensin-induced inflammation and atherogenesis. J. Clin. Investig. 2019, 129, 406–421. [Google Scholar] [CrossRef]
- Gusev, E.; Sarapultsev, A. Exploring the Pathophysiology of Long COVID: The Central Role of Low-Grade Inflammation and Multisystem Involvement. Int. J. Mol. Sci. 2024, 25, 6389. [Google Scholar] [CrossRef]
- Jin, Y.; Ji, W.; Yang, H.; Chen, S.; Zhang, W.; Duan, G. Endothelial activation and dysfunction in COVID-19: From basic mechanisms to potential therapeutic approaches. Signal Transduct. Target. Ther. 2020, 5, 293. [Google Scholar] [CrossRef]
- Karki, R.; Kanneganti, T.D. Innate immunity, cytokine storm, and inflammatory cell death in COVID-19. J. Transl. Med. 2022, 20, 542. [Google Scholar] [CrossRef]
- Shabani, Z.; Liu, J.; Su, H. Vascular Dysfunctions Contribute to the Long-Term Cognitive Deficits Following COVID-19. Biology 2023, 12, 1106. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Connolly, R.; Brennan, D.; Laffan, A.; O’Keeffe, E.; Zaporojan, L.; O’Callaghan, J.; Thomson, B.; Connolly, E.; Argue, R.; et al. Blood-brain barrier disruption and sustained systemic inflammation in individuals with long COVID-associated cognitive impairment. Nat. Neurosci. 2024, 27, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Zhang, C.; Israelow, B.; Lu-Culligan, A.; Prado, A.V.; Skriabine, S.; Lu, P.; Weizman, O.E.; Liu, F.; Dai, Y.; et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med. 2021, 218, e20202135. [Google Scholar] [CrossRef]
- Welcome, M.O.; Mastorakis, N.E. Neuropathophysiology of coronavirus disease 2019: Neuroinflammation and blood brain barrier disruption are critical pathophysiological processes that contribute to the clinical symptoms of SARS-CoV-2 infection. Inflammopharmacology 2021, 29, 939–963. [Google Scholar] [CrossRef]
- Bodnar, B.; Patel, K.; Ho, W.; Luo, J.J.; Hu, W. Cellular mechanisms underlying neurological/neuropsychiatric manifestations of COVID-19. J. Med. Virol. 2021, 93, 1983–1998. [Google Scholar] [CrossRef] [PubMed]
- Charfeddine, S.; Ibn Hadj Amor, H.; Jdidi, J.; Torjmen, S.; Kraiem, S.; Hammami, R.; Bahloul, A.; Kallel, N.; Moussa, N.; Touil, I.; et al. Long COVID 19 Syndrome: Is It Related to Microcirculation and Endothelial Dysfunction? Insights From TUN-EndCOV Study. Front. Cardiovasc. Med. 2021, 8, 745758. [Google Scholar] [CrossRef]
- Katsoularis, I.; Fonseca-Rodriguez, O.; Farrington, P.; Jerndal, H.; Lundevaller, E.H.; Sund, M.; Lindmark, K.; Fors Connolly, A.M. Risks of deep vein thrombosis, pulmonary embolism, and bleeding after COVID-19: Nationwide self-controlled cases series and matched cohort study. BMJ 2022, 377, e069590. [Google Scholar] [CrossRef]
- Pretorius, E.; Vlok, M.; Venter, C.; Bezuidenhout, J.A.; Laubscher, G.J.; Steenkamp, J.; Kell, D.B. Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc. Diabetol. 2021, 20, 172. [Google Scholar] [CrossRef]
- Kubankova, M.; Hohberger, B.; Hoffmanns, J.; Furst, J.; Herrmann, M.; Guck, J.; Krater, M. Physical phenotype of blood cells is altered in COVID-19. Biophys. J. 2021, 120, 2838–2847. [Google Scholar] [CrossRef]
- Patra, T.; Meyer, K.; Geerling, L.; Isbell, T.S.; Hoft, D.F.; Brien, J.; Pinto, A.K.; Ray, R.B.; Ray, R. SARS-CoV-2 spike protein promotes IL-6 trans-signaling by activation of angiotensin II receptor signaling in epithelial cells. PLoS Pathog. 2020, 16, e1009128. [Google Scholar] [CrossRef] [PubMed]
- Akwii, R.G.; Sajib, M.S.; Zahra, F.T.; Mikelis, C.M. Role of Angiopoietin-2 in Vascular Physiology and Pathophysiology. Cells 2019, 8, 471. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L.; et al. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef]
- Parekh, R.U.; Sriramula, S. Activation of Kinin B1R Upregulates ADAM17 and Results in ACE2 Shedding in Neurons. Int. J. Mol. Sci. 2020, 22, 145. [Google Scholar] [CrossRef]
- Lobov, I.B.; Brooks, P.C.; Lang, R.A. Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 11205–11210. [Google Scholar] [CrossRef] [PubMed]
- Robles, J.P.; Zamora, M.; Adan-Castro, E.; Siqueiros-Marquez, L.; Martinez de la Escalera, G.; Clapp, C. The spike protein of SARS-CoV-2 induces endothelial inflammation through integrin alpha5beta1 and NF-kappaB signaling. J. Biol. Chem. 2022, 298, 101695. [Google Scholar] [CrossRef]
- DeOre, B.J.; Tran, K.A.; Andrews, A.M.; Ramirez, S.H.; Galie, P.A. SARS-CoV-2 Spike Protein Disrupts Blood-Brain Barrier Integrity via RhoA Activation. J. Neuroimmune Pharmacol. 2021, 16, 722–728. [Google Scholar] [CrossRef]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4, a006049. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.W.; Savani, R.C.; Zaki, H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-kappaB pathway. Elife 2021, 10, e68563. [Google Scholar] [CrossRef]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef]
