
Molecular Epidemiology of SARS-CoV-2 in Bangladesh

 , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Setting and Data Sources
2.2. Nucleotide Extraction and Sequencing
2.3. Bioinformatic Analysis for Generating Sequencing Data
2.4. Nucleotide Substitution Analysis and Phylogeny
2.5. Lineage Assignment
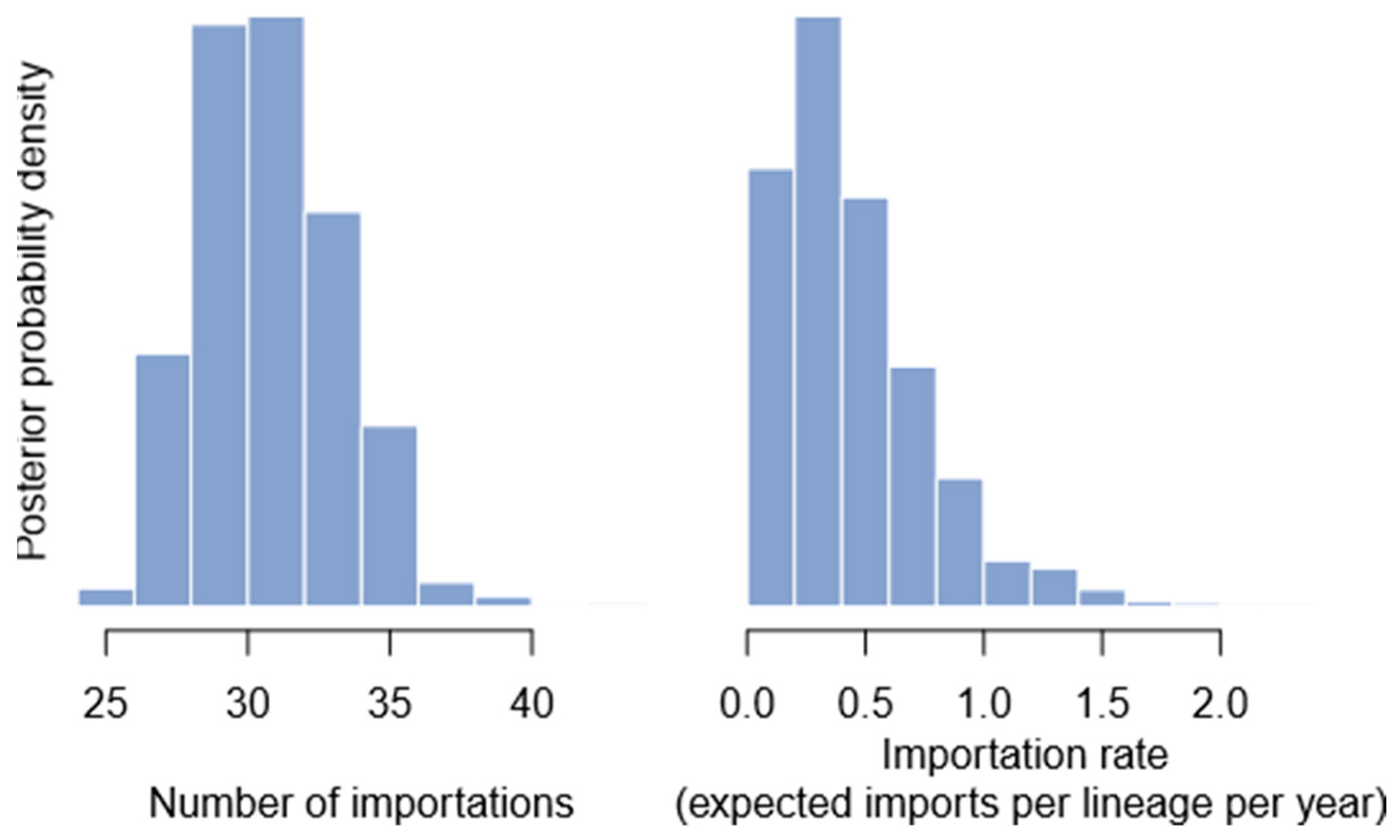
2.6. Number of Importation Events
3. Results
3.1. Characterization of Samples
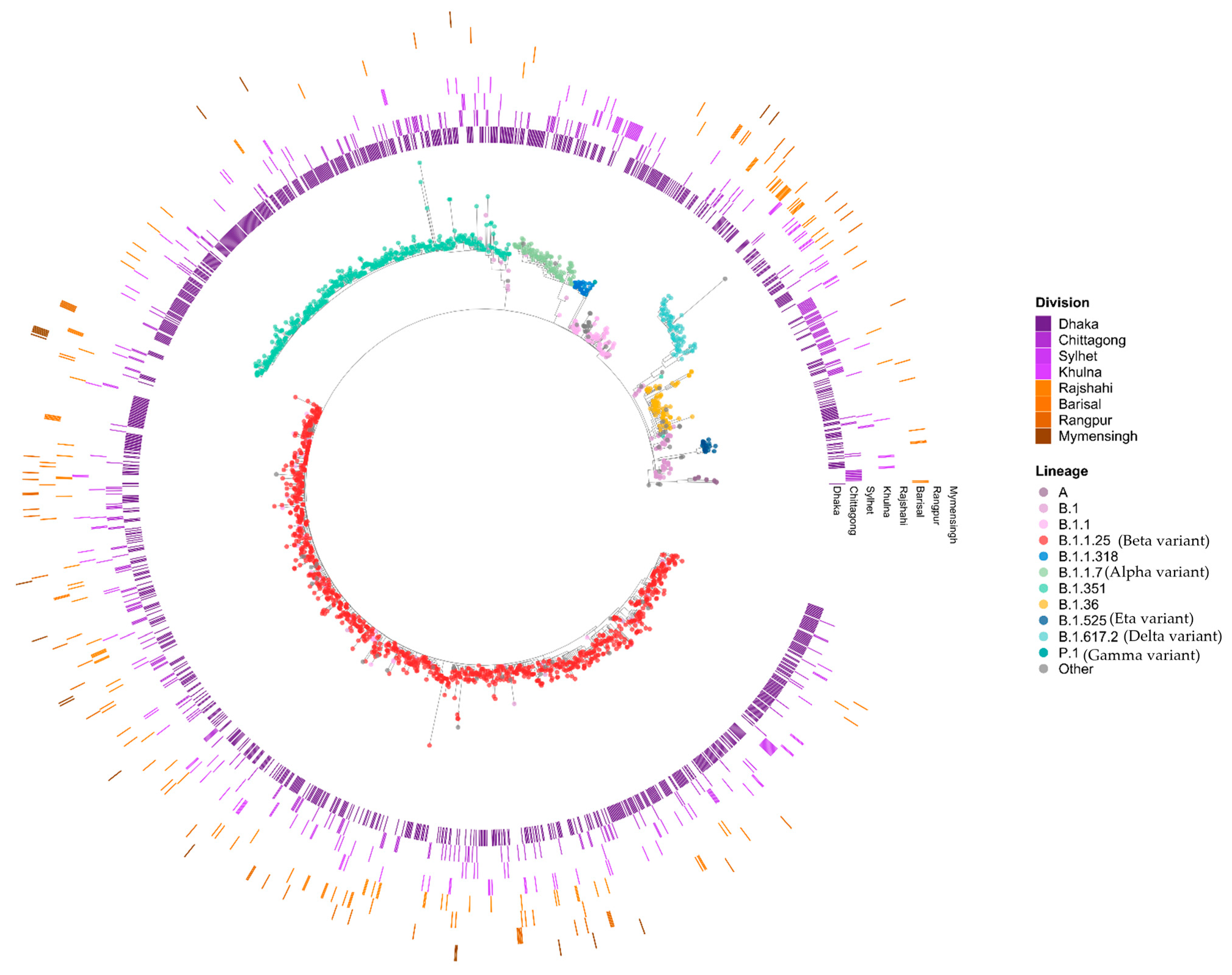
3.2. Phylogenetic Analysis of SARS-CoV-2
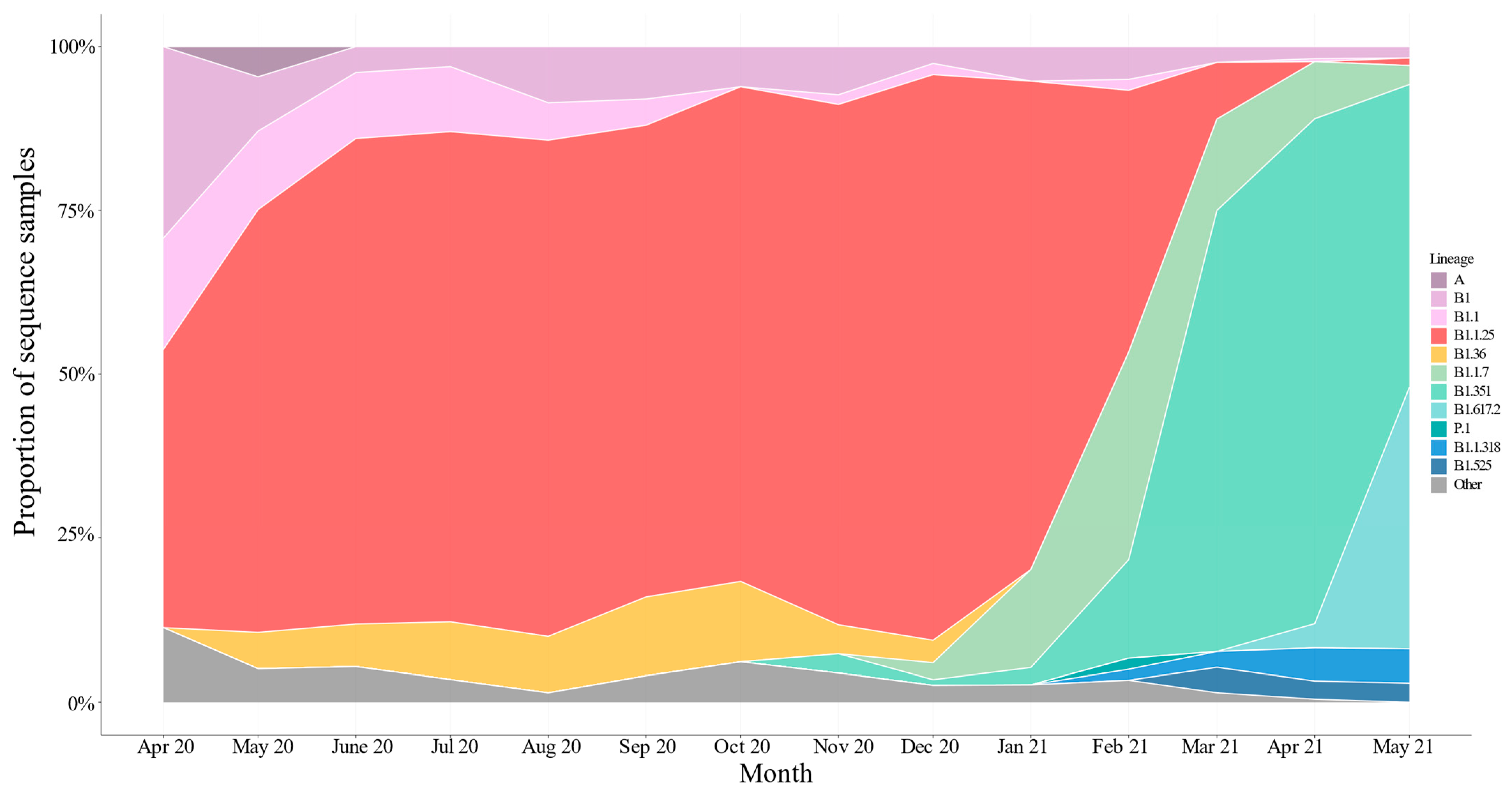
3.3. Genomic Variations in SARS-CoV-2
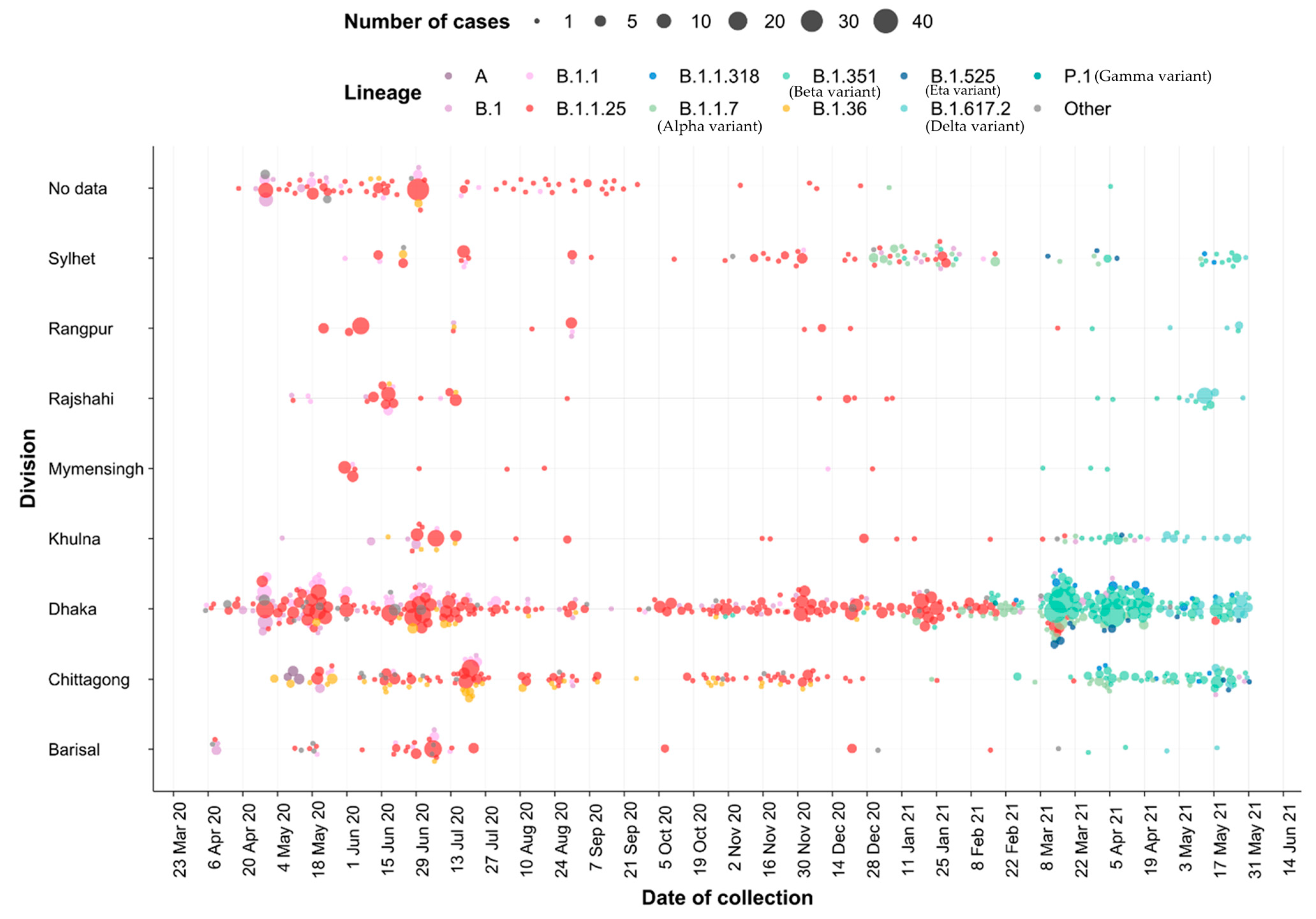
3.4. Importation Dynamics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Bangladesh COVID-19 Morbidity and Mortality Weekly Update (MMWU); WHO: Geneva, Switzerland, 2021; Volume 70. [Google Scholar]
- Mohmmad Mahmud, A.S.; Taznin, T.; Hasan Sarkar, M.M.; Uzzaman, M.S.; Osman, E.; Habib, M.A.; Akter, S.; Banu, T.A.; Goswami, B.; Jahan, I.; et al. The genetic variant analyses of SARS-CoV-2 strains; circulating in Bangladesh. bioRxiv 2020. [Google Scholar] [CrossRef]
- Parvin, R.; Afrin, S.Z.; Begum, J.A.; Ahmed, S.; Nooruzzaman, M.; Chowdhury, E.H.; Pohlmann, A.; Paul, S.K. Molecular Analysis of SARS-CoV-2 Circulating in Bangladesh during 2020 Revealed Lineage Diversity and Potential Mutations. Microorganisms 2021, 9, 1035. [Google Scholar] [CrossRef]
- Fraser, C.; Riley, S.; Anderson, R.M.; Ferguson, N.M. Factors that make an infectious disease outbreak controllable. Proc. Natl. Acad. Sci. USA 2004, 101, 6146–6151. [Google Scholar]
