
Whole Genome Sequencing of Lumpy Skin Disease Virus from 2021–2023 in Eastern Eurasia Reveals No More Recombination Signals in the Circulating Pool of Strains

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains
2.2. Sequencing
2.3. Bioinformatics
2.4. Phylogenetic Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Azeem, S.; Sharma, B.; Shabir, S.; Akbar, H.; Venter, E. Lumpy skin disease is expanding its geographic range: A challenge for Asian livestock management and food security. Vet. J. 2022, 279, 105785. [Google Scholar] [CrossRef] [PubMed]
- Chouhan, C.S.; Parvin, M.S.; Ali, M.Y.; Sadekuzzaman, M.; Chowdhury, M.G.A.; Ehsan, M.A.; Islam, M.T. Epidemiology and economic impact of lumpy skin disease of cattle in Mymensingh and Gaibandha districts of Bangladesh. Transbound. Emerg. Dis. 2022, 69, 3405–3418. [Google Scholar] [CrossRef] [PubMed]
- Sprygin, A.; Mazloum, A.; van Schalkwyk, A.; Babiuk, S. Capripoxviruses, leporipoxviruses, and orthopoxviruses: Occurrences of recombination. Front. Microbiol. 2022, 13, 978829. [Google Scholar] [CrossRef]
- Roy, P.; Jaisree, S.; Balakrishnan, S.; Senthilkumar, K.; Mahaprabhu, R.; Mishra, A.; Maity, B.; Ghosh, T.K.; Karmakar, A.P. Molecular epidemiology of goat pox viruses. Transbound. Emerg. Dis. 2018, 65, 32–36. [Google Scholar] [CrossRef]
- Sprygin, A.; Babin, Y.; Pestova, Y.; Kononova, S.; Wallace, D.B.; Van Schalkwyk, A.; Byadovskaya, O.; Diev, V.; Lozovoy, D.; Kononov, A. Analysis and insights into recombination signals in lumpy skin disease virus recovered in the field. PLoS ONE 2018, 13, e0207480. [Google Scholar] [CrossRef]
- Shumilova, I.; Sprygin, A.; Mazloum, A.; Pronin, V.; Byadovskaya, O.; Babiuk, S.; Donnik, I.; Chvala, I. Comparison of Gross Pathology Between Classical and Recombinant Lumpy Skin Disease Viruses. Viruses 2023, 15, 1883. [Google Scholar] [CrossRef]
- Adamu, K.; Abayneh, T.; Getachew, B.; Mohammed, H.; Deresse, G.; Zekarias, M.; Chala, W.; Gelaye, E. Lumpy skin disease virus isolation, experimental infection, and evaluation of disease development in a calf. Sci. Rep. 2024, 14, 20460. [Google Scholar] [CrossRef]
- Parvin, R.; Chowdhury, E.H.; Islam, M.T.; Begum, J.A.; Nooruzzaman, M.; Globig, A.; Dietze, K.; Hoffmann, B.; Tuppurainen, E. Clinical Epidemiology, Pathology, and Molecular Investigation of Lumpy Skin Disease Outbreaks in Bangladesh During 2020–2021 Indicate the Re-Emergence of an Old African Strain. Viruses 2022, 14, 2529. [Google Scholar] [CrossRef]
- Van Schalkwyk, A.; Kara, P.; Last, R.D.; Romito, M.; Wallace, D.B. Detection and Genome Sequencing of Lumpy Skin Disease Viruses in Wildlife Game Species in South Africa. Viruses 2024, 16, 172. [Google Scholar] [CrossRef]
- Dao, T.D.; Tran, L.H.; Nguyen, H.D.; Hoang, T.T.; Nguyen, G.H.; Tran, K.V.D.; Nguyen, H.X.; Van Dong, H.; Bui, A.N.; Bui, V.N. Characterization of lumpy skin disease virus isolated from a giraffe in Vietnam. Transbound. Emerg. Dis. 2022, 69, e3268–e3272. [Google Scholar] [CrossRef]
