
Genetic and Phenotypic Investigations of Viral Subpopulations Detected in Different Tissues of Laying Hens Following Infectious Bronchitis Virus Infection

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Sample Collection
2.3. Extraction of RNA, cDNA Synthesis, and Viral RNA Load Quantification
2.4. Amplification of Full S1, Library Preparation, and Next-Generation Sequencing
2.5. Bioinformatic Analysis of Sequencing Data
2.6. Analysis of Genetic Variation
2.7. Structural Modelling and Mapping of Amino Acid Mutations
2.8. Statistical Analysis and Data Visualization
3. Results
3.1. Amplification of S1
3.2. Quality Control of Sequencing Data
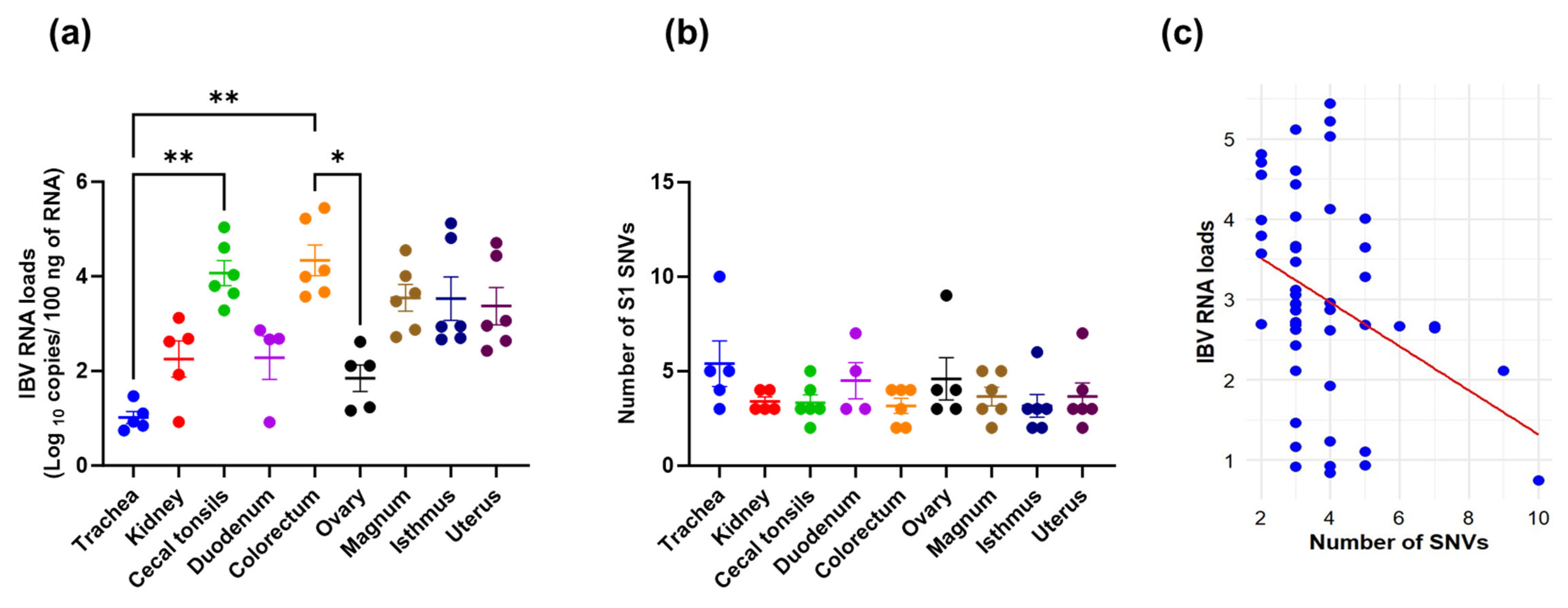
3.3. Viral RNA Loads, SNVs Occurrence, and Their Correlation
3.4. S1 Subpopulations in Different Tissues
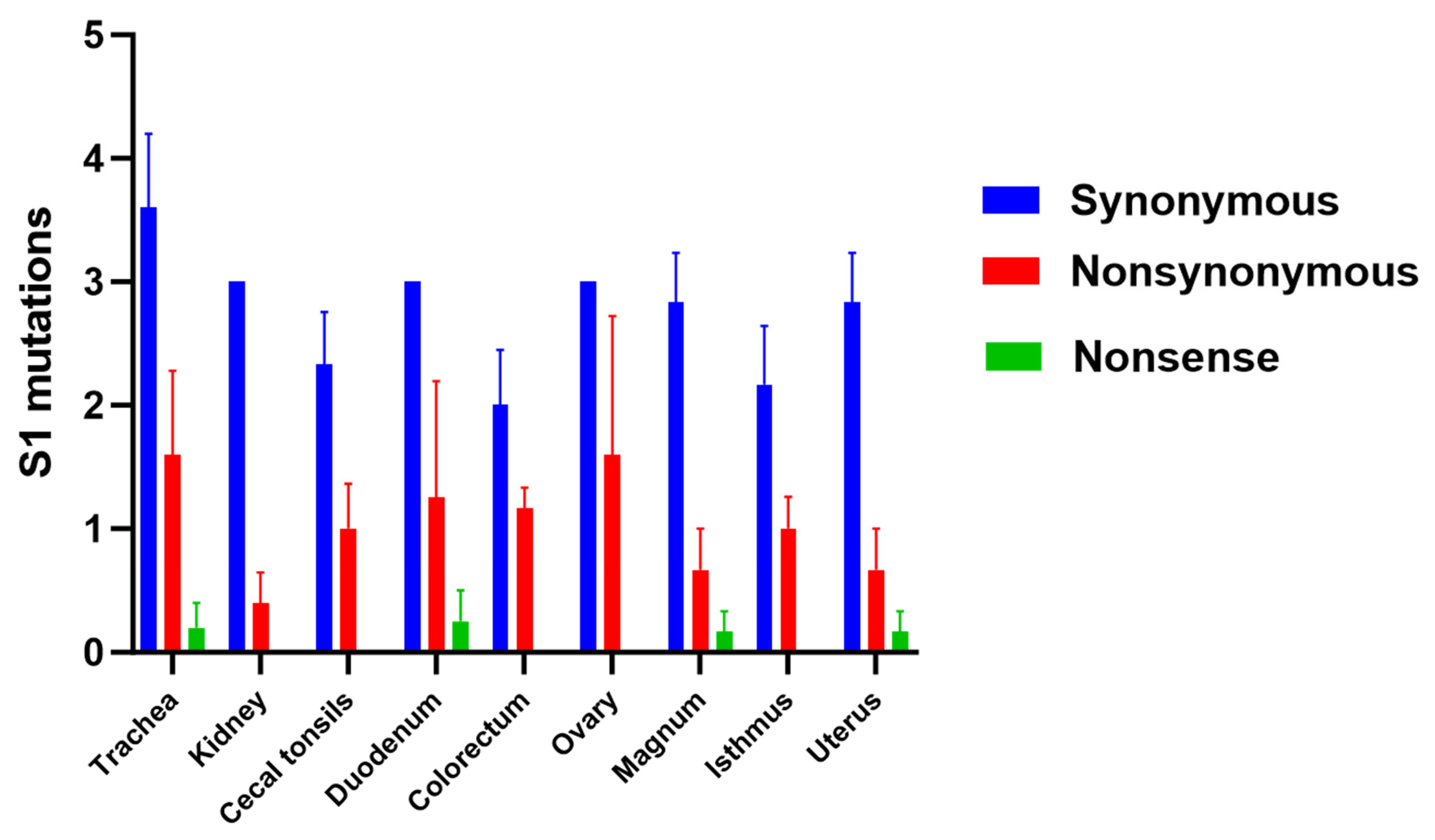
3.5. Genetic Variation Analysis
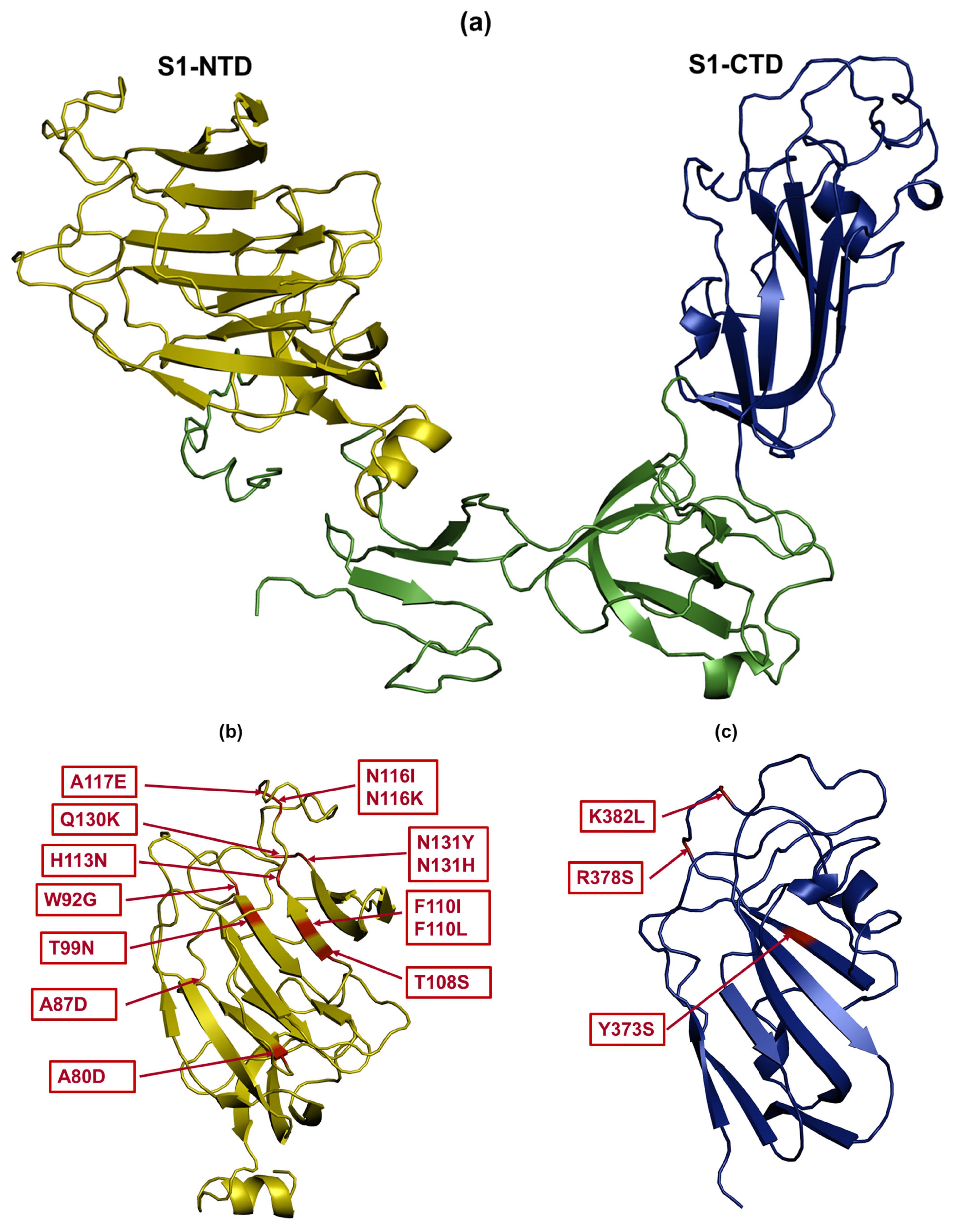
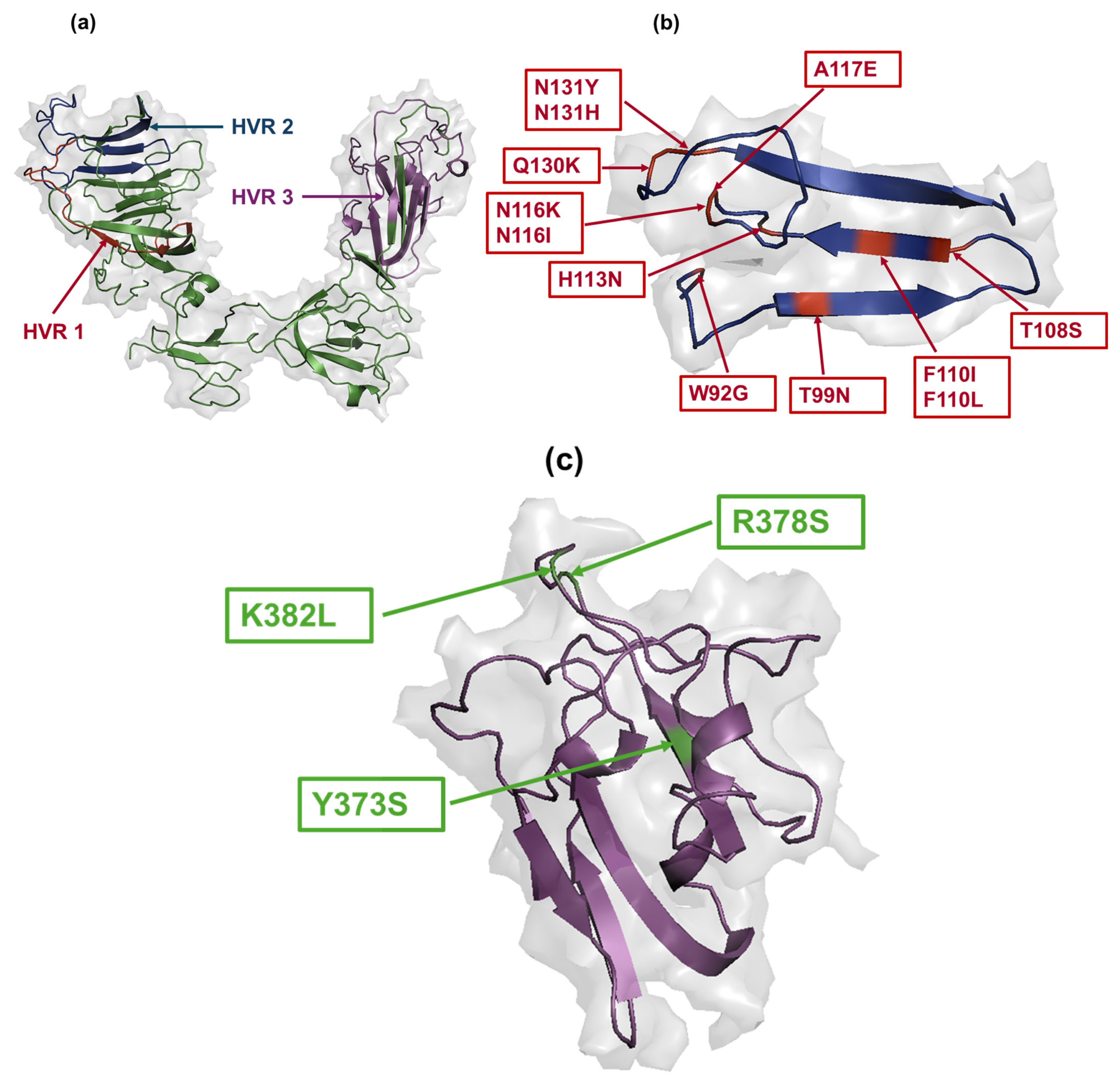
3.6. Amino Acid Changes in S1 Glycoprotein
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cook, J.K.; Jackwood, M.; Jones, R.C. The long view: 40 years of infectious bronchitis research. Avian Pathol. 2012, 41, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Bande, F.; Arshad, S.S.; Omar, A.R.; Hair-Bejo, M.; Mahmuda, A.; Nair, V. Global distributions and strain diversity of avian infectious bronchitis virus: A review. Anim. Health Res. Rev. 2017, 18, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Sjaak de Wit, J.J.; Cook, J.K.; van der Heijden, H.M. Infectious bronchitis virus variants: A review of the history, current situation and control measures. Avian Pathol. 2011, 40, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. In Coronaviruses; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 1282, pp. 1–23. [Google Scholar] [CrossRef]
- Armesto, M.; Cavanagh, D.; Britton, P. The replicase gene of avian coronavirus infectious bronchitis virus is a determinant of pathogenicity. PLoS ONE 2009, 4, e7384. [Google Scholar] [CrossRef]
- Quinteros, J.A.; Markham, P.F.; Lee, S.W.; Hewson, K.A.; Hartley, C.A.; Legione, A.R.; Coppo, M.J.; Vaz, P.K.; Browning, G.F. Analysis of the complete genomic sequences of two virus subpopulations of the Australian infectious bronchitis virus vaccine VicS. Avian Pathol. 2015, 44, 182–191. [Google Scholar] [CrossRef]
- Wickramasinghe, I.N.; van Beurden, S.J.; Weerts, E.A.; Verheije, M.H. The avian coronavirus spike protein. Virus Res. 2014, 194, 37–48. [Google Scholar] [CrossRef]
- McKinley, E.T.; Jackwood, M.W.; Hilt, D.A.; Kissinger, J.C.; Robertson, J.S.; Lemke, C.; Paterson, A.H. Attenuated live vaccine usage affects accurate measures of virus diversity and mutation rates in avian coronavirus infectious bronchitis virus. Virus Res. 2011, 158, 225–234. [Google Scholar] [CrossRef]