- Anderson, R.M.; May, R.M. Infectious Diseases of Humans: Dynamics and Control; Oxford University Press: Oxford, UK, 1992. [Google Scholar]
- Johansson, M.A.; Quandelacy, T.M.; Kada, S.; Prasad, P.V.; Steele, M.; Brooks, J.T.; Slayton, R.B.; Biggerstaff, M.; Butler, J.C. SARS-CoV-2 transmission from people without COVID-19 symptoms. JAMA Netw. Open 2021, 4, e2035057. [Google Scholar]
- Qiu, X.; Nergiz, A.I.; Maraolo, A.E.; Bogoch, I.I.; Low, N.; Cevik, M. The role of asymptomatic and pre-symptomatic infection in SARS-CoV-2 transmission—A living systematic review. Clin. Microbiol. Infect. 2021, 27, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Nasrullah, M.; Hosen, M.J. COVID-19 and Bangladesh: Challenges and How to Address Them. Front. Public Health 2020, 8, 154. [Google Scholar]
- Seemann, T.; Lane, C.R.; Sherry, N.L.; Duchene, S.; da Silva, A.G.; Caly, L.; Sait, M.; Ballard, S.A.; Horan, K.; Schultz, M.B.; et al. Tracking the COVID-19 pandemic in Australia using genomics. Nat. Commun. 2020, 11, 4376. [Google Scholar] [PubMed]
- Hamadani, J.D.; Hasan, M.I.; Baldi, A.J.; Hossain, S.J.; Shiraji, S.; Bhuiyan, M.S.A.; Mehrin, S.F.; Fisher, J.; Tofail, F.; Tipu, S.M.M.U.; et al. Immediate impact of stay-at-home orders to control COVID-19 transmission on socioeconomic conditions, food insecurity, mental health, and intimate partner violence in Bangladeshi women and their families: An interrupted time series. Lancet Glob. Health 2020, 8, e1380–e1389. [Google Scholar]