- Kumar, A.; Venkatesan, G.; Kushwaha, A.; Poulinlu, G.; Saha, T.; Ramakrishnan, M.A.; Dhar, P.; Kumar, G.S.; Singh, R.K. Genomic characterization of Lumpy Skin Disease virus (LSDV) from India: Circulation of Kenyan-like LSDV strains with unique kelch-like proteins. Acta Trop. 2023, 241, 106838. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, S.B.; Mishra, N.; Kalaiyarasu, S.; Ahirwar, K.; Chatterji, S.; Parihar, O.; Singh, V.P.; Sanyal, A. Lumpy Skin Disease Virus Infection in Free-Ranging Indian Gazelles (Gazella bennettii), Rajasthan, India. Emerg. Infect Dis. 2023, 29, 1407–1410. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, Y.; Zeng, Z.; Li, K.; Rehman, M.U.; Nawaz, S.; Kulyar, M.F.-E.-A.; Hu, M.; Zhang, W.; Zhang, Z.; An, M.; et al. Detection of Culex tritaeniorhynchus Giles and Novel recombinant strain of lumpy skin disease virus causes high mortality in yaks. Viruses 2023, 15, 880. [Google Scholar] [CrossRef] [PubMed]
- Porco, A.; Chea, S.; Sours, S.; Nou, V.; Groenenberg, M.; Agger, C.; Tum, S.; Chhuon, V.; Sorn, S.; Hong, C.; et al. Case report: Lumpy skin disease in an endangered wild banteng (Bos javanicus) and initiation of a vaccination campaign in domestic livestock in Cambodia. Front. Vet. Sci. 2023, 10, 1228505. [Google Scholar] [CrossRef]
- Ul-Rahman, A.; Shabbir, M.Z.; Raza, M.A.; Rossiter, P. The expanding host range of lumpy skin disease virus in wild and domestic animals. Trop. Anim. Health Prod. 2024, 56, 269. [Google Scholar] [CrossRef]
- Sprygin, A.; Pestova, Y.; Wallace, D.B.; Tuppurainen, E.; Kononov, A.V. Transmission of lumpy skin disease virus: A short review. Virus Res. 2019, 269, 197637. [Google Scholar] [CrossRef]
- Sohier, C.; Haegeman, A.; Mostin, L.; De Leeuw, I.; Campe, W.V.; De Vleeschauwer, A.; Tuppurainen, E.S.M.; van den Berg, T.; De Regge, N.; De Clercq, K. Experimental evidence of mechanical lumpy skin disease virus transmission by Stomoxys calcitrans biting flies and Haematopota spp. horseflies. Sci. Rep. 2019, 9, 20076. [Google Scholar] [CrossRef]
- Ali, S.; Ahmad, A.S.; Ashraf, K.; Khan, J.A.; Rashid, M.I. Insights into the involvement of male Hyalomma anatolicum ticks in transmitting Anaplasma marginale, lumpy skin disease virus and Theileria annulata. Trop. Anim. Health Prod. 2024, 56, 167. [Google Scholar] [CrossRef]
- Sprygin, A.; Van Schalkwyk, A.; Shumilova, I.; Nesterov, A.; Kononova, S.; Prutnikov, P.; Byadovskaya, O.; Kononov, A. Full-length genome characterization of a novel recombinant vaccine-like lumpy skin disease virus strain detected during the climatic winter in Russia, 2019. Arch. Virol. 2020, 165, 2675–2677. [Google Scholar] [CrossRef]
- Nesterov, A.; Mazloum, A.; Byadovskaya, O.; Shumilova, I.; Van Schalkwyk, A.; Krotova, A.; Kirpichenko, V.; Donnik, I.; Chvala, I.; Sprygin, A. Experimentally controlled study indicates that the naturally occurring recombinant vaccine-like lumpy skin disease strain Udmurtiya/2019, detected during freezing winter in northern latitudes, is transmitted via indirect contact. Front. Vet. Sci. 2022, 9, 1001426. [Google Scholar] [CrossRef]
