

Self-Amplifying RNA: Advantages and Challenges of a Versatile Platform for Vaccine Development

Abstract
1. Introduction
2. Background and History of Nucleic Acid Vaccines
2.1. The Principle of Genetic Immunization and DNA Vaccines
2.2. mRNA Constructs: From Concept to Vaccine Application
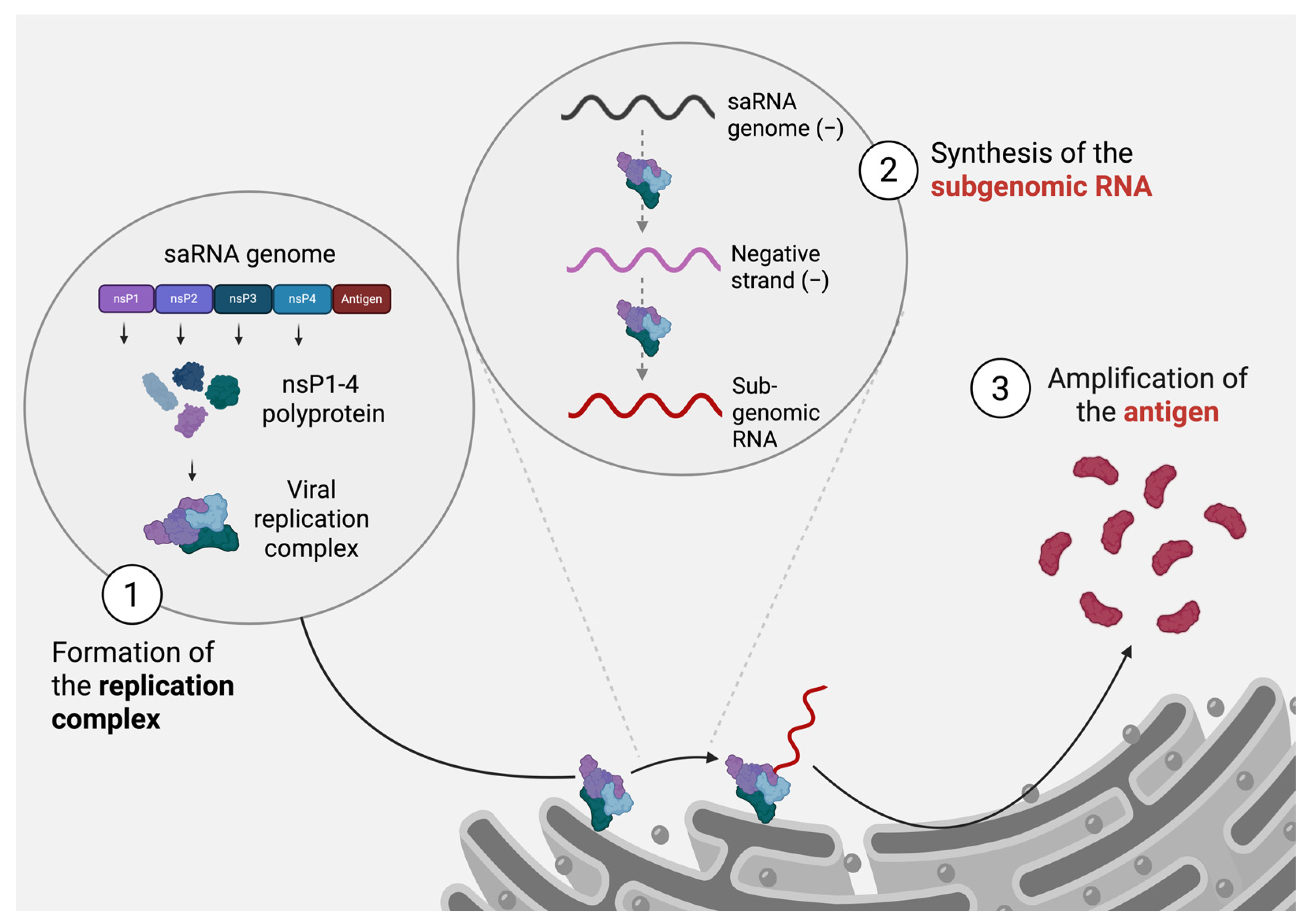
3. Self-Amplifying RNA Innovation
3.1. Advantages of saRNA over mRNA Technology
3.2. Other Key Roles Played by Alphavirus nsPs That May Affect the Function of saRNA as a Therapeutic Vector
3.3. Limitations and Optimization Strategies
3.3.1. Recombination Potential
3.3.2. Cytopathogenicity
3.3.3. Immunogenicity
3.3.4. Stability
3.4. saRNA Vaccines
3.4.1. Preclinical and Clinical Trials
3.4.2. Regulatory Obstacles
- Manufacturing challenges
- Approval process
4. Discussion
- Advantages of saRNA across vaccine platforms
- Optimization methods of saRNA constructs
- Other saRNA platforms
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APCs | Antigen-presenting cells |
| CHIKV | Chikungunya virus |
| LNPs | Lipid nanoparticles |
| LRP | Liposome-protamine-RNA |
| nsPs | Non-structural proteins |
| ORF | Open reading frame |
| saRNA | Self-amplifying RNA |
| SFV | Semliki Forest virus |
| SINV | Sindbis virus |
| taRNA | Trans-amplifying RNA |
| VEEV | Venezuelan equine encephalitis virus |
| WEEV | Western equine encephalitis virus |
| ZIKV | Zika virus |
References
- Bhadoria, P.; Gupta, G.; Agarwal, A. Viral Pandemics in the Past Two Decades: An Overview. J. Fam. Med. Prim. Care 2021, 27, 2745–2750. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; chu DeVit, M.; Johnston, S. A Genetic immunization is a simple method for eliciting an immune response. Nature 1992, 356, 152–154. [Google Scholar] [CrossRef]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct Gene Transfer into Mouse Muscle In Vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef]
- Gote, V.; Bolla, P.K.; Kommineni, N.; Butreddy, A.; Nukala, P.K.; Palakurthi, S.S.; Khan, W. A Comprehensive Review of mRNA Vaccines. Int. J. Mol. Sci. 2023, 31, 2700. [Google Scholar] [CrossRef] [PubMed]
- Chemaitelly, H.; Abu-Raddad, L.J. Waning effectiveness of COVID-19 vaccines. Lancet 2022, 399, 771–773. [Google Scholar] [CrossRef]
- Naaber, P.; Tserel, L.; Kangro, K.; Sepp, E.; Jürjenson, V.; Adamson, A.; Haljasmägi, L.; Rumm, A.P.; Maruste, R.; Kärner, J.; et al. Dynamics of antibody response to BNT162b2 vaccine after six months: A longitudinal prospective study. Lancet Reg. Health Eur. 2021, 10, 100208. [Google Scholar] [CrossRef] [PubMed]
- Frolov, I.; Hoffman, T.A.; Prágai, B.M.; Dryga, S.A.; Huang, H.V.; Schlesinger, S.; Rice, C.M. Alphavirus-based expression vectors: Strategies and applications. Proc. Natl. Acad. Sci. USA 1996, 93, 11371–11377. [Google Scholar] [CrossRef] [PubMed]
- Oda, Y.; Kumagai, Y.; Kanai, M.; Iwama, Y.; Okura, I.; Minamida, T.; Yagi, Y.; Kurosawa, T.; Greener, B.; Zhang, Y.; et al. Immunogenicity and safety of a booster dose of a self-amplifying RNA COVID-19 vaccine (ARCT-154) versus BNT162b2 mRNA COVID-19 vaccine: A double-blind, multicentre, randomised, controlled, phase 3, non-inferiority trial. Lancet Infect. Dis. 2024, 24, 351–360. [Google Scholar] [CrossRef]
- Liu, M.A. DNA vaccines: A review. J. Intern. Med. 2003, 253, 402–410. [Google Scholar] [CrossRef]