- Biswas, R.K.; Afiaz, A.; Huq, S. Underreporting COVID-19: The curious case of the Indian subcontinent. Epidemiol. Infect. 2020, 148, e207. [Google Scholar]
- Attwood, S.W.; Hill, S.C.; Aanensen, D.M.; Connor, T.R.; Pybus, O.G. Phylogenetic and phylodynamic approaches to understanding and combating the early SARS-CoV-2 pandemic. Nat. Rev. Genet. 2022, 23, 547–562. [Google Scholar] [PubMed]
- Kalantar, K.L.; Carvalho, T.; de Bourcy, C.F.A.; Dimitrov, B.; Dingle, G.; Egger, R.; Han, J.; Holmes, O.B.; Juan, Y.F.; King, R.; et al. IDseq—An Open Source Cloud-based Pipeline and Analysis Service for Metagenomic Pathogen Detection and Monitoring. bioRxiv 2020. [Google Scholar] [CrossRef]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantara, L.C.; Vanden Eynden, E.; Vandamme, A.-M.; et al. Genome Detective: An automated system for virus identification from high-throughput sequencing data. Bioinformatics 2019, 35, 871–873. [Google Scholar]
- National Genomics Data Center Members and Partners. Database Resources of the National Genomics Data Center in 2020. Nucleic Acids Res. 2019, 48, D24–D33. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar]
- Steenwyk, J.L.; Buida, T.J., 3rd; Li, Y.; Shen, X.X.; Rokas, A. ClipKIT: A multiple sequence alignment trimming software for accurate phylogenomic inference. PLoS Biol. 2020, 18, e3001007. [Google Scholar]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar]