- Thor, S.W.; Hilt, D.A.; Kissinger, J.C.; Paterson, A.H.; Jackwood, M.W. Recombination in avian gamma-coronavirus infectious bronchitis virus. Viruses 2011, 3, 1777–1799. [Google Scholar] [CrossRef]
- Moreno, A.; Franzo, G.; Massi, P.; Tosi, G.; Blanco, A.; Antilles, N.; Biarnes, M.; Majó, N.; Nofrarías, M.; Dolz, R.; et al. A novel variant of the infectious bronchitis virus resulting from recombination events in Italy and Spain. Avian Pathol. 2017, 46, 28–35. [Google Scholar] [CrossRef]
- Franzo, G.; Massi, P.; Tucciarone, C.M.; Barbieri, I.; Tosi, G.; Fiorentini, L.; Ciccozzi, M.; Lavazza, A.; Cecchinato, M.; Moreno, A. Think globally, act locally: Phylodynamic reconstruction of infectious bronchitis virus (IBV) QX genotype (GI-19 lineage) reveals different population dynamics and spreading patterns when evaluated on different epidemiological scales. PLoS ONE 2017, 12, e0184401. [Google Scholar] [CrossRef]
- Sniegowski, P.D.; Gerrish, P.J.; Johnson, T.; Shaver, A. The evolution of mutation rates: Separating causes from consequences. Bioessays 2000, 22, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018, 16, e3000003. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Martin, V.; Perales, C.; Grande-Pérez, A.; García-Arriaza, J.; Arias, A. Viruses as quasispecies: Biological implications. In Quasispecies: Concept and Implications for Virology; Springer: Berlin/Heidelberg, Germany, 2006; Volume 299, pp. 51–82. [Google Scholar] [CrossRef]
- Eigen, M.; Gardiner, W.; Schuster, P.; Winkler-Oswatitsch, R. The origin of genetic information. Sci. Am. 1981, 244, 88–92, 96, et passim. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Zheng, Y.; Yang, Y.; Liu, C.; Geng, Q.; Luo, C.; Zhang, W.; Li, F. Cryo-EM structure of infectious bronchitis coronavirus spike protein reveals structural and functional evolution of coronavirus spike proteins. PLoS Pathog. 2018, 14, e1007009. [Google Scholar] [CrossRef]
- Cavanagh, D.; Mawditt, K.; Adzhar, A.; Gough, R.E.; Picault, J.P.; Naylor, C.J.; Haydon, D.; Shaw, K.; Britton, P. Does IBV change slowly despite the capacity of the spike protein to vary greatly? Adv. Exp. Med. Biol. 1998, 440, 729–734. [Google Scholar] [CrossRef]
- Koch, G.; Hartog, L.; Kant, A.; van Roozelaar, D.J. Antigenic domains on the peplomer protein of avian infectious bronchitis virus: Correlation with biological functions. J. Gen. Virol. 1990, 71 Pt 9, 1929–1935. [Google Scholar] [CrossRef]