- Takeuchi, O.; Hoshino, K.; Kawai, T.; Sanjo, H.; Takada, H.; Ogawa, T.; Takeda, K.; Akira, S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 1999, 11, 443–451. [Google Scholar] [CrossRef]
- Kawai, T.; Ikegawa, M.; Ori, D.; Akira, S. Decoding Toll-like receptors: Recent insights and perspectives in innate immunity. Immunity 2024, 57, 649–673. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Kawai, T.; Muhlradt, P.F.; Morr, M.; Radolf, J.D.; Zychlinsky, A.; Takeda, K.; Akira, S. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 2001, 13, 933–940. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef]
- Mukherjee, S.; Karmakar, S.; Babu, S.P. TLR2 and TLR4 mediated host immune responses in major infectious diseases: A review. Braz. J. Infect. Dis. 2016, 20, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Akira, S. Toll-Like Receptor Signaling and Its Inducible Proteins. Microbiol. Spectr. 2016, 4, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Nascimento, L.; Massari, P.; Wetzler, L.M. The Role of TLR2 in Infection and Immunity. Front. Immunol. 2012, 3, 79. [Google Scholar] [CrossRef]
- Kagan, J.C.; Su, T.; Horng, T.; Chow, A.; Akira, S.; Medzhitov, R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat. Immunol. 2008, 9, 361–368. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef] [PubMed]
- Al-Qahtani, A.A.; Pantazi, I.; Alhamlan, F.S.; Alothaid, H.; Matou-Nasri, S.; Sourvinos, G.; Vergadi, E.; Tsatsanis, C. SARS-CoV-2 modulates inflammatory responses of alveolar epithelial type II cells via PI3K/AKT pathway. Front. Immunol. 2022, 13, 1020624. [Google Scholar] [CrossRef]
- Abate, W.; Alghaithy, A.A.; Parton, J.; Jones, K.P.; Jackson, S.K. Surfactant lipids regulate LPS-induced interleukin-8 production in A549 lung epithelial cells by inhibiting translocation of TLR4 into lipid raft domains. J. Lipid Res. 2010, 51, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Kuronuma, K.; Mitsuzawa, H.; Takeda, K.; Nishitani, C.; Chan, E.D.; Kuroki, Y.; Nakamura, M.; Voelker, D.R. Anionic pulmonary surfactant phospholipids inhibit inflammatory responses from alveolar macrophages and U937 cells by binding the lipopolysaccharide-interacting proteins CD14 and MD-2. J. Biol. Chem. 2009, 284, 25488–25500. [Google Scholar] [CrossRef]
- Voelker, D.R.; Numata, M. Phospholipid regulation of innate immunity and respiratory viral infection. J. Biol. Chem. 2019, 294, 4282–4289. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef]
- Frantz, S.; Kobzik, L.; Kim, Y.D.; Fukazawa, R.; Medzhitov, R.; Lee, R.T.; Kelly, R.A. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J. Clin. Investig. 1999, 104, 271–280. [Google Scholar] [CrossRef]
- Shirato, K.; Kizaki, T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon 2021, 7, e06187. [Google Scholar] [CrossRef]
- Alves, V.S.; Santos, S.; Leite-Aguiar, R.; Paiva-Pereira, E.; Dos Reis, R.R.; Calazans, M.L.; Fernandes, G.G.; Antonio, L.S.; de Lima, E.V.; Kurtenbach, E.; et al. SARS-CoV-2 Spike protein alters microglial purinergic signaling. Front. Immunol. 2023, 14, 1158460. [Google Scholar] [CrossRef]
- Yue, Z.; Zhang, X.; Gu, Y.; Liu, Y.; Lan, L.M.; Liu, Y.; Li, Y.; Yang, G.; Wan, P.; Chen, X. Regulation and functions of the NLRP3 inflammasome in RNA virus infection. Front. Cell. Infect. Microbiol. 2023, 13, 1309128. [Google Scholar] [CrossRef]
- Lecuyer, D.; Nardacci, R.; Tannous, D.; Gutierrez-Mateyron, E.; Deva Nathan, A.; Subra, F.; Di Primio, C.; Quaranta, P.; Petit, V.; Richetta, C.; et al. The purinergic receptor P2X7 and the NLRP3 inflammasome are druggable host factors required for SARS-CoV-2 infection. Front. Immunol. 2023, 14, 1270081. [Google Scholar] [CrossRef] [PubMed]
- Pupovac, A.; Geraghty, N.J.; Watson, D.; Sluyter, R. Activation of the P2X7 receptor induces the rapid shedding of CD23 from human and murine B cells. Immunol. Cell Biol. 2015, 93, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Koff, J.L.; Shao, M.X.; Ueki, I.F.; Nadel, J.A. Multiple TLRs activate EGFR via a signaling cascade to produce innate immune responses in airway epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L1068–L1075. [Google Scholar] [CrossRef]
- Katakia, Y.T.; Thakkar, N.P.; Thakar, S.; Sakhuja, A.; Goyal, R.; Sharma, H.; Dave, R.; Mandloi, A.; Basu, S.; Nigam, I.; et al. Dynamic alterations of H3K4me3 and H3K27me3 at ADAM17 and Jagged-1 gene promoters cause an inflammatory switch of endothelial cells. J. Cell. Physiol. 2022, 237, 992–1012. [Google Scholar] [CrossRef] [PubMed]
- Tyrkalska, S.D.; Martinez-Lopez, A.; Pedoto, A.; Candel, S.; Cayuela, M.L.; Mulero, V. The Spike protein of SARS-CoV-2 signals via Tlr2 in zebrafish. Dev. Comp. Immunol. 2023, 140, 104626. [Google Scholar] [CrossRef]
- Frank, M.G.; Nguyen, K.H.; Ball, J.B.; Hopkins, S.; Kelley, T.; Baratta, M.V.; Fleshner, M.; Maier, S.F. SARS-CoV-2 spike S1 subunit induces neuroinflammatory, microglial and behavioral sickness responses: Evidence of PAMP-like properties. Brain Behav. Immun. 2022, 100, 267–277. [Google Scholar] [CrossRef]