- Yang, Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: Approximate methods. J. Mol. Evol. 1994, 39, 306–314. [Google Scholar] [PubMed]
- Junier, T.; Zdobnov, E.M. The Newick utilities: High-throughput phylogenetic tree processing in the UNIX shell. Bioinformatics 2010, 26, 1669–1670. [Google Scholar]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar]
- Duchene, S.; Featherstone, L.; Freiesleben de Blasio, B.; Holmes, E.C.; Bohlin, J.; Pettersson, J.H. The impact of public health interventions in the Nordic countries during the first year of SARS-CoV-2 transmission and evolution. EuroSurveillance 2021, 26, 2001996. [Google Scholar]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar]
- Wilton, T.; Bujaki, E.; Klapsa, D.; Majumdar, M.; Zambon, M.; Fritzsche, M.; Mate, R.; Martin, J. Rapid Increase of SARS-CoV-2 Variant B.1.1.7 Detected in Sewage Samples from England between October 2020 and January 2021. mSystems 2021, 6, e0035321. [Google Scholar] [CrossRef]
- Benvenuto, D.; Giovanetti, M.; Ciccozzi, A.; Spoto, S.; Angeletti, S.; Ciccozzi, M. The 2019-new coronavirus epidemic: Evidence for virus evolution. J. Med. Virol. 2020, 92, 455–459. [Google Scholar]
- Flores-Alanis, A.; Cruz-Rangel, A.; Rodríguez-Gómez, F.; González, J.; Torres-Guerrero, C.A.; Delgado, G.; Cravioto, A.; Morales-Espinosa, R. Molecular Epidemiology Surveillance of SARS-CoV-2: Mutations and Genetic Diversity One Year after Emerging. Pathogens 2021, 10, 184. [Google Scholar] [CrossRef]