- Kant, A.; Koch, G.; van Roozelaar, D.J.; Kusters, J.G.; Poelwijk, F.A.; van der Zeijst, B.A. Location of antigenic sites defined by neutralizing monoclonal antibodies on the S1 avian infectious bronchitis virus glycopolypeptide. J. Gen. Virol. 1992, 73 Pt 3, 591–596. [Google Scholar] [CrossRef]
- Cavanagh, D.; Davis, P.J.; Cook, J.K.; Li, D.; Kant, A.; Koch, G. Location of the amino acid differences in the S1 spike glycoprotein subunit of closely related serotypes of infectious bronchitis virus. Avian Pathol. 1992, 21, 33–43. [Google Scholar] [CrossRef]
- McKinley, E.T.; Hilt, D.A.; Jackwood, M.W. Avian coronavirus infectious bronchitis attenuated live vaccines undergo selection of subpopulations and mutations following vaccination. Vaccine 2008, 26, 1274–1284. [Google Scholar] [CrossRef]
- Ndegwa, E.N.; Joiner, K.S.; Toro, H.; van Ginkel, F.W.; van Santen, V.L. The proportion of specific viral subpopulations in attenuated Arkansas Delmarva poultry industry infectious bronchitis vaccines influences vaccination outcome. Avian Dis. 2012, 56, 642–653. [Google Scholar] [CrossRef]
- Van Santen, V.L.; Toro, H. Rapid selection in chickens of subpopulations within ArkDPI-derived infectious bronchitis virus vaccines. Avian Pathol. 2008, 37, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, I.N.; de Vries, R.P.; Gröne, A.; de Haan, C.A.; Verheije, M.H. Binding of avian coronavirus spike proteins to host factors reflects virus tropism and pathogenicity. J. Virol. 2011, 85, 8903–8912. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, R.A.; van Santen, V.L.; Toro, H. Host intraspatial selection of infectious bronchitis virus populations. Avian Dis. 2010, 54, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Farooq, M.; Altakrouni, D.; Najimudeen, S.M.; Hassan, M.S.H.; Isham, I.M.; Shalaby, A.A.; Gallardo, R.A.; Abdul-Careem, M.F. Comparative pathogenicity of CA1737/04 and Mass infectious bronchitis virus genotypes in laying chickens. Front. Vet. Sci. 2024, 11, 1338563. [Google Scholar] [CrossRef]
- Callison, S.A.; Hilt, D.A.; Boynton, T.O.; Sample, B.F.; Robison, R.; Swayne, D.E.; Jackwood, M.W. Development and evaluation of a real-time Taqman RT-PCR assay for the detection of infectious bronchitis virus from infected chickens. J. Virol. Methods 2006, 138, 60–65. [Google Scholar] [CrossRef]
- Isham, I.M.; Najimudeen, S.M.; Cork, S.C.; Gupta, A.; Abdul-Careem, M.F. Comparison of quantitative PCR and digital PCR assays for quantitative detection of infectious bronchitis virus (IBV) genome. J. Virol. Methods 2024, 324, 114859. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v6: Recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 2024, 52, W78–W82. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Jackwood, M.W.; Hilt, D.A.; Callison, S.A. Detection of infectious bronchitis virus by real-time reverse transcriptase-polymerase chain reaction and identification of a quasispecies in the Beaudette strain. Avian Dis. 2003, 47, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hasoksuz, M.; Spiro, D.; Halpin, R.; Wang, S.; Vlasova, A.; Janies, D.; Jones, L.R.; Ghedin, E.; Saif, L.J. Quasispecies of bovine enteric and respiratory coronaviruses based on complete genome sequences and genetic changes after tissue culture adaptation. Virology 2007, 363, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Ojkic, D.; Elshafiee, E.A.; Shany, S.; El-Safty, M.M.; Shalaby, A.A.; Abdul-Careem, M.F. Genotyping and In Silico Analysis of Delmarva (DMV/1639) Infectious Bronchitis Virus (IBV) Spike 1 (S1) Glycoprotein. Genes 2022, 13, 1617. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.; Ali, A.; Hassan, M.S.H.; Abdul-Careem, M.F. Nucleotide and Amino Acid Analyses of Unique Infectious Bronchitis Virus (IBV) Variants from Canadian Poultry Flocks with Drop in Egg Production. Genes 2024, 15, 1480. [Google Scholar] [CrossRef]
- Huang, Y.P.; Wang, C.H. Sequence changes of infectious bronchitis virus isolates in the 3′ 7.3 kb of the genome after attenuating passage in embryonated eggs. Avian Pathol. 2007, 36, 59–67. [Google Scholar] [CrossRef]
- Cavanagh, D.; Picault, J.P.; Gough, R.; Hess, M.; Mawditt, K.; Britton, P. Variation in the spike protein of the 793/B type of infectious bronchitis virus, in the field and during alternate passage in chickens and embryonated eggs. Avian Pathol. 2005, 34, 20–25. [Google Scholar] [CrossRef]
- Liu, S.; Han, Z.; Chen, J.; Liu, X.; Shao, Y.; Kong, X.; Tong, G.; Rong, J. S1 gene sequence heterogeneity of a pathogenic infectious bronchitis virus strain and its embryo-passaged, attenuated derivatives. Avian Pathol. 2007, 36, 231–234. [Google Scholar] [CrossRef]
- Cavanagh, D. Coronavirus avian infectious bronchitis virus. Vet. Res. 2007, 38, 281–297. [Google Scholar] [CrossRef]
- Ramazzotti, D.; Angaroni, F.; Maspero, D.; Mauri, M.; D’Aliberti, D.; Fontana, D.; Antoniotti, M.; Elli, E.M.; Graudenzi, A.; Piazza, R. Large-scale analysis of SARS-CoV-2 synonymous mutations reveals the adaptation to the human codon usage during the virus evolution. Virus Evol. 2022, 8, veac026. [Google Scholar] [CrossRef]
- Barroso da Silva, F.L.; Giron, C.C.; Laaksonen, A. Electrostatic Features for the Receptor Binding Domain of SARS-COV-2 Wildtype and Its Variants. Compass to the Severity of the Future Variants with the Charge-Rule. J. Phys. Chem. B 2022, 126, 6835–6852. [Google Scholar] [CrossRef]