- Ferraro, B.; Morrow, M.P.; Hutnick, N.A.; Shin, T.H.; Lucke, C.E.; Weiner, D.B. Clinical Applications of DNA Vaccines: Current Progress. Clin. Infect. Dis. 2011, 53, 296–302. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Pahl, H.L.; Baeuerle, P.A. Expression of influenza virus hemagglutinin activates transcription factor NF-kappa B. J. Virol. 1995, 69, 1480–1484. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Pei, J.; Xu, S.; Liu, J.; Yu, J. Recent advances in mRNA cancer vaccines: Meeting challenges and embracing opportunities. Front. Immunol. 2023, 14, 1246682. [Google Scholar] [CrossRef] [PubMed]
- Montana, G.; Bondì, M.L.; Carrotta, R.; Picone, P.; Craparo E., F.; San Biagio P., L.; Giammona, G.; Di Carloa, M. Employment of Cationic Solid-Lipid Nanoparticles as RNA Carriers. Bioconjugate Chem. 2007, 18, 302–308. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of Pseudouridine Into mRNA Yields Superior Nonimmunogenic Vector with Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Andries, O.; Mc Cafferty, S.; De Smedt, S.C.; Weiss, R.; Sanders, N.N.; Kitada, T. N1-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control. Release 2015, 217, 337–344. [Google Scholar] [CrossRef]
- Gallie, D.R. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. 1991, 5, 2108–2116. [Google Scholar] [CrossRef]
- Wu, X.; Shan, K.; Zan, F.; Tang, X.; Qian, Z.; Lu, J. Optimization and Deoptimization of Codons in SARS-CoV-2 and Related Implications for Vaccine Development. Adv. Sci. 2023, 10, 2205445. [Google Scholar] [CrossRef]
- Hammer, S.M.; Sobieszczyk, M.E.; Janes, H.; Karuna, S.T.; Mulligan, M.J.; Grove, D.; Koblin, B.A.; Buchbinder, S.P.; Keefer, M.C.; Tomaras, G.D.; et al. Efficacy Trial of a DNA/rAd5 HIV-1 Preventive Vaccine. N. Engl. J. Med. 2013, 369, 2083–2092. [Google Scholar] [CrossRef]
- Sekaly, R.P. The failed HIV Merck vaccine study: A step back or a launching point for future vaccine development? J. Exp. Med. 2008, 205, 7–12. [Google Scholar] [CrossRef]
- Lorentzen, C.L.; Haanen, J.B.; Met, Ö.; Svane, I.M. Clinical advances and ongoing trials of mRNA vaccines for cancer treatment. Lancet Oncol. 2022, 23, e450–e458. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Muik, A.; Derhovanessian, E.; Vogler, I.; Kranz, L.M.; Vormehr, M.; Baum, A.; Pascal, K.; Quandt, J.; Maurus, D.; et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature 2020, 586, 594–599. [Google Scholar] [CrossRef]
- Altmann, D.M.; Boyton, R.J. Waning immunity to SARS-CoV-2: Implications for vaccine booster strategies. Lancet Respir. Med. 2021, 9, 1356–1358. [Google Scholar] [CrossRef]
- Goel, R.R.; Apostolidis, S.A.; Painter, M.M.; Mathew, D.; Pattekar, A.; Kuthuru, O.; Gouma, S.; Hicks, P.; Meng, W.; Rosenfeld, A.M.; et al. Distinct antibody and memory B cell responses in SARS-CoV-2 naïve and recovered individuals after mRNA vaccination. Sci. Immunol. 2021, 6, eabi6950. [Google Scholar] [CrossRef]
- Cameroni, E.; Bowen, J.E.; Rosen, L.E.; Saliba, C.; Zepeda, S.K.; Culap, K.; Pinto, D.; VanBlargan, L.A.; De Marco, A.; di Iulio, J.; et al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature 2022, 602, 664–670. [Google Scholar] [CrossRef]
- Sadoff, J.; Gray, G.; Vandebosch, A.; Cárdenas, V.; Shukarev, G.; Grinsztejn, B.; Goepfert, P.A.; Truyers, C.; Fennema, H.; Spiessens, B.; et al. Safety and Efficacy of Single-Dose Ad26.COV2.S Vaccine against COVID-19. N. Engl. J. Med. 2021, 384, 2187–2201. [Google Scholar]
- Zeng, G.; Wu, Q.; Pan, H.; Li, M.; Yang, J.; Wang, L.; Wu, Z.; Jiang, D.; Deng, X.; Chu, K.; et al. Immunogenicity and safety of a third dose of CoronaVac, and immune persistence of a two-dose schedule, in healthy adults: Interim results from two single-centre, double-blind, randomised, placebo-controlled phase 2 clinical trials. Lancet Infect. Dis. 2022, 22, 483–495. [Google Scholar] [CrossRef]