- Shumilova, I.; Prutnikov, P.; Mazloum, A.; Krotova, A.; Tenitilov, N.; Byadovskaya, O.; Chvala, I.; Prokhvatilova, L.; Sprygin, A. Subclinical infection caused by a recombinant vaccine-like strain poses high risks of lumpy skin disease virus transmission. Front. Vet. Sci. 2024, 11, 1330657. [Google Scholar] [CrossRef]
- Mazloum, A.; Van Schalkwyk, A.; Babiuk, S.; Venter, E.; Wallace, D.B.; Sprygin, A. Lumpy skin disease: History, current understanding and research gaps in the context of recent geographic expansion. Front. Microbiol. 2023, 14, 1266759. [Google Scholar] [CrossRef]
- Van Borm, S.; Dellicour, S.; Martin, D.P.; Lemey, P.; Agianniotaki, E.I.; Chondrokouki, E.D.; Vidanovic, D.; Vaskovic, N.; Petroviċ, T.; Laziċ, S.; et al. Complete genome reconstruction of the global and European regional dispersal history of the lumpy skin disease virus. J. Virol. 2023, 97, e0139423. [Google Scholar] [CrossRef]
- Van Schalkwyk, A.; Kara, P.; Heath, L. Phylogenomic characterization of historic lumpy skin disease virus isolates from South Africa. Arch. Virol. 2022, 167, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- Krotova, A.; Byadovskaya, O.; Shumilova, I.; van Schalkwyk, A.; Sprygin, A. An in-depth bioinformatic analysis of the novel recombinant lumpy skin disease virus strains: From unique patterns to established lineage. BMC Genom. 2022, 23, 396. [Google Scholar] [CrossRef]
- Byadovskaya, O.; Prutnikov, P.; Shalina, K.; Babiuk, S.; Perevozchikova, N.; Korennoy, F.; Chvala, I.; Kononov, A.; Sprygin, A. The changing epidemiology of lumpy skin disease in Russia since the first introduction from 2015 to 2020. Transbound. Emerg. Dis. 2022, 69, e2551–e2562. [Google Scholar] [CrossRef]
- Tran, A.T.; Tran, H.T.T.; Truong, A.D.; Dinh, V.T.; Dang, A.K.; Chu, N.T.; Phan, L.; Phan, H.T.; Nguyen, H.T.; To, N.B.T.; et al. Molecular characterization of lumpy skin disease virus in North Central Vietnam during 2021 and early 2022. Vet. Ital. 2024, 60. [Google Scholar] [CrossRef]
- Bich, T.N.; Trung, L.Q.; Hieu, T.V.; Huyen, V.T.K.; Hieu, T.Q.; Giang, H.T.; Chien, N.T.P. Characterization and molecular identification of the lumpy skin disease virus in cattle in the Mekong Delta of Vietnam. Open Vet. J. 2024, 14, 1877–1895. [Google Scholar] [CrossRef]
- Manzoor, S.; Abubakar, M.; Ul-Rahman, A.; Syed, Z.; Ahmad, K.; Afzal, M. Molecular characterization of lumpy skin disease virus from recent outbreaks in Pakistan. Arch. Virol. 2023, 168, 297. [Google Scholar] [CrossRef]
- Wang, J.; Wan, S.; Liu, S.; Wang, Z.; Ding, X.; Wu, Q.; Liu, X.; Chen, Z.; Chen, L.; Wang, H.; et al. Prevalence of the novel recombinant LSDV in east and Southeast Asia: Inducing skin and testicular damage in golden hamsters. Microb. Pathog. 2024, 197, 107057. [Google Scholar] [CrossRef]
- World Organization for Animal Health (OIE). Lumpy Skin Disease Technical Disease Card. 2017. Available online: https://www.woah.org/app/uploads/2021/03/lumpy-skin-disease.pdf (accessed on 26 March 2023).