- Nie, C.; Sahoo, A.K.; Netz, R.R.; Herrmann, A.; Ballauff, M.; Haag, R. Charge Matters: Mutations in Omicron Variant Favor Binding to Cells. Chembiochem 2022, 23, e202100681. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kearns, F.L.; Rosenfeld, M.A.; Votapka, L.; Casalino, L.; Papanikolas, M.; Amaro, R.E.; Freeman, R. SARS-CoV-2 evolved variants optimize binding to cellular glycocalyx. Cell Rep. Phys. Sci. 2023, 4, 101346. [Google Scholar] [CrossRef] [PubMed]
- Pénzes, Z.; Wroe, C.; Brown, T.D.; Britton, P.; Cavanagh, D. Replication and packaging of coronavirus infectious bronchitis virus defective RNAs lacking a long open reading frame. J. Virol. 1996, 70, 8660–8668. [Google Scholar] [CrossRef] [PubMed]
- Imamichi, H.; Dewar, R.L.; Adelsberger, J.W.; Rehm, C.A.; O’Doherty, U.; Paxinos, E.E.; Fauci, A.S.; Lane, H.C. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2016, 113, 8783–8788. [Google Scholar] [CrossRef]
- Ho, Y.C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef]
- Ammayappan, A.; Upadhyay, C.; Gelb, J., Jr.; Vakharia, V.N. Identification of sequence changes responsible for the attenuation of avian infectious bronchitis virus strain Arkansas DPI. Arch. Virol. 2009, 154, 495–499. [Google Scholar] [CrossRef]
- Fang, S.G.; Shen, S.; Tay, F.P.; Liu, D.X. Selection of and recombination between minor variants lead to the adaptation of an avian coronavirus to primate cells. Biochem. Biophys. Res. Commun. 2005, 336, 417–423. [Google Scholar] [CrossRef]
- Moore, K.M.; Jackwood, M.W.; Hilt, D.A. Identification of amino acids involved in a serotype and neutralization specific epitope within the s1 subunit of avian infectious bronchitis virus. Arch. Virol. 1997, 142, 2249–2256. [Google Scholar] [CrossRef]
- Niesters, H.G.; Bleumink-Pluym, N.M.; Osterhaus, A.D.; Horzinek, M.C.; van der Zeijst, B.A. Epitopes on the peplomer protein of infectious bronchitis virus strain M41 as defined by monoclonal antibodies. Virology 1987, 161, 511–519. [Google Scholar] [CrossRef]
- Santos Fernando, F.; Coelho Kasmanas, T.; Diniz Lopes, P.; da Silva Montassier, M.F.; Zanella Mores, M.A.; Casagrande Mariguela, V.; Pavani, C.; Moreira Dos Santos, R.; Assayag, M.S., Jr.; Montassier, H.J. Assessment of molecular and genetic evolution, antigenicity and virulence properties during the persistence of the infectious bronchitis virus in broiler breeders. J. Gen. Virol. 2017, 98, 2470–2481. [Google Scholar] [CrossRef]
- Shan, D.; Fang, S.; Han, Z.; Ai, H.; Zhao, W.; Chen, Y.; Jiang, L.; Liu, S. Effects of hypervariable regions in spike protein on pathogenicity, tropism, and serotypes of infectious bronchitis virus. Virus Res. 2018, 250, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Ignjatovic, J.; Sapats, S. Identification of previously unknown antigenic epitopes on the S and N proteins of avian infectious bronchitis virus. Arch. Virol. 2005, 150, 1813–1831. [Google Scholar] [CrossRef] [PubMed]