- Vogel, A.B.; Lambert, L.; Kinnear, E.; Busse, D.; Erbar, S.; Reuter, K.C.; Wicke, L.; Perkovic, M.; Beissert, T.; Haas, H.; et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol. Ther. 2018, 26, 446–455. [Google Scholar] [CrossRef]
- McKay, P.F.; Hu, K.; Blakney, A.K.; Samnuan, K.; Brown, J.C.; Penn, R.; Zhou, J.; Bouton, C.R.; Rogers, P.; Polra, K.; et al. Self-amplifying RNA SARS-CoV-2 lipid nanoparticle vaccine candidate induces high neutralizing antibody titers in mice. Nat. Commun. 2020, 11, 3523. [Google Scholar] [CrossRef] [PubMed]
- Akahata, W.; Sekida, T.; Nogimori, T.; Ode, H.; Tamura, T.; Kono, K.; Kazami, Y.; Washizaki, A.; Masuta, Y.; Suzuki, R.; et al. Safety and immunogenicity of SARS-CoV-2 self-amplifying RNA vaccine expressing an anchored RBD: A randomized, observer-blind phase 1 study. Cell Rep. Med. 2023, 4, 101134. [Google Scholar] [CrossRef] [PubMed]
- Cu, Y.; Broderick, K.E.; Banerjee, K.; Hickman, J.; Otten, G.; Barnett, S.; Kichaev, G.; Sardesai, N.Y.; Ulmer, J.B.; Geall, A. Enhanced Delivery and Potency of Self-Amplifying mRNA Vaccines by Electroporation in Situ. Vaccines 2013, 1, 367–383. [Google Scholar] [CrossRef]
- Kanechi, R.; Shishido, T.; Tachikawa, M.; Nishimura, T.; Sawada, A.; Okade, H.; Ishikawa, D.; Yamaguchi, H.; Araki, M. Differential clearance rate of proteins encoded on a self-amplifying mRNA vaccine in muscle and lymph nodes. Mol. Biol. 2024, 10, 615730. Available online: http://biorxiv.org/lookup/doi/10.1101/2024.10.07.615730 (accessed on 30 March 2025). [CrossRef]
- Kaplan, G.; Racaniello, V.R. Construction and characterization of poliovirus subgenomic replicons. J. Virol. 1988, 62, 1687–1696. [Google Scholar] [CrossRef]
- Fernandes, R.S.; Freire, M.C.L.C.; Bueno, R.V.; Godoy, A.S.; Gil, L.; Oliva, G. Reporter Replicons for Antiviral Drug Discovery against Positive Single-Stranded RNA Viruses. Viruses 2020, 12, 598. [Google Scholar] [CrossRef] [PubMed]
- Liljeström, P.; Garoff, H. A New Generation of Animal Cell Expression Vectors Based on the Semliki Forest Virus Replicon. Nat. Biotechnol. 1991, 9, 1356–1361. [Google Scholar] [CrossRef]
- Ehrengruber, M.U.; Schlesinger, S.; Lundstrom, K. Alphaviruses: Semliki Forest Virus and Sindbis Virus Vectors for Gene Transfer into Neurons. Curr. Protoc. Neurosci. 2011, 57, ns0422s57. Available online: https://currentprotocols.onlinelibrary.wiley.com/doi/10.1002/0471142301.ns0422s57 (accessed on 9 April 2024). [CrossRef]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol Rev. 1994, 58, 491–562. [Google Scholar] [CrossRef]
- Singh, I.R.; Suomalainen, M.; Varadarajan, S.; Garoff, H.; Helenius, A. Multiple Mechanisms for the Inhibition of Entry and Uncoating of Superinfecting Semliki Forest Virus. Virology 1997, 231, 59–71. [Google Scholar] [CrossRef]
- Akhrymuk, I.; Kulemzin, S.V.; Frolova, E.I. Evasion of the Innate Immune Response: The Old World Alphavirus nsP2 Protein Induces Rapid Degradation of Rpb1, a Catalytic Subunit of RNA Polymerase II. J. Virol. 2012, 86, 7180–7191. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, L.; Sanz, M.; González-Almela, E. The Regulation of Translation in Alphavirus-Infected Cells. Viruses 2018, 10, 70. [Google Scholar] [CrossRef]
- Ahola, T.; Laakkonen, P.; Vihinen, H.; Kääriäinen, L. Critical residues of Semliki Forest virus RNA capping enzyme involved in methyltransferase and guanylyltransferase-like activities. J. Virol. 1997, 71, 392–397. [Google Scholar] [CrossRef]