- Sprygin, A.; van Schalkwyk, A.; Mazloum, A.; Byadovskaya, O.; Chvala, I. Genome sequence characterization of the unique recombinant vaccine-like lumpy skin disease virus strain Kurgan/2018. Arch. Virol. 2024, 169, 23. [Google Scholar] [CrossRef] [PubMed]
- Breman, F.C.; Haegeman, A.; Krešić, N.; Philips, W.; De Regge, N. Lumpy Skin Disease Virus Genome Sequence Analysis: Putative Spatio-Temporal Epidemiology, Single Gene Versus Whole Genome Phylogeny and Genomic Evolution. Viruses 2023, 15, 1471. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Xie, J.; Chen, Y.; Cai, G.; Cai, R.; Hu, Z.; Wang, H. Tree Visualization by One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023, 51, W587–W592. [Google Scholar] [CrossRef]
- Gershon, P.D.; Kitching, R.P.; Hammond, J.M.; Black, D.N. Poxvirus genetic recombination during natural virus transmission. J. Gen. Virol. 1989, 70 Pt 2, 485–489. [Google Scholar] [CrossRef]
- Vandenbussche, F.; Mathijs, E.; Philips, W.; Saduakassova, M.; De Leeuw, I.; Sultanov, A.; Haegeman, A.; De Clercq, K. Recombinant LSDV Strains in Asia: Vaccine Spillover or Natural Emergence? Viruses 2022, 14, 1429. [Google Scholar] [CrossRef] [PubMed]
- Haegeman, A.; De Leeuw, I.; Saduakassova, M.; Van Campe, W.; Aerts, L.; Philips, W.; Sultanov, A.; Mostin, L.; De Clercq, K. The Importance of Quality Control of LSDV Live Attenuated Vaccines for Its Safe Application in the Field. Vaccines 2021, 9, 1019. [Google Scholar] [CrossRef] [PubMed]
- Sprygin, A.; Babin, Y.; Pestova, Y.; Kononova, S.; Byadovskaya, O.; Kononov, A. Complete Genome Sequence of the Lumpy Skin Disease Virus Recovered from the First Outbreak in the Northern Caucasus Region of Russia in 2015. Microbiol. Resour. Announc. 2019, 8, e01733-18. [Google Scholar] [CrossRef] [PubMed]
- Van Schalkwyk, A.; Byadovskaya, O.; Shumilova, I.; Wallace, D.B.; Sprygin, A. Estimating evolutionary changes between highly passaged and original parental lumpy skin disease virus strains. Transbound. Emerg. Dis. 2022, 69, e486–e496. [Google Scholar] [CrossRef]
- Tran, H.T.T.; Truong, A.D.; Dang, A.K.; Ly, D.V.; Nguyen, C.T.; Chu, N.T.; Hoang, T.V.; Nguyen, H.T.; Nguyen, V.T.; Dang, H.V. Lumpy skin disease outbreaks in Vietnam, 2020. Transbound. Emerg. Dis. 2021, 68, 977–980. [Google Scholar] [CrossRef]
- Arjkumpa, O.; Suwannaboon, M.; Boonrawd, M.; Punyawan, I.; Laobannu, P.; Yantaphan, S.; Bungwai, A.; Ponyium, V.; Suwankitwat, N.; Boonpornprasert, P.; et al. First emergence of lumpy skin disease in cattle in Thailand, 2021. Transbound. Emerg. Dis. 2021, 68, 3002–3004. [Google Scholar] [CrossRef]
- Ma, J.; Yuan, Y.; Shao, J.; Sun, M.; He, W.; Chen, J.; Liu, Q. Genomic characterization of lumpy skin disease virus in southern China. Transbound. Emerg. Dis. 2022, 69, 2788–2799. [Google Scholar] [CrossRef]
- Lu, G.; Xie, J.; Luo, J.; Shao, R.; Jia, K.; Li, S. Lumpy skin disease outbreaks in China, since 3 August 2019. Transbound. Emerg. Dis. 2021, 68, 216–219. [Google Scholar] [CrossRef]