- Kusters, J.G.; Jager, E.J.; Lenstra, J.A.; Koch, G.; Posthumus, W.P.; Meloen, R.H.; van der Zeijst, B.A. Analysis of an immunodominant region of infectious bronchitis virus. J. Immunol. 1989, 143, 2692–2698. [Google Scholar] [PubMed]
- Henn, M.R.; Boutwell, C.L.; Charlebois, P.; Lennon, N.J.; Power, K.A.; Macalalad, A.R.; Berlin, A.M.; Malboeuf, C.M.; Ryan, E.M.; Gnerre, S.; et al. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 2012, 8, e1002529. [Google Scholar] [CrossRef]
- Wang, C.; Mitsuya, Y.; Gharizadeh, B.; Ronaghi, M.; Shafer, R.W. Characterization of mutation spectra with ultra-deep pyrosequencing: Application to HIV-1 drug resistance. Genome Res. 2007, 17, 1195–1201. [Google Scholar] [CrossRef]
- Metzner, K.J.; Giulieri, S.G.; Knoepfel, S.A.; Rauch, P.; Burgisser, P.; Yerly, S.; Günthard, H.F.; Cavassini, M. Minority quasispecies of drug-resistant HIV-1 that lead to early therapy failure in treatment-naive and -adherent patients. Clin. Infect. Dis. 2009, 48, 239–247. [Google Scholar] [CrossRef]
- Alkan, C.; Sajjadian, S.; Eichler, E.E. Limitations of next-generation genome sequence assembly. Nat. Methods 2011, 8, 61–65. [Google Scholar] [CrossRef]
- De Wit, J.J. Detection of infectious bronchitis virus. Avian Pathol. 2000, 29, 71–93. [Google Scholar] [CrossRef]
- Hassan, M.S.H.; Ali, A.; Buharideen, S.M.; Goldsmith, D.; Coffin, C.S.; Cork, S.C.; van der Meer, F.; Boulianne, M.; Abdul-Careem, M.F. Pathogenicity of the Canadian Delmarva (DMV/1639) Infectious Bronchitis Virus (IBV) on Female Reproductive Tract of Chickens. Viruses 2021, 13, 2488. [Google Scholar] [CrossRef]
- Hassan, M.S.H.; Najimudeen, S.M.; Ali, A.; Altakrouni, D.; Goldsmith, D.; Coffin, C.S.; Cork, S.C.; van der Meer, F.; Abdul-Careem, M.F. Immunopathogenesis of the Canadian Delmarva (DMV/1639) infectious bronchitis virus (IBV): Impact on the reproductive tract in layers. Microb. Pathog. 2022, 166, 105513. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence (5′-3′) a | Amplicon Size |
|---|---|---|
| Forward primer 1 Reverse primer 1 | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGTGTGGTAAGTTGCTGGTAAGA GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGTGCGAATATGATTCTGTGGTACA | 426 bp |
| Forward primer 2 Reverse primer 2 | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTGCCCTACAACAGGTTT GTCTCGTGGGCTC GGAGATGTGTATAAGAGACAGCTCCCGTAAACAATGAACTTCTG | 425 bp |
| Forward primer 3 Reverse primer 3 | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGACACCTAGAGGTTTATTAGCATGTC GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGCTGCTTACACCCACCTTGAA | 411 bp |