- Decroly, E.; Ferron, F.; Lescar, J.; Canard, B. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 2012, 10, 51–65. [Google Scholar] [CrossRef]
- Law, M.C.Y.; Zhang, K.; Tan, Y.B.; Nguyen, T.M.; Luo, D. Chikungunya virus nonstructural protein 1 is a versatile RNA capping and decapping enzyme. J. Biol. Chem. 2023, 299, 105415. [Google Scholar] [CrossRef]
- Spuul, P.; Salonen, A.; Merits, A.; Jokitalo, E.; Kääriäinen, L.; Ahola, T. Role of the Amphipathic Peptide of Semliki Forest Virus Replicase Protein nsP1 in Membrane Association and Virus Replication. J. Virol. 2007, 81, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Lark, T.; Keck, F.; Narayanan, A. Interactions of Alphavirus nsP3 Protein with Host Proteins. Front Microbiol. 2018, 8, 2652. [Google Scholar] [CrossRef]
- Mathur, K.; Anand, A.; Dubey, S.K.; Sanan-Mishra, N.; Bhatnagar, R.K.; Sunil, S. Analysis of chikungunya virus proteins reveals that non-structural proteins nsP2 and nsP3 exhibit RNA interference (RNAi) suppressor activity. Sci. Rep. 2016, 6, 38065. [Google Scholar] [CrossRef] [PubMed]
- Fros, J.J.; Domeradzka, N.E.; Baggen, J.; Geertsema, C.; Flipse, J.; Vlak, J.M.; Pijlman, G.P. Chikungunya Virus nsP3 Blocks Stress Granule Assembly by Recruitment of G3BP into Cytoplasmic Foci. J. Virol. 2012, 86, 10873–10879. [Google Scholar] [CrossRef]
- Aldon, Y.; McKay, P.F.; Herrero, J.M.; Vogel, A.B.; Lévai, R.; Maisonnasse, P.; Dereuddre-Bosquet, N.; Haas, H.; Fábián, K.; Le Grand, R.; et al. Immunogenicity of stabilized HIV-1 Env trimers delivered by self-amplifying mRNA. Mol. Ther. Nucleic Acids 2021, 25, 483–493. [Google Scholar] [CrossRef]
- Hick, T.A.; Geertsema, C.; Nguyen, W.; Bishop, C.R.; van Oosten, L.; Abbo, S.R.; Dumenil, T.; van Kuppeveld, F.J.; Langereis, M.A.; Rawle, D.J.; et al. Safety concern of recombination between self-amplifying mRNA vaccines and viruses is mitigated in vivo. Mol. Ther. 2024, 32, 2519–2534. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.B.; Stallknecht, D.E.; Holmes, E.C. Evolutionary genetics and vector adaptation of recombinant viruses of the western equine encephalitis antigenic complex provides new insights into alphavirus diversity and host switching. Virology 2015, 474, 154–162. [Google Scholar] [CrossRef]
- Hyvärinen, A.; Yongabi, F.; Mäkinen, K.; Wahlfors, J.; Pellinen, R. Recombination of replicon and helper RNAs and the emergence of propagation-competent vectors upon Sindbis virus vector production. Int. J. Mol. Med. 2013, 32, 410–422. [Google Scholar] [CrossRef]
- Beissert, T.; Perkovic, M.; Vogel, A.; Erbar, S.; Walzer, K.C.; Hempel, T.; Brill, S.; Haefner, E.; Becker, R.; Türeci, Ö.; et al. A Trans-amplifying RNA Vaccine Strategy for Induction of Potent Protective Immunity. Mol. Ther. 2020, 28, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Zaher, H.S.; Unrau, P.J. Selection of an improved RNA polymerase ribozyme with superior extension and fidelity. RNA 2007, 13, 1017–1026. [Google Scholar] [CrossRef]
- Brennan, J.W.; Sun, Y. Defective viral genomes: Advances in understanding their generation, function, and impact on infection outcomes. mBio 2024, 15, e00692-24. [Google Scholar] [CrossRef]
- Wang, E.; Volkova, E.; Adams, A.P.; Forrester, N.; Xiao, S.Y.; Frolov, I.; Weaver, S.C. Chimeric alphavirus vaccine candidates for chikungunya. Vaccine 2008, 26, 5030–5039. [Google Scholar] [CrossRef]
- Yeh, M.T.; Bujaki, E.; Dolan, P.T.; Smith, M.; Wahid, R.; Konz, J.; Weiner, A.J.; Bandyopadhyay, A.S.; Van Damme, P.; De Coster, I.; et al. Engineering the Live-Attenuated Polio Vaccine to Prevent Reversion to Virulence. Cell Host Microbe 2020, 27, 736–751.e8. [Google Scholar] [CrossRef] [PubMed]