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. More than 50 long-term effects of COVID-19: A systematic review and meta-analysis. Sci. Rep. 2021, 11, 16144. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Landolina, N.; Ricci, B.; Veneziani, I.; Alicata, C.; Mariotti, F.R.; Pelosi, A.; Quatrini, L.; Mortari, E.P.; Carsetti, R.; Vacca, P.; et al. TLR2/4 are novel activating receptors for SARS-CoV-2 spike protein on NK cells. Front. Immunol. 2024, 15, 1368946. [Google Scholar] [CrossRef]
- Wilson, A.M.; Shao, Z.; Grenier, V.; Mawambo, G.; Daudelin, J.F.; Dejda, A.; Pilon, F.; Popovic, N.; Boulet, S.; Parinot, C.; et al. Neuropilin-1 expression in adipose tissue macrophages protects against obesity and metabolic syndrome. Sci. Immunol. 2018, 3, eaan4626. [Google Scholar] [CrossRef]
- Kofler, N.; Simons, M. The expanding role of neuropilin: Regulation of transforming growth factor-beta and platelet-derived growth factor signaling in the vasculature. Curr. Opin. Hematol. 2016, 23, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Bag, A.K.; Singh, R.K.; Talmadge, J.E.; Batra, S.K.; Datta, K. Multifaceted Role of Neuropilins in the Immune System: Potential Targets for Immunotherapy. Front. Immunol. 2017, 8, 1228. [Google Scholar] [CrossRef]
- Bruder, D.; Probst-Kepper, M.; Westendorf, A.M.; Geffers, R.; Beissert, S.; Loser, K.; von Boehmer, H.; Buer, J.; Hansen, W. Neuropilin-1: A surface marker of regulatory T cells. Eur. J. Immunol. 2004, 34, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Alnomasy, S.F. Virus-receptor interactions of SARS-CoV-2 spikereceptor-binding domain and human neuropilin-1 b1 domain. Saudi J. Biol. Sci. 2021, 28, 3926–3928. [Google Scholar] [CrossRef]
- Wang, H.-B.; Zhang, H.; Zhang, J.-P.; Li, Y.; Zhao, B.; Feng, G.-K.; Du, Y.; Xiong, D.; Zhong, Q.; Liu, W.-L.; et al. Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nat. Commun. 2015, 6, 6240. [Google Scholar] [CrossRef] [PubMed]
- Sultan, R.H.; Abdallah, M.; Ali, T.M.; Ahmed, A.E.; Assal, H.H.; Elesawy, B.H.; Ahmed, O.M. The Associations between Cytokine Levels, Kidney and Heart Function Biomarkers, and Expression Levels of Angiotensin-Converting Enzyme-2 and Neuropilin-1 in COVID-19 Patients. Vaccines 2022, 10, 1045. [Google Scholar] [CrossRef]
- Pellet-Many, C.; Mehta, V.; Fields, L.; Mahmoud, M.; Lowe, V.; Evans, I.; Ruivo, J.; Zachary, I. Neuropilins 1 and 2 mediate neointimal hyperplasia and re-endothelialization following arterial injury. Cardiovasc. Res. 2015, 108, 288–298. [Google Scholar] [CrossRef]
- Raimondi, C.; Ruhrberg, C. Neuropilin signalling in vessels, neurons and tumours. Semin. Cell Dev. Biol. 2013, 24, 172–178. [Google Scholar] [CrossRef]
- Mercurio, A.M. VEGF/Neuropilin Signaling in Cancer Stem Cells. Int. J. Mol. Sci. 2019, 20, 490. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- El-Arabey, A.A.; Abdalla, M. Neuropilin-1 may be responsible for retinal findings in patients with COVID-19. Hum. Cell 2021, 34, 1280–1281. [Google Scholar] [CrossRef] [PubMed]
- Issitt, T.; Bosseboeuf, E.; De Winter, N.; Dufton, N.; Gestri, G.; Senatore, V.; Chikh, A.; Randi, A.M.; Raimondi, C. Neuropilin-1 Controls Endothelial Homeostasis by Regulating Mitochondrial Function and Iron-Dependent Oxidative Stress. iScience 2019, 11, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, C.; Lechien, J.R.; Saussez, S. More that ACE2? NRP1 may play a central role in the underlying pathophysiological mechanism of olfactory dysfunction in COVID-19 and its association with enhanced survival. Med. Hypotheses 2021, 146, 110406. [Google Scholar] [CrossRef]
- Davies, J.; Randeva, H.S.; Chatha, K.; Hall, M.; Spandidos, D.A.; Karteris, E.; Kyrou, I. Neuropilin-1 as a new potential SARS-CoV-2 infection mediator implicated in the neurologic features and central nervous system involvement of COVID-19. Mol. Med. Rep. 2020, 22, 4221–4226. [Google Scholar] [CrossRef]
- Mone, P.; Gambardella, J.; Wang, X.; Jankauskas, S.S.; Matarese, A.; Santulli, G. miR-24 Targets the Transmembrane Glycoprotein Neuropilin-1 in Human Brain Microvascular Endothelial Cells. Noncoding RNA 2021, 7, 9. [Google Scholar] [CrossRef]
- Moutal, A.; Martin, L.F.; Boinon, L.; Gomez, K.; Ran, D.; Zhou, Y.; Stratton, H.J.; Cai, S.; Luo, S.; Gonzalez, K.B.; et al. SARS-CoV-2 spike protein co-opts VEGF-A/neuropilin-1 receptor signaling to induce analgesia. Pain 2021, 162, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Perez-Miller, S.; Patek, M.; Moutal, A.; Duran, P.; Cabel, C.R.; Thorne, C.A.; Campos, S.K.; Khanna, R. Novel Compounds Targeting Neuropilin Receptor 1 with Potential To Interfere with SARS-CoV-2 Virus Entry. ACS Chem. Neurosci. 2021, 12, 1299–1312. [Google Scholar] [CrossRef]