- Isabel, S.; Graña-Miraglia, L.; Gutierrez, J.M.; Bundalovic-Torma, C.; Groves, H.E.; Isabel, M.R.; Eshaghi, A.; Patel, S.N.; Gubbay, J.B.; Poutanen, T.; et al. Evolutionary and structural analyses of SARS-CoV-2 D614G spike protein mutation are now documented worldwide. Sci. Rep. 2020, 10, 14031. [Google Scholar]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar]
- Syed, A.M.; Taha, T.Y.; Tabata, T.; Chen, I.P.; Ciling, A.; Khalid, M.M.; Sreekumar, B.; Chen, P.Y.; Hayashi, J.M.; Soczek, K.M.; et al. Rapid assessment of SARS-CoV-2–evolved variants using virus-like particles. Science 2021, 374, 1626–1632. [Google Scholar] [CrossRef]
- Pehrsson, E.C.; Tsukayama, P.; Patel, S.; Mejía-Bautista, M.; Sosa-Soto, G.; Navarrete, K.M.; Calderon, M.; Cabrera, L.; Hoyos-Arango, W.; Bertoli, M.T.; et al. Interconnected microbiomes and resistomes in low-income human habitats. Nature 2016, 533, 212–216. [Google Scholar] [CrossRef]
- Saito, A.; Irie, T.; Suzuki, R.; Maemura, T.; Nasser, H.; Uriu, K.; Kosugi, Y.; Shirakawa, K.; Sadamasu, K.; Kimura, I.; et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature 2022, 602, 300–306. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. Cell Rep. 2022, 39, 110829. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dutta, S.; Xiong, S.; Chan, M.; Chan, K.K.; Fan, T.M.; Bailey, K.L.; Lindeblad, M.; Cooper, L.M.; Rong, L.; et al. Engineered ACE2 decoy mitigates lung injury and death induced by SARS-CoV-2 variants. Nat. Chem. Biol. 