- Garmashova, N.; Gorchakov, R.; Frolova, E.; Frolov, I. Sindbis Virus Nonstructural Protein nsP2 Is Cytotoxic and Inhibits Cellular Transcription. J. Virol. 2006, 80, 5686–5696. [Google Scholar] [CrossRef]
- Treffers, E.E.; Tas, A.; Scholte, F.E.M.; de Ru, A.H.; Snijder, E.J.; van Veelen, P.A.; van Hemert, M.J. The alphavirus nonstructural protein 2 NTPase induces a host translational shut-off through phosphorylation of eEF2 via cAMP-PKA-eEF2K signaling. PLoS Pathog. 2023, 19, e1011179. [Google Scholar] [CrossRef]
- Sharma, A.; Knollmann-Ritschel, B. Current Understanding of the Molecular Basis of Venezuelan Equine Encephalitis Virus Pathogenesis and Vaccine Development. Viruses 2019, 11, 164. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K.; Abenavoli, A.; Malgaroli, A.; Ehrengruber, M.U. Novel semliki forest virus vectors with reduced cytotoxicity and temperature sensitivity for long-term enhancement of transgene expression. Mol. Ther. 2003, 7, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Yong, D.; Liu, G.; Xu, J.; Ding, J.; Jia, W. A Novel Self-Amplifying mRNA with Decreased Cytotoxicity and Enhanced Protein Expression by Macrodomain Mutations. Adv. Sci. 2024, 11, 2402936. [Google Scholar] [CrossRef]
- Cherkashchenko, L.; Rausalu, K.; Basu, S.; Alphey, L.; Merits, A. Expression of Alphavirus Nonstructural Protein 2 (nsP2) in Mosquito Cells Inhibits Viral RNA Replication in Both a Protease Activity-Dependent and -Independent Manner. Viruses 2022, 14, 1327. [Google Scholar] [CrossRef]
- Varjak, M.; Žusinaite, E.; Merits, A. Novel Functions of the Alphavirus Nonstructural Protein nsP3 C-Terminal Region. J. Virol. 2010, 84, 2352–2364. [Google Scholar] [CrossRef]
- Geiss, B.J.; Shimonkevitz, L.H.; Sackal, C.I.; Olson, K.E. Recombination-ready Sindbis replicon expression vectors for transgene expression. Virol. J. 2007, 4, 112. [Google Scholar] [CrossRef]
- Kim, D.Y.; Atasheva, S.; McAuley, A.J.; Plante, J.A.; Frolova, E.I.; Beasley, D.W.C.; Frolov, I. Enhancement of protein expression by alphavirus replicons by designing self-replicating subgenomic RNAs. Proc. Natl. Acad. Sci. USA 2014, 111, 10708–10713. [Google Scholar] [CrossRef] [PubMed]
- Lemm, J.A.; Durbin, R.K.; Stollar, V.; Rice, C.M. Mutations which alter the level or structure of nsP4 can affect the efficiency of Sindbis virus replication in a host-dependent manner. J. Virol. 1990, 64, 3001–3011. [Google Scholar] [CrossRef]
- Lello, L.S.; Bartholomeeusen, K.; Wang, S.; Coppens, S.; Fragkoudis, R.; Alphey, L.; Ariën, K.K.; Merits, A.; Utt, A. nsP4 Is a Major Determinant of Alphavirus Replicase Activity and Template Selectivity. J. Virol. 2021, 95, e0035521. [Google Scholar] [CrossRef]
- McCafferty, S.; Haque, A.A.; Vandierendonck, A.; Weidensee, B.; Plovyt, M.; Stuchlíková, M.; François, N.; Valembois, S.; Heyndrickx, L.; Michiels, J.; et al. A dual-antigen self-amplifying RNA SARS-CoV-2 vaccine induces potent humoral and cellular immune responses and protects against SARS-CoV-2 variants through T cell-mediated immunity. Mol. Ther. 2022, 30, 2968–2983. [Google Scholar] [CrossRef]
- Biddlecome, A.; Habte, H.H.; McGrath, K.M.; Sambanthamoorthy, S.; Wurm, M.; Sykora, M.M.; Knobler, C.M.; Lorenz, I.C.; Lasaro, M.; Elbers, K.; et al. Delivery of self-amplifying RNA vaccines in in vitro reconstituted virus-like particles. PLoS ONE 2019, 14, e0215031. [Google Scholar] [CrossRef] [PubMed]