- Hanson, A.L.; Mule, M.P.; Ruffieux, H.; Mescia, F.; Bergamaschi, L.; Pelly, V.S.; Turner, L.; Kotagiri, P.; Cambridge Institute of Therapeutic Immunology; Infectious Disease–National Institute for Health Research (CITIID–NIHR) COVID BioResource Collaboration; et al. Iron dysregulation and inflammatory stress erythropoiesis associates with long-term outcome of COVID-19. Nat. Immunol. 2024, 25, 471–482. [Google Scholar] [CrossRef]
- Berentschot, J.C.; Drexhage, H.A.; Aynekulu Mersha, D.G.; Wijkhuijs, A.J.M.; GeurtsvanKessel, C.H.; Koopmans, M.P.G.; Voermans, J.J.C.; Hendriks, R.W.; Nagtzaam, N.M.A.; de Bie, M.; et al. Immunological profiling in long COVID: Overall low grade inflammation and T-lymphocyte senescence and increased monocyte activation correlating with increasing fatigue severity. Front. Immunol. 2023, 14, 1254899. [Google Scholar] [CrossRef]
- Dhawan, M.; Rabaan, A.A.; Alwarthan, S.; Alhajri, M.; Halwani, M.A.; Alshengeti, A.; Najim, M.A.; Alwashmi, A.S.S.; Alshehri, A.A.; Alshamrani, S.A.; et al. Regulatory T Cells (Tregs) and COVID-19: Unveiling the Mechanisms, and Therapeutic Potentialities with a Special Focus on Long COVID. Vaccines 2023, 11, 699. [Google Scholar] [CrossRef]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef]
- Haunhorst, S.; Bloch, W.; Javelle, F.; Kruger, K.; Baumgart, S.; Drube, S.; Lemhofer, C.; Reuken, P.; Stallmach, A.; Muller, M.; et al. A scoping review of regulatory T cell dynamics in convalescent COVID-19 patients—Indications for their potential involvement in the development of Long COVID? Front. Immunol. 2022, 13, 1070994. [Google Scholar] [CrossRef]
- Boonacker, E.; Van Noorden, C.J. The multifunctional or moonlighting protein CD26/DPPIV. Eur. J. Cell Biol. 2003, 82, 53–73. [Google Scholar] [CrossRef]
- Aliyari Serej, Z.; Ebrahimi Kalan, A.; Mehdipour, A.; Nozad Charoudeh, H. Regulation and roles of CD26/DPPIV in hematopoiesis and diseases. Biomed. Pharmacother. 2017, 91, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, T.; Tsuda, H.; Morimoto, C. Good or evil: CD26 and HIV infection. J. Dermatol. Sci. 2000, 22, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Solerte, S.B.; Di Sabatino, A.; Galli, M.; Fiorina, P. Dipeptidyl peptidase-4 (DPP4) inhibition in COVID-19. Acta Diabetol. 2020, 57, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Tisi, A.; Zerti, D.; Genitti, G.; Vicentini, M.T.; Baccante, M.; Flati, V.; Maccarone, R. Characterization of SARS-CoV-2 Entry Factors’ Expression in Corneal and Limbal Tissues of Adult Human Donors Aged from 58 to 85. J. Ocul. Pharmacol. Ther. 2022, 38, 56–65. [Google Scholar] [CrossRef]
- Bardaweel, S.K.; Hajjo, R.; Sabbah, D.A. Sitagliptin: A potential drug for the treatment of COVID-19? Acta Pharm. 2021, 71, 175–184. [Google Scholar] [CrossRef]
- Drucker, D.J. The biology of incretin hormones. Cell Metab. 2006, 3, 153–165. [Google Scholar] [CrossRef]
- Zheng, T.; Gao, Y.; Baskota, A.; Chen, T.; Ran, X.; Tian, H. Increased plasma DPP4 activity is predictive of prediabetes and type 2 diabetes onset in Chinese over a four-year period: Result from the China National Diabetes and Metabolic Disorders Study. J. Clin. Endocrinol. Metab. 2014, 99, E2330–E2334. [Google Scholar] [CrossRef]
- Sarkar, J.; Nargis, T.; Tantia, O.; Ghosh, S.; Chakrabarti, P. Increased Plasma Dipeptidyl Peptidase-4 (DPP4) Activity Is an Obesity-Independent Parameter for Glycemic Deregulation in Type 2 Diabetes Patients. Front. Endocrinol. 2019, 10, 505. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Huang, J.; Zhu, G.; Wang, Q.; Lv, Q.; Huang, Y.; Yu, Y.; Si, X.; Yi, H.; Wang, C.; et al. Elevation of blood glucose level predicts worse outcomes in hospitalized patients with COVID-19: A retrospective cohort study. BMJ Open Diabetes Res. Care 2020, 8, e001476. [Google Scholar] [CrossRef]
- Nadasdi, A.; Sinkovits, G.; Bobek, I.; Lakatos, B.; Forhecz, Z.; Prohaszka, Z.Z.; Reti, M.; Arato, M.; Cseh, G.; Masszi, T.; et al. Decreased circulating dipeptidyl peptidase-4 enzyme activity is prognostic for severe outcomes in COVID-19 inpatients. Biomark. Med. 2022, 16, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Sebastián-Martín, A.; Sánchez, B.G.; Mora-Rodríguez, J.M.; Bort, A.; Díaz-Laviada, I. Role of Dipeptidyl Peptidase-4 (DPP4) on COVID-19 Physiopathology. Biomedicines 2022, 10, 2026. [Google Scholar] [CrossRef] [PubMed]
- Spanakis, E.K.; Yoo, A.; Ajayi, O.N.; Siddiqui, T.; Khan, M.M.; Seliger, S.L.; Klonoff, D.C.; Feng, Z.; Sorkin, J.D. Excess Mortality in COVID-19-Positive Versus COVID-19-Negative Inpatients With Diabetes: A Nationwide Study. Diabetes Care 2021, 44, e169–e170. [Google Scholar] [CrossRef]
- Kazakou, P.; Lambadiari, V.; Ikonomidis, I.; Kountouri, A.; Panagopoulos, G.; Athanasopoulos, S.; Korompoki, E.; Kalomenidis, I.; Dimopoulos, M.A.; Mitrakou, A. Diabetes and COVID-19; A Bidirectional Interplay. Front. Endocrinol. 2022, 13, 780663. [Google Scholar] [CrossRef]