2022, 18, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Alizon, S.; Hurford, A.; Mideo, N.; Van Baalen, M. Virulence evolution and the trade-off hypothesis: History, the current state of affairs and the future. J. Evol. Biol. 2009, 22, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Cowley, L.A.; Afrad, M.H.; Rahman, S.I.A.; Mamun, M.M.A.; Chin, T.; Mahmud, A.; Rahman, M.Z.; Billah, M.M.; Khan, M.H.; Sultana, S.; et al. Genomics, social media, and mobile phone data enable mapping of SARS-CoV-2 lineages to inform health policy in Bangladesh. Nat. Microbiol. 2021, 6, 1271–1278. [Google Scholar] [CrossRef]
- Lane, C.R.; Sherry, N.L.; Porter, A.F.; Duchene, S.; Horan, K.; Andersson, P.; Wilmot, M.; Turner, A.; Dougall, S.; Johnson, S.A.; et al. Genomics-informed responses in the elimination of COVID-19 in Victoria, Australia: An observational, genomic epidemiological study. Lancet Public Health 2021, 6, e547–e556, Correction in Lancet Public Health 2021, 6, e708. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Division | * BCSIR Dataset | ** Total Dataset | Population | COVID-19 Cases |
|---|---|---|---|---|
| Barishal | 26 (3.9%) | 70 (3.6%) | 9,100,102 (5.5%) | 2.4% |
| Chittagong | 86 (13.0%) | 295 (15.0%) | 33,202,326 (20.1%) | 13.4% |
| Dhaka | 390 (59.1%) | 1098 (55.9%) | 44,215,107 (26.8%) | 63.9% |
| Khulna | 23 (3.5%) | 91 (4.6%) | 17,416,645 (10.5%) | 6.1% |
| Mymensingh | 14 (2.1%) | 22 (1.1%) | 12,225,498 (7.4%) | 1.8% |