- Minnaert, A.-K.; Vanluchene, H.; Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Raemdonck, K.; Sanders, N.N.; Remaut, K. Strategies for controlling the innate immune activity of conventional and self-amplifying mRNA therapeutics: Getting the message across. Adv. Drug Deliv. Rev. 2021, 176, 113900. [Google Scholar] [CrossRef] [PubMed]
- Azizi, H.; Renner, T.M.; Agbayani, G.; Simard, B.; Dudani, R.; A Harrison, B.; Iqbal, U.; Jia, Y.; McCluskie, M.J.; Akache, B. Self-amplifying RNAs generated with the modified nucleotides 5-methylcytidine and 5-methyluridine mediate strong expression and immunogenicity in vivo. NAR Mol. Med. 2024, 1, ugae004. [Google Scholar] [CrossRef]
- Hồ, N.T.; Hughes, S.G.; Ta, V.T.; Phan, L.T.; Đỗ, Q.; Nguyễn, T.V.; Phạm, A.T.V.; Đặng, M.T.N.; Nguyễn, L.V.; Trịnh, Q.V.; et al. Safety, immunogenicity and efficacy of the self-amplifying mRNA ARCT-154 COVID-19 vaccine: Pooled phase 1, 2, 3a and 3b randomized, controlled trials. Nat. Commun. 2024, 15, 4081. [Google Scholar] [CrossRef]
- Low, J.G.; de Alwis, R.; Chen, S.; Kalimuddin, S.; Leong, Y.S.; Mah, T.K.L.; Yuen, N.; Tan, H.C.; Zhang, S.L.; Sim, J.X.Y.; et al. A phase I/II randomized, double-blinded, placebo-controlled trial of a self-amplifying COVID-19 mRNA vaccine. NPJ Vaccines 2022, 7, 161. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.D.; Scallan, C.D.; Tardif, L.D.K.; Kachura, M.A.; Rappaport, A.R.; Koralek, D.O.; Uriel, A.; Gitlin, L.; Klein, J.; Davis, M.J.; et al. GRT-R910: A self-amplifying mRNA SARS-CoV-2 vaccine boosts immunity for ≥6 months in previously-vaccinated older adults. Nat. Commun. 2023, 14, 3274. [Google Scholar] [CrossRef]
- Pollock, K.M.; Cheeseman, H.M.; Szubert, A.J.; Libri, V.; Boffito, M.; Owen, D.; Bern, H.; McFarlane, L.R.; O’Hara, J.; Lemm, N.-M.; et al. Safety and immunogenicity of a self-amplifying RNA vaccine against COVID-19: COVAC1, a phase I.; dose-ranging trial. eClinicalMedicine 2022, 44, 101262. [Google Scholar] [CrossRef]
- Szubert, A.J.; Pollock, K.M.; Cheeseman, H.M.; Alagaratnam, J.; Bern, H.; Bird, O.; Boffito, M.; Byrne, R.; Cole, T.; Cosgrove, C.A.; et al. COVAC1 phase 2a expanded safety and immunogenicity study of a self-amplifying RNA vaccine against SARS-CoV-2. eClinicalMedicine 2023, 56, 101823. [Google Scholar] [CrossRef]
- Saraf, A.; Gurjar, R.; Kaviraj, S.; Kulkarni, A.; Kumar, D.; Kulkarni, R.; Virkar, R.; Krishnan, J.; Yadav, A.; Baranwal, E.; et al. An Omicron-specific, self-amplifying mRNA booster vaccine for COVID-19: A phase 2/3 randomized trial. Nat. Med. 2024, 30, 1363–1372. [Google Scholar] [CrossRef]
- Kumar, D.; Gaikwad, K.; Gunnale, R.; Vishwakarma, S.; Shukla, S.; Srivastava, S.; Gopal, J.; Vaidya, B.; Saraf, A.; Gurjar, R.; et al. Cellular immune breadth of an Omicron-specific, self-amplifying monovalent mRNA vaccine booster for COVID-19. NPJ Vaccines 2025, 10, 42. [Google Scholar] [CrossRef]
- Chang, C.; Patel, H.; Ferrari, A.; Scalzo, T.; Petkov, D.; Xu, H.; Rossignol, E.; Palladino, G.; Wen, Y. sa-mRNA influenza vaccine raises a higher and more durable immune response than mRNA vaccine in preclinical models. Vaccine 2025, 51, 126883. [Google Scholar] [CrossRef] [PubMed]
- Stokes, A.; Pion, J.; Binazon, O.; Laffont, B.; Bigras, M.; Dubois, G.; Blouin, K.; Young, J.K.; Ringenberg, M.A.; Ben Abdeljelil, N.; et al. Nonclinical safety assessment of repeated administration and biodistribution of a novel rabies self-amplifying mRNA vaccine in rats. Regul. Toxicol. Pharmacol. 2020, 113, 104648. [Google Scholar] [CrossRef] [PubMed]