- Omeragic, A.; Hoque, M.T.; Choi, U.Y.; Bendayan, R. Peroxisome proliferator-activated receptor-gamma: Potential molecular therapeutic target for HIV-1-associated brain inflammation. J. Neuroinflamm. 2017, 14, 183. [Google Scholar] [CrossRef]
- Wang, Z.; Pekarskaya, O.; Bencheikh, M.; Chao, W.; Gelbard, H.A.; Ghorpade, A.; Rothstein, J.D.; Volsky, D.J. Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology 2003, 312, 60–73. [Google Scholar] [CrossRef]
- Wang, J.; Yang, G.; Wang, X.; Wen, Z.; Shuai, L.; Luo, J.; Wang, C.; Sun, Z.; Liu, R.; Ge, J.; et al. SARS-CoV-2 uses metabotropic glutamate receptor subtype 2 as an internalization factor to infect cells. Cell Discov. 2021, 7, 119. [Google Scholar] [CrossRef]
- de Oliveira, L.G.; de Souza Angelo, Y.; Yamamoto, P.; Carregari, V.C.; Crunfli, F.; Reis-de-Oliveira, G.; Costa, L.; Vendramini, P.H.; Duque, E.A.; Dos Santos, N.B.; et al. SARS-CoV-2 infection impacts carbon metabolism and depends on glutamine for replication in Syrian hamster astrocytes. J. Neurochem. 2022, 163, 113–132. [Google Scholar] [CrossRef]
- World Health Organization (WHO). A Clinical Case Definition of Post COVID-19 Condition by a Delphi Consensus, 6 October 2021; World Health Organization (WHO): Geneva, Switzerland, 2021. [Google Scholar]
- Bharadwaj, S.; Singh, M.; Kirtipal, N.; Kang, S.G. SARS-CoV-2 and Glutamine: SARS-CoV-2 Triggered Pathogenesis via Metabolic Reprograming of Glutamine in Host Cells. Front. Mol. Biosci. 2020, 7, 627842. [Google Scholar] [CrossRef] [PubMed]
- Andrews, M.G.; Mukhtar, T.; Eze, U.C.; Simoneau, C.R.; Ross, J.; Parikshak, N.; Wang, S.; Zhou, L.; Koontz, M.; Velmeshev, D.; et al. Tropism of SARS-CoV-2 for human cortical astrocytes. Proc. Natl. Acad. Sci. USA 2022, 119, e2122236119. [Google Scholar] [CrossRef] [PubMed]
- Constantino, F.B.; Cury, S.S.; Nogueira, C.R.; Carvalho, R.F.; Justulin, L.A. Prediction of Non-canonical Routes for SARS-CoV-2 Infection in Human Placenta Cells. Front. Mol. Biosci. 2021, 8, 614728. [Google Scholar] [CrossRef]
- Banazadeh, M.; Olangian-Tehrani, S.; Sharifi, M.; Malek-Ahmadi, M.; Nikzad, F.; Doozandeh-Nargesi, N.; Mohammadi, A.; Stephens, G.J.; Shabani, M. Mechanisms of COVID-19-induced cerebellitis. Curr. Med. Res. Opin. 2022, 38, 2109–2118. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Isath, A.; Aronow, W.S. Cardiovascular complications of diabetes. Expert Rev. Endocrinol. Metab. 2022, 17, 383–388. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Matsui, T.; Maeda, S.; Higashimoto, Y.; Yamagishi, S. Advanced glycation end products evoke endothelial cell damage by stimulating soluble dipeptidyl peptidase-4 production and its interaction with mannose 6-phosphate/insulin-like growth factor II receptor. Cardiovasc. Diabetol. 2013, 12, 125. [Google Scholar] [CrossRef]
- Glinka, Y.; Prud’homme, G.J. Neuropilin-1 is a receptor for transforming growth factor beta-1, activates its latent form, and promotes regulatory T cell activity. J. Leukoc. Biol. 2008, 84, 302–310. [Google Scholar] [CrossRef]
- Xiong, L.; Edwards, C.K., 3rd; Zhou, L. The biological function and clinical utilization of CD147 in human diseases: A review of the current scientific literature. Int. J. Mol. Sci. 2014, 15, 17411–17441. [Google Scholar] [CrossRef]
- Pistol, G.; Matache, C.; Calugaru, A.; Stavaru, C.; Tanaseanu, S.; Ionescu, R.; Dumitrache, S.; Stefanescu, M. Roles of CD147 on T lymphocytes activation and MMP-9 secretion in systemic lupus erythematosus. J. Cell. Mol. Med. 2007, 11, 339–348. [Google Scholar] [CrossRef]
- Zhao, P.; Zhang, W.; Wang, S.J.; Yu, X.L.; Tang, J.; Huang, W.; Li, Y.; Cui, H.Y.; Guo, Y.S.; Tavernier, J.; et al. HAb18G/CD147 promotes cell motility by regulating annexin II-activated RhoA and Rac1 signaling pathways in hepatocellular carcinoma cells. Hepatology 2011, 54, 2012–2024. [Google Scholar] [CrossRef]
- Bernard, S.C.; Simpson, N.; Join-Lambert, O.; Federici, C.; Laran-Chich, M.P.; Maissa, N.; Bouzinba-Segard, H.; Morand, P.C.; Chretien, F.; Taouji, S.; et al. Pathogenic Neisseria meningitidis utilizes CD147 for vascular colonization. Nat. Med. 2014, 20, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, T. Basigin (CD147), a multifunctional transmembrane glycoprotein with various binding partners. J. Biochem. 2016, 159, 481–490. [Google Scholar] [CrossRef]
- Pushkarsky, T.; Zybarth, G.; Dubrovsky, L.; Yurchenko, V.; Tang, H.; Guo, H.; Toole, B.; Sherry, B.; Bukrinsky, M. CD147 facilitates HIV-1 infection by interacting with virus-associated cyclophilin A. Proc. Natl. Acad. Sci. USA 2001, 98, 6360–6365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; Zhang, Y.; Wu, X.D.; Zhang, K.; Lin, P.; Bian, H.J.; Qin, M.M.; Huang, W.; Wei, D.; Zhang, Z.; et al. Disrupting CD147-RAP2 interaction abrogates erythrocyte invasion by Plasmodium falciparum. Blood 2018, 131, 1111–1121. [Google Scholar] [CrossRef]