| Forward primer 4 Reverse primer 4 | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGTGGCTTGTGGTTTAATTCACTTTC GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGTGTCTATAGCACCTGACGTATCT | 407 bp |
| Forward primer 5 Reverse primer 5 | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGTATGGCAGAGTTGGACAAGG GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGAACTAACATATGGACAATTAGTAACATT | 343 bp |
| Tissue | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| SNVs | Trachea | Kidney | Cecal Tonsils | Duodenum | Colorectum | Ovary | Magnum | Isthmus | Uterus |
| A138G | 5/5 a | 5/5 | 5/6 | 4/4 | 3/6 | 5/5 | 5/6 | 2/6 | 5/6 |
| A300T | 5/5 | 5/5 | 4/6 | 4/4 | 3/6 | 5/5 | 5/6 | 2/6 | 5/6 |
| T1296G | 5/5 | 5/5 | 6/6 | 4/4 | 6/6 | 5/5 | 6/6 | 6/6 | 6/6 |
| A391T | 2/5 | 0/5 | 3/6 | 2/4 | 3/6 | 1/5 | 3/6 | 5/6 | 2/6 |
| C388A | 2/5 | 2/5 | 1/6 | 0/4 | 1/6 | 2/5 | 1/6 | 2/6 | 0/6 |
| C803T | 0/5 | 0/5 | 1/6 | 1/4 | 2/6 | 0/5 | 0/6 | 0/6 | 0/6 |
| T328A | 0/5 | 0/5 | 0/6 | 0/4 | 0/6 | 1/5 | 0/6 | 0/6 | 1/6 |
| T274G | 0/5 | 0/5 | 1/6 | 0/4 | 0/6 | 0/5 | 0/6 | 1/6 | 0/6 |
| C284A | 1/5 | 0/5 | 0/6 | 1/4 | 0/6 | 0/5 | 0/6 | 0/6 | 1/6 |
| A1089T | 1/5 | 0/5 | 0/6 | 0/4 | 0/6 | 0/5 | 0/6 | 0/6 | 0/6 |
| T1107C | 1/5 | 0/5 | 0/6 | 0/4 | 0/6 | 0/5 | 0/6 | 0/6 | 0/6 |
| A1118C | 1/5 | 0/5 | 0/6 | 0/4 | 0/6 | 0/5 | 0/6 | 0/6 | 0/6 |
| A1134C | 1/5 | 0/5 | 0/6 | 0/4 | 0/6 | 0/5 | 0/6 | 0/6 | 0/6 |
| A1144T | 1/5 | 0/5 | 0/6 | 0/4 | 0/6 | 0/5 | 0/6 | 0/6 | 0/6 |
| A1145T | 1/5 | 0/5 | 0/6 | 0/4 | 0/6 | 0/5 | 0/6 | 0/6 | 0/6 |
| A391C | 1/5 | 0/5 | 0/6 | 0/4 | 0/6 | 0/5 | 0/6 | 0/6 | 0/6 |
| T330A | 0/5 | 0/5 | 0/5 | 1/4 | 0/6 | 0/5 | 0/6 | 0/6 | 0/6 |
| T348G | 0/5 | 0/5 | 0/5 | 1/4 | 0/6 | 0/5 | 0/6 | 0/6 | 0/6 |
| A347T | 0/5 | 0/5 | 0/5 | 0/4 | 1/6 | 0/5 | 0/6 | 0/6 | 0/6 |
| C260A | 0/5 | 0/5 | 0/5 | 0/4 | 0/6 | 1/5 | 0/6 | 0/6 | 0/6 |
| C239A | 0/5 | 0/5 | 0/5 | 0/4 | 0/6 | 1/5 | 0/6 | 0/6 | 0/6 |
| C296A | 0/5 | 0/5 | 0/5 | 0/4 | 0/6 | 1/5 | 0/6 | 0/6 | 0/6 |
| C337A | 0/5 | 0/5 | 0/5 | 0/4 | 0/6 | 1/5 | 0/6 | 0/6 | 0/6 |
| C286T | 0/5 | 0/5 | 0/5 | 0/4 | 0/6 | 0/5 | 1/6 | 0/6 | 0/6 |
| A322T | 0/5 | 0/5 | 0/5 | 0/4 | 0/6 | 0/5 | 1/6 | 0/6 | 0/6 |
| C261T | 0/5 | 0/5 | 0/5 | 0/4 | 0/6 | 0/5 | 0/6 | 1/6 | 0/6 |
| C350A | 0/5 | 0/5 | 0/5 | 0/4 | 0/6 | 0/5 | 0/6 | 0/6 | 1/6 |
| Designation | SNVs | Tissue |
|---|---|---|
| SP1 | A138G, A300T, T1296G | All tissues |
| SP2 | A138G, A300T, T1296G, a A391T, C388A | All tissues, except kidney, duodenum and uterus |
| SP3 | A138G, A300T, T1296G, C803T | Cecal tonsils, Duodenum, colorectum |
| SP4 | A138G, A300T, T1296G, T328A | Uterus, ovary |
| SP5 | A138G, A300T, T1296G, T274G | Isthmus, cecal tonsils |