- Maine, C.J.; Miyake-Stoner, S.J.; Spasova, D.S.; Picarda, G.; Chou, A.C.; Brand, E.D.; Olesiuk, M.D.; Domingo, C.C.; Little, H.J.; Goodman, T.T.; et al. Safety and immunogenicity of an optimized self-replicating RNA platform for low dose or single dose vaccine applications: A randomized, open label Phase I study in healthy volunteers. Nat. Commun. 2025, 16, 456. [Google Scholar] [CrossRef]
- Kis, Z.; Tak, K.; Ibrahim, D.; Papathanasiou, M.M.; Chachuat, B.; Shah, N.; Kontoravdi, C. Pandemic-response adenoviral vector and RNA vaccine manufacturing. NPJ Vaccines 2022, 7, 29. Available online: http://medrxiv.org/lookup/doi/10.1101/2021.08.20.21262370 (accessed on 30 March 2025). [CrossRef] [PubMed]
- Mehta, M.; Bui, T.A.; Yang, X.; Aksoy, Y.; Goldys, E.M.; Deng, W. Lipid-Based Nanoparticles for Drug/Gene Delivery: An Overview of the Production Techniques and Difficulties Encountered in Their Industrial Development. ACS Mater. Au. 2023, 3, 600–619. [Google Scholar] [CrossRef]
- Battisti, P.; Ykema, M.R.; Kasal, D.N.; Jennewein, M.F.; Beaver, S.; Weight, A.E.; Hanson, D.; Singh, J.; Bakken, J.; Cross, N.; et al. A bivalent self-amplifying RNA vaccine against yellow fever and Zika viruses. Immunology 2025, 31, 635934. Available online: http://biorxiv.org/lookup/doi/10.1101/2025.01.31.635934 (accessed on 30 March 2025).
- Fausther-Bovendo, H.; Kobinger, G.P. Pre-existing immunity against Ad vectors: Humoral, cellular, and innate response, what’s important? Hum. Vaccines Immunother. 2014, 10, 2875–2884. [Google Scholar] [CrossRef]
- Keech, C.; Albert, G.; Cho, I.; Robertson, A.; Reed, P.; Neal, S.; Plested, J.S.; Zhu, M.; Cloney-Clark, S.; Zhou, H.; et al. Phase 1–2 Trial of a SARS-CoV-2 Recombinant Spike Protein Nanoparticle Vaccine. N. Engl. J. Med. 2020, 383, 2320–2332. [Google Scholar] [CrossRef]
- Heydenreich, F.M.; Miljuš, T.; Jaussi, R.; Benoit, R.; Milić, D.; Veprintsev, D.B. High-throughput mutagenesis using a two-fragment PCR approach. Sci. Rep. 2017, 7, 6787. [Google Scholar] [CrossRef]
- Mauro, V.P.; Chappell, S.A. A critical analysis of codon optimization in human therapeutics. Trends Mol. Med. 2014, 20, 604–613. [Google Scholar] [CrossRef]
- Goulet, D.R.; Yan, Y.; Agrawal, P.; Waight, A.B.; Mak, A.N.-S.; Zhu, Y. Codon Optimization Using a Recurrent Neural Network. J. Comput. Biol. 2023, 30, 70–81. [Google Scholar] [CrossRef]
- Zarnack, K.; Eyras, E. Artificial intelligence and machine learning in RNA biology. Brief. Bioinform. 2023, 24, bbad415. [Google Scholar] [CrossRef]
- He, S.; Gao, B.; Sabnis, R.; Sun, Q. RNAdegformer: Accurate prediction of mRNA degradation at nucleotide resolution with deep learning. Brief. Bioinform. 2023, 24, bbac581. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Hu, Z.; Sun, S.; Liu, D.; Wong, F.; Wang, J.; Chen, J.; Wang, Y.; Hong, L.; Xiao, J.; et al. Accurate RNA 3D structure prediction using a language model-based deep learning approach. Nat. Methods 2024, 21, 2287–2298. [Google Scholar] [CrossRef] [PubMed]
- Selisko, B.; Wang, C.; Harris, E.; Canard, B. Regulation of Flavivirus RNA synthesis and replication. Curr. Opin. Virol. 2014, 9, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zou, S.; Chen, H.; Higazy, D.; Gao, X.; Zhang, Y.; Cao, S.; Cui, M. Zika virus replication on endothelial cells and invasion into the central nervous system by inhibiting interferon β translation. Virology 2023, 582, 23–34. [Google Scholar] [CrossRef]
- Markoff, L. 5′- and 3′-noncoding regions in flavivirus RNA. Adv. Virus Res. 2003, 59, 177–228. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0065352703590066 (accessed on 30 March 2025).