- Kalejaiye, T.D.; Bhattacharya, R.; Burt, M.A.; Travieso, T.; Okafor, A.E.; Mou, X.; Blasi, M.; Musah, S. SARS-CoV-2 Employ BSG/CD147 and ACE2 Receptors to Directly Infect Human Induced Pluripotent Stem Cell-Derived Kidney Podocytes. Front. Cell Dev. Biol. 2022, 10, 855340. [Google Scholar] [CrossRef]
- Ragotte, R.J.; Pulido, D.; Donnellan, F.R.; Hill, M.L.; Gorini, G.; Davies, H.; Brun, J.; McHugh, K.; King, L.D.W.; Skinner, K.; et al. Human Basigin (CD147) Does Not Directly Interact with SARS-CoV-2 Spike Glycoprotein. mSphere 2021, 6, e0064721. [Google Scholar] [CrossRef]
- Badeti, S.; Jiang, Q.; Naghizadeh, A.; Tseng, H.C.; Bushkin, Y.; Marras, S.A.E.; Nisa, A.; Tyagi, S.; Chen, F.; Romanienko, P.; et al. Development of a novel human CD147 knock-in NSG mouse model to test SARS-CoV-2 viral infection. Cell Biosci. 2022, 12, 88. [Google Scholar] [CrossRef] [PubMed]
- Asgari, R.; Vaisi-Raygani, A.; Aleagha, M.S.E.; Mohammadi, P.; Bakhtiari, M.; Arghiani, N. CD147 and MMPs as key factors in physiological and pathological processes. Biomed. Pharmacother. 2023, 157, 113983. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Aleya, L.; Sehgal, A.; Singh, S.; Sharma, N.; Bhatia, S.; Al-Harrasi, A.; Bungau, S. CD147-spike protein interaction in COVID-19: Get the ball rolling with a novel receptor and therapeutic target. Sci. Total Environ. 2022, 808, 152072. [Google Scholar] [CrossRef]
- Grass, G.D.; Toole, B.P. How, with whom and when: An overview of CD147-mediated regulatory networks influencing matrix metalloproteinase activity. Biosci. Rep. 2015, 36, e00283. [Google Scholar] [CrossRef]
- Fenizia, C.; Galbiati, S.; Vanetti, C.; Vago, R.; Clerici, M.; Tacchetti, C.; Daniele, T. SARS-CoV-2 Entry: At the Crossroads of CD147 and ACE2. Cells 2021, 10, 1434. [Google Scholar] [CrossRef] [PubMed]
- Cavezzi, A.; Menicagli, R.; Troiani, E.; Corrao, S. COVID-19, Cation Dysmetabolism, Sialic Acid, CD147, ACE2, Viroporins, Hepcidin and Ferroptosis: A Possible Unifying Hypothesis. F1000Research 2022, 11, 102. [Google Scholar] [CrossRef]
- Wang, R.; Ahmed, J.; Wang, G.; Hassan, I.; Strulovici-Barel, Y.; Salit, J.; Mezey, J.G.; Crystal, R.G. Airway epithelial expression of TLR5 is downregulated in healthy smokers and smokers with chronic obstructive pulmonary disease. J. Immunol. 2012, 189, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Andrews, M.G.; Mukhtar, T.; Eze, U.C.; Simoneau, C.R.; Perez, Y.; Mostajo-Radji, M.A.; Wang, S.; Velmeshev, D.; Salma, J.; Kumar, G.R.; et al. Tropism of SARS-CoV-2 for Developing Human Cortical Astrocytes. bioRxiv 2021. [Google Scholar] [CrossRef]
- Geng, J.; Chen, L.; Yuan, Y.; Wang, K.; Wang, Y.; Qin, C.; Wu, G.; Chen, R.; Zhang, Z.; Wei, D.; et al. CD147 antibody specifically and effectively inhibits infection and cytokine storm of SARS-CoV-2 and its variants delta, alpha, beta, and gamma. Signal Transduct. Target. Ther. 2021, 6, 347. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. DPP-4 inhibition and COVID-19: From initial concerns to recent expectations. Diabetes Metab. 2021, 47, 101213. [Google Scholar] [CrossRef]
- Kawabata, H. Transferrin and transferrin receptors update. Free Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef]
- Rizzollo, F.; More, S.; Vangheluwe, P.; Agostinis, P. The lysosome as a master regulator of iron metabolism. Trends Biochem. Sci. 2021, 46, 960–975. [Google Scholar] [CrossRef]
- Nemeth, E.; Ganz, T. Hepcidin-Ferroportin Interaction Controls Systemic Iron Homeostasis. Int. J. Mol. Sci. 2021, 22, 6493. [Google Scholar] [CrossRef]
- Dassler, K.; Kaup, M.; Tauber, R.; Fuchs, H. Mutational suppression of transferrin receptor shedding can be compensated by distinct metalloproteases acting on alternative sites. FEBS Lett. 2003, 536, 25–29. [Google Scholar] [CrossRef]
- Gaiatto, A.C.M.; Bibo, T.A.; de Godoy Moreira, N.; Raimundo, J.R.S.; da Costa Aguiar Alves, B.; Gascon, T.; Carvalho, S.S.; Pereira, E.C.; Fonseca, F.L.A.; da Veiga, G.L. COVID-19 compromises iron homeostasis: Transferrin as a target of investigation. J. Trace Elem. Med. Biol. 2023, 76, 127109. [Google Scholar] [CrossRef] [PubMed]
- Kaup, M.; Dassler, K.; Weise, C.; Fuchs, H. Shedding of the transferrin receptor is mediated constitutively by an integral membrane metalloprotease sensitive to tumor necrosis factor alpha protease inhibitor-2. J. Biol. Chem. 2002, 277, 38494–38502. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Qian, Z.M.; Sheng, Y.; Zheng, J.; Liu, Y. Angiotensin II down-regulates transferrin receptor 1 and ferroportin 1 expression in Neuro-2a cells via activation of type-1 receptor. Neurosci. Lett. 2020, 716, 134684. [Google Scholar] [CrossRef] [PubMed]
- Nai, A.; Lore, N.I.; Pagani, A.; De Lorenzo, R.; Di Modica, S.; Saliu, F.; Cirillo, D.M.; Rovere-Querini, P.; Manfredi, A.A.; Silvestri, L. Hepcidin levels predict COVID-19 severity and mortality in a cohort of hospitalized Italian patients. Am. J. Hematol. 2021, 96, E32–E35. [Google Scholar] [CrossRef]