| Rajshahi | 38 (5.8%) | 78 (4.0%) | 20,353,119 (12.3%) | 5.6% |
| Rangpur | 25 (3.8%) | 44 (2.2%) | 17,610,956 (10.7%) | 3.4% |
| Sylhet | 58 (8.8%) | 116 (5.9%) | 11,034,863 (6.7%) | 3.4% |
| No data | 151 (7.7%) | |||
| Total | 660 | 165,158,616 † | 327,349 ^ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammad Mahmud, A.S.; Andersson, P.; Bulach, D.; Duchene, S.; da Silva, A.G.; Lin, C.; Seemann, T.; Howden, B.P.; Stinear, T.P.; Taznin, T.; et al. Molecular Epidemiology of SARS-CoV-2 in Bangladesh. Viruses 2025, 17, 517. https://doi.org/10.3390/v17040517
Mohammad Mahmud AS, Andersson P, Bulach D, Duchene S, da Silva AG, Lin C, Seemann T, Howden BP, Stinear TP, Taznin T, et al. Molecular Epidemiology of SARS-CoV-2 in Bangladesh. Viruses. 2025; 17(4):517. https://doi.org/10.3390/v17040517
Chicago/Turabian StyleMohammad Mahmud, Abu Sayeed, Patiyan Andersson, Dieter Bulach, Sebastian Duchene, Anders Goncalves da Silva, Chantel Lin, Torsten Seemann, Benjamin P. Howden, Timothy P. Stinear, Tarannum Taznin, and et al. 2025. "Molecular Epidemiology of SARS-CoV-2 in Bangladesh" Viruses 17, no. 4: 517. https://doi.org/10.3390/v17040517
APA StyleMohammad Mahmud, A. S., Andersson, P., Bulach, D., Duchene, S., da Silva, A. G., Lin, C., Seemann, T., Howden, B. P., Stinear, T. P., Taznin, T., Habib, M. A., Akter, S., Banu, T. A., Sarkar, M. M. H., Goswami, B., Jahan, I., & Khan, M. S. (2025). Molecular Epidemiology of SARS-CoV-2 in Bangladesh. Viruses, 17(4), 517. https://doi.org/10.3390/v17040517