{kind=link}
| Limitation | Risk | Optimization Strategy |
|---|---|---|
| Recombination Potential | Presence of co-infecting viruses leading to novel viral strains | Non-replicating helper RNA systems |
| Trans-amplifying RNA (taRNA) | ||
| High-fidelity RNA polymerases or ribozymes | ||
| Cytopathogenicity | Cellular damage due to viral replication | Point mutations in nsP4 and nsP3 |
| Truncation of nsP2 and nsP3 domains | ||
| Optimization of subgenomic promoter strength | ||
| Alteration of replication kinetics via nsP4 modifications | ||
| Immunogenicity | Skewing of immune response toward cellular immunity | Dual-antigen saRNA design |
| Optimizing adjuvants or modifications to enhance B-cell activation alongside T-cell responses | ||
| Stability | Degradation and immunogenicity issues with modified nucleotides | Use CleanCAP AU for improved capping efficiency |
| 5-methylcytidine, 5-methyluridine, or 5-hydroxymethylcytidine instead of pseudouridine |
| Vaccine | Targeted Virus | Backbone | Key Features | Stage of Development | Clinical Trials (ClinicalTrials.gov Identifier) |
|---|---|---|---|---|---|
| ARCT-154 | SARS-CoV-2 | VEEV | Modified VEEV replicon for enhanced replication and antigen expression | Clinical Trials (Phase 3) | NCT05012943 |
| ARCT-165 | SARS-CoV-2 | VEEV | Combination of ARCT-154 and ARCT-021 vaccines | Clinical Trials (Phase 1/2) | NCT05037097 |
| saRNA-LNP | SARS-CoV-2 | VEEV | Lipid nanoparticles formulated saRNA vaccine | Clinical Trials (Phase 1) | NCT04776317 |
| PF-07852351, PF-07836391, PF-07836394, PF-07836395, PF-07836396, PF-07867426 | Influenza | VEEV | saRNA targeting various influenza strains | Clinical Trials (Phase 1) | NCT05227001 |
| AAHI-SC2 | SARS-CoV-2 | VEEV | saRNA encoding the spike protein, delivered by a nanostructured lipid carrier | Clinical Trials (Phase 1) | NCT05370040 |
| GRT-R910 | SARS-CoV-2 | NA 1 | saRNA booster | Clinical Trials (Phase 1) | NCT05148962 |
| HGC019 | SARS-CoV-2 | NA1 | - | Clinical Trials (Phase 1/2) | CTRI/2020/11/028476 |
| GEMCOVAC | SARS-CoV-2 | VEEV | - | Clinical Trials (Phase 2/3) | CTRI/2022/10/046475 |
| RBI-4000 | Rabies | VEEV | srRNA encoding rabies glycoprotein, optimized for low-dose delivery | Clinical Trials (Phase 1) | NCT06048770 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallet, T.; Vignuzzi, M. Self-Amplifying RNA: Advantages and Challenges of a Versatile Platform for Vaccine Development. Viruses 2025, 17, 566. https://doi.org/10.3390/v17040566
Vallet T, Vignuzzi M. Self-Amplifying RNA: Advantages and Challenges of a Versatile Platform for Vaccine Development. Viruses. 2025; 17(4):566. https://doi.org/10.3390/v17040566
Chicago/Turabian StyleVallet, Thomas, and Marco Vignuzzi. 2025. "Self-Amplifying RNA: Advantages and Challenges of a Versatile Platform for Vaccine Development" Viruses 17, no. 4: 566. https://doi.org/10.3390/v17040566
APA StyleVallet, T., & Vignuzzi, M. (2025). Self-Amplifying RNA: Advantages and Challenges of a Versatile Platform for Vaccine Development. Viruses, 17(4), 566. https://doi.org/10.3390/v17040566