- Sonnweber, T.; Boehm, A.; Sahanic, S.; Pizzini, A.; Aichner, M.; Sonnweber, B.; Kurz, K.; Koppelstatter, S.; Haschka, D.; Petzer, V.; et al. Persisting alterations of iron homeostasis in COVID-19 are associated with non-resolving lung pathologies and poor patients’ performance: A prospective observational cohort study. Respir. Res. 2020, 21, 276. [Google Scholar] [CrossRef]
- Wang, X.; Wen, Z.; Cao, H.; Luo, J.; Shuai, L.; Wang, C.; Ge, J.; Wang, X.; Bu, Z.; Wang, J. Transferrin Receptor Protein 1 Is an Entry Factor for Rabies Virus. J. Virol. 2023, 97, e0161222. [Google Scholar] [CrossRef]
- Hogg, R.C.; Raggenbass, M.; Bertrand, D. Nicotinic acetylcholine receptors: From structure to brain function. Rev. Physiol. Biochem. Pharmacol. 2003, 147, 1–46. [Google Scholar] [CrossRef]
- Wonnacott, S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997, 20, 92–98. [Google Scholar] [CrossRef]
- Gharpure, A.; Noviello, C.M.; Hibbs, R.E. Progress in nicotinic receptor structural biology. Neuropharmacology 2020, 171, 108086. [Google Scholar] [CrossRef]
- Cecchini, M.; Changeux, J.P. The nicotinic acetylcholine receptor and its prokaryotic homologues: Structure, conformational transitions & allosteric modulation. Neuropharmacology 2015, 96, 137–149. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Hone, A.J.; McIntosh, J.M. Nicotinic acetylcholine receptors in neuropathic and inflammatory pain. FEBS Lett. 2018, 592, 1045–1062. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. Reflex control of immunity. Nat. Rev. Immunol. 2009, 9, 418–428. [Google Scholar] [CrossRef]
- Piovesana, R.; Salazar Intriago, M.S.; Dini, L.; Tata, A.M. Cholinergic Modulation of Neuroinflammation: Focus on alpha7 Nicotinic Receptor. Int. J. Mol. Sci. 2021, 22, 4912. [Google Scholar] [CrossRef]
- Li, S.; Qi, D.; Li, J.N.; Deng, X.Y.; Wang, D.X. Vagus nerve stimulation enhances the cholinergic anti-inflammatory pathway to reduce lung injury in acute respiratory distress syndrome via STAT3. Cell Death Discov. 2021, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.M.; Pimentel, V.E.; Fuzo, C.A.; da Silva-Neto, P.V.; Toro, D.M.; Fraga-Silva, T.F.C.; Gardinassi, L.G.; Oliveira, C.N.S.; Souza, C.O.S.; Torre-Neto, N.T.; et al. Acetylcholine, Fatty Acids, and Lipid Mediators Are Linked to COVID-19 Severity. J. Immunol. 2022, 209, 250–261. [Google Scholar] [CrossRef]
- Tillman, T.S.; Chen, Q.; Bondarenko, V.; Coleman, J.A.; Xu, Y.; Tang, P. SARS-CoV-2 Spike Protein Downregulates Cell Surface alpha7nAChR through a Helical Motif in the Spike Neck. ACS Chem. Neurosci. 2023, 14, 689–698. [Google Scholar] [CrossRef]
- Hone, A.J.; Santiago, U.; Harvey, P.J.; Tekarli, B.; Gajewiak, J.; Craik, D.J.; Camacho, C.J.; McIntosh, J.M. Design, Synthesis, and Structure-Activity Relationships of Novel Peptide Derivatives of the Severe Acute Respiratory Syndrome-Coronavirus-2 Spike-Protein that Potently Inhibit Nicotinic Acetylcholine Receptors. J. Med. Chem. 2024, 67, 9587–9598. [Google Scholar] [CrossRef]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Strom, A.; Treuter, E.; Warner, M.; et al. Estrogen receptors: How do they signal and what are their targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef]
- Libby, A.E.; Jones, B.; Lopez-Santiago, I.; Rowland, E.; Levi, M. Nuclear receptors in the kidney during health and disease. Mol. Aspects Med. 2021, 78, 100935. [Google Scholar] [CrossRef]
- Isola, J.V.V.; Ko, S.; Ocanas, S.R.; Stout, M.B. Role of Estrogen Receptor alpha in Aging and Chronic Disease. Adv. Geriatr. Med. Res. 2023, 5, e230005. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, B.; Ou-Yang, L. Role of estrogen receptors in health and disease. Front. Endocrinol. 2022, 13, 839005. [Google Scholar] [CrossRef]
- Douin-Echinard, V.; Laffont, S.; Seillet, C.; Delpy, L.; Krust, A.; Chambon, P.; Gourdy, P.; Arnal, J.F.; Guery, J.C. Estrogen receptor alpha, but not beta, is required for optimal dendritic cell differentiation and [corrected] CD40-induced cytokine production. J. Immunol. 2008, 180, 3661–3669. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Takahashi, T.; Wood, J.; Lu, P.; Tabachnikova, A.; Gehlhausen, J.R.; Greene, K.; Bhattacharjee, B.; Monteiro, V.S.; Lucas, C.; et al. Sex differences in symptomatology and immune profiles of Long COVID. medRxiv 2024. [Google Scholar] [CrossRef]
- Barbieri, S.S.; Cattani, F.; Sandrini, L.; Grillo, M.M.; Amendola, A.; Valente, C.; Talarico, C.; Iaconis, D.; Turacchio, G.; Lucariello, M.; et al. Relevance of Spike/Estrogen Receptor-alpha interaction for endothelial-based coagulopathy induced by SARS-CoV-2. Signal Transduct. Target. Ther. 2023, 8, 203. [Google Scholar] [CrossRef]