| SP6 | A138G, A300T, T1296G, C284A | Trachea, uterus, duodenum |
| SP7 | A138G, A300T, T1296G, A1089T, T1107C, A1118C, A1134C, A1144T, A1145T | Trachea |
| SP8 | A138G, A300T, T1296G, A391C | Trachea |
| SP9 | A138G, A300T, T1296G, T330A, T348G | Duodenum |
| SP10 | A138G, A300T, T1296G, A347T | Colorectum |
| SP11 | A138G, A300T, T1296G, C239A, C260A, C296A, C337A | Ovary |
| SP12 | A138G, A300T, T1296G, C286T | Magnum |
| SP13 | A138G, A300T, T1296G, A322T | Magnum |
| SP14 | A138G, A300T, T1296G, C261T | Isthmus |
| SP15 | A138G, A300T, T1296G, C350A | Uterus |
| Subpopulation | Synonymous | Nonsense | Nonsynonymous |
|---|---|---|---|
| SP1 | A138G, A300T, T1296G (Core SNVs) | - | - a |
| SP2 | Core SNVs | - | C388A (Q130K) b A391T (N131Y) |
| SP3 | Core SNVs | - | C803T (T268I) |
| SP4 | Core SNVs | - | T328A (F110I) |
| SP5 | Core SNVs | - | T274G (W92G) |
| SP6 | Core SNVs | C284A (S95 *) | - |
| SP7 | Core SNVs, A1089T, T1107C | - | A1118C (Y373S) A1134C (R378S) A1144T, A1145T (K382L) |
| SP8 | Core SNVs | - | A391C (N131H) |
| SP9 | Core SNVs | - | T330A (F110L) T348G (N116K) |
| SP10 | Core SNVs | - | A347T (N116I) |
| SP11 | Core SNVs | - | C239A (A80D) C260A (A87D) C296A (T99N) C337A (H113N) |
| SP12 | Core SNVs | C286T (Q96 *) | - |
| SP13 | Core SNVs | - | A322T (T108S) |
| SP14 | Core SNVs, C261T | - | - |
| SP15 | Core SNVs | - | C350A (A117E) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.; Rahimi, R.; Mahmoud, M.E.; Shalaby, A.A.; Gallardo, R.A.; Abdul-Careem, M.F. Genetic and Phenotypic Investigations of Viral Subpopulations Detected in Different Tissues of Laying Hens Following Infectious Bronchitis Virus Infection. Viruses 2025, 17, 527. https://doi.org/10.3390/v17040527
Ali A, Rahimi R, Mahmoud ME, Shalaby AA, Gallardo RA, Abdul-Careem MF. Genetic and Phenotypic Investigations of Viral Subpopulations Detected in Different Tissues of Laying Hens Following Infectious Bronchitis Virus Infection. Viruses. 2025; 17(4):527. https://doi.org/10.3390/v17040527
Chicago/Turabian StyleAli, Ahmed, Ryan Rahimi, Motamed Elsayed Mahmoud, Adel A. Shalaby, Rodrigo A. Gallardo, and Mohamed Faizal Abdul-Careem. 2025. "Genetic and Phenotypic Investigations of Viral Subpopulations Detected in Different Tissues of Laying Hens Following Infectious Bronchitis Virus Infection" Viruses 17, no. 4: 527. https://doi.org/10.3390/v17040527
APA StyleAli, A., Rahimi, R., Mahmoud, M. E., Shalaby, A. A., Gallardo, R. A., & Abdul-Careem, M. F. (2025). Genetic and Phenotypic Investigations of Viral Subpopulations Detected in Different Tissues of Laying Hens Following Infectious Bronchitis Virus Infection. Viruses, 17(4), 527. https://doi.org/10.3390/v17040527