- Solis, O.; Beccari, A.R.; Iaconis, D.; Talarico, C.; Ruiz-Bedoya, C.A.; Nwachukwu, J.C.; Cimini, A.; Castelli, V.; Bertini, R.; Montopoli, M.; et al. The SARS-CoV-2 spike protein binds and modulates estrogen receptors. Sci. Adv. 2022, 8, eadd4150. [Google Scholar] [CrossRef]
- Leach, D.A.; Brooke, G.N.; Bevan, C.L. Roles of steroid receptors in the lung and COVID-19. Essays Biochem. 2021, 65, 1025–1038. [Google Scholar] [CrossRef]
- Stelzig, K.E.; Canepa-Escaro, F.; Schiliro, M.; Berdnikovs, S.; Prakash, Y.S.; Chiarella, S.E. Estrogen regulates the expression of SARS-CoV-2 receptor ACE2 in differentiated airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L1280–L1281. [Google Scholar] [CrossRef]
- Costa, A.J.; Lemes, R.M.R.; Bartolomeo, C.S.; Nunes, T.A.; Pereira, G.C.; Oliveira, R.B.; Gomes, A.L.; Smaili, S.S.; Maciel, R.M.B.; Newson, L.; et al. Overexpression of estrogen receptor GPER1 and G1 treatment reduces SARS-CoV-2 infection in BEAS-2B bronchial cells. Mol. Cell. Endocrinol. 2022, 558, 111775. [Google Scholar] [CrossRef]
- Blume, C.; Jackson, C.L.; Spalluto, C.M.; Legebeke, J.; Nazlamova, L.; Conforti, F.; Perotin, J.M.; Frank, M.; Butler, J.; Crispin, M.; et al. A novel ACE2 isoform is expressed in human respiratory epithelia and is upregulated in response to interferons and RNA respiratory virus infection. Nat. Genet. 2021, 53, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Onabajo, O.O.; Banday, A.R.; Stanifer, M.L.; Yan, W.; Obajemu, A.; Santer, D.M.; Florez-Vargas, O.; Piontkivska, H.; Vargas, J.M.; Ring, T.J.; et al. Interferons and viruses induce a novel truncated ACE2 isoform and not the full-length SARS-CoV-2 receptor. Nat. Genet. 2020, 52, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, W.; Yang, L.; You, R. Physiological and pathological regulation of ACE2, the SARS-CoV-2 receptor. Pharmacol. Res. 2020, 157, 104833. [Google Scholar] [CrossRef]
- Stilhano, R.S.; Costa, A.J.; Nishino, M.S.; Shams, S.; Bartolomeo, C.S.; Breithaupt-Faloppa, A.C.; Silva, E.A.; Ramirez, A.L.; Prado, C.M.; Ureshino, R.P. SARS-CoV-2 and the possible connection to ERs, ACE2, and RAGE: Focus on susceptibility factors. FASEB J. 2020, 34, 14103–14119. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spike-Cell Receptor Interaction | Pathophysiological Effects in Long COVID (LC) | Affected System | References |
|---|---|---|---|
| Spike-Ace2/integrin β1 | Vasculopathy and persistent inflammation | Cardiovascular, neurological, and respiratory systems | [61,62,63,64] |
| Spike-TLR2/4 | Neuroinflammation and immunodysregulation | Central nervous system (CNS) and immunological systems | [4,108] |
| Spike-NRP1 | Ferroptosis-cellular senescence and immune dysregulation | Cardiovascular, neurological, and immunological systems | [123,130,131] |
| Spike-DPP4 | Metabolic dysregulation | Vascular and neurological systems | [146,153] |
| Spike-CD147 | Systemic inflammation, metabolic dysregulation, and mitochondrial dysfunction | Vascular system | [171,174] |
| Spike-TfR | Ferroptosis-cellular senescence | Cardiovascular, neurological, immunological, and reproductive systems | [129,183,188] |
| Spike-nAchR | Decreased free acholine acetyltransferase and increased release of pro-inflammatory cytokines | Central nervous system (CNS) | [10,199] |
| Spike-ERα | Coagulopathy activation and viral infection | Cardiovascular, pulmonary, neurological systems | [208,209,210,211,212] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Melo, B.P.; da Silva, J.A.M.; Rodrigues, M.A.; Palmeira, J.d.F.; Amato, A.A.; Argañaraz, G.A.; Argañaraz, E.R. SARS-CoV-2 Spike Protein and Long COVID—Part 2: Understanding the Impact of Spike Protein and Cellular Receptor Interactions on the Pathophysiology of Long COVID Syndrome. Viruses 2025, 17, 619. https://doi.org/10.3390/v17050619
de Melo BP, da Silva JAM, Rodrigues MA, Palmeira JdF, Amato AA, Argañaraz GA, Argañaraz ER. SARS-CoV-2 Spike Protein and Long COVID—Part 2: Understanding the Impact of Spike Protein and Cellular Receptor Interactions on the Pathophysiology of Long COVID Syndrome. Viruses. 2025; 17(5):619. https://doi.org/10.3390/v17050619
Chicago/Turabian Stylede Melo, Bruno Pereira, Jhéssica Adriane Mello da Silva, Mariana Alves Rodrigues, Julys da Fonseca Palmeira, Angélica Amorim Amato, Gustavo Adolfo Argañaraz, and Enrique Roberto Argañaraz. 2025. "SARS-CoV-2 Spike Protein and Long COVID—Part 2: Understanding the Impact of Spike Protein and Cellular Receptor Interactions on the Pathophysiology of Long COVID Syndrome" Viruses 17, no. 5: 619. https://doi.org/10.3390/v17050619
APA Stylede Melo, B. P., da Silva, J. A. M., Rodrigues, M. A., Palmeira, J. d. F., Amato, A. A., Argañaraz, G. A., & Argañaraz, E. R. (2025). SARS-CoV-2 Spike Protein and Long COVID—Part 2: Understanding the Impact of Spike Protein and Cellular Receptor Interactions on the Pathophysiology of Long COVID Syndrome. Viruses, 17(5), 619. https://doi.org/10.3390/v17050619