- Morgenstern, M.; Klement, E. The Effect of Vaccination with Live Attenuated Neethling Lumpy Skin Disease Vaccine on Milk Production and Mortality—An Analysis of 77 Dairy Farms in Israel. Vaccines 2020, 8, 324. [Google Scholar] [CrossRef]
- Kononova, S.; Kononov, A.; Shumilova, I.; Byadovskaya, O.; Nesterov, A.; Prutnikov, P.; Babiuk, S.; Sprygin, A. A lumpy skin disease virus which underwent a recombination event demonstrates more aggressive growth in primary cells and cattle than the classical field isolate. Transbound. Emerg. Dis. 2021, 68, 1377–1383. [Google Scholar] [CrossRef]
- Shumilova, I.; Shalina, K.; Abed Alhussen, M.; Prutnikov, P.; Krotova, A.; Byadovskaya, O.; Prokhvatilova, L.; Chvala, I.; Sprygin, A. An Attenuated Vaccine Virus of the Neethling Lineage Protects Cattle against the Virulent Recombinant Vaccine-like Isolate of the Lumpy Skin Disease Virus Belonging to the Currently Established Cluster 2.5. Vaccines 2024, 12, 598. [Google Scholar] [CrossRef] [PubMed]
- Krotova, A.; Shalina, K.; Mazloum, A.; Kwon, D.; Van Schalkwyk, A.; Byadovskaya, O.; Sprygin, A. Genetic characterization of sheep pox virus strains from outbreaks in Central Russia in 2018–2019. Transbound. Emerg. Dis. 2022, 69, e3430–e3435. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, L.; Yang, J.; Shi, M.; Nie, F.; Liu, S.; Wang, Z.; Huang, D.; Wu, H.; Li, D.; et al. Analysis of vaccine-like lumpy skin disease virus from flies near the western border of China. Transbound. Emerg. Dis. 2022, 69, 1813–1823. [Google Scholar] [CrossRef] [PubMed]
- Shumilova, I.; Krotova, A.; Nesterov, A.; Byadovskaya, O.; van Schalkwyk, A.; Sprygin, A. Overwintering of recombinant lumpy skin disease virus in northern latitudes, Russia. Transbound. Emerg. Dis. 2022, 69, e3239–e3243. [Google Scholar] [CrossRef]
- Sudhakar, S.B.; Mishra, N.; Kalaiyarasu, S.; Jhade, S.K.; Singh, V.P. Genetic and phylogenetic analysis of lumpy skin disease viruses (LSDV) isolated from the first and subsequent field outbreaks in India during 2019 reveals close proximity with unique signatures of historical Kenyan NI-2490/Kenya/KSGP-like field strains. Transbound. Emerg. Dis. 2022, 69, e451–e462. [Google Scholar] [CrossRef]
- Ren, S.; Chen, H.; Yuan, L.; Yang, X.; Afera, T.B.; Rehman, Z.U.; Wang, H.; Wang, X.; Ma, C.; Lin, Y.; et al. Phylogenetic and pathogenic characterization of lumpy skin disease virus circulating in China. Virology 2023, 585, 127–138. [Google Scholar] [CrossRef]
- Gari, G.; Grosbois, V.; Waret-Szkuta, A.; Babiuk, S.; Jacquiet, P.; Roger, F. Lumpy skin disease in Ethiopia: Seroprevalence study across different agroclimate zones. Acta Trop. 2012, 123, 101–106. [Google Scholar] [CrossRef]
- Bianchini, J.; Simons, X.; Humblet, M.F.; Saegerman, C. Lumpy Skin Disease: A Systematic Review of Mode of Transmission, Risk of Emergence and Risk Entry Pathway. Viruses 2023, 15, 1622. [Google Scholar] [CrossRef]
- Sudhakar, S.B.; Mishra, N.; Kalaiyarasu, S.; Puri, R.; Ghule, P.; Agarwal, F.; Mustare, A.; Pawar, S.J.; Pathan, Y.K.; Sanyal, A. Evidence of natural lumpy skin disease virus (LSDV) infection and genetic characterization of LSDV strains from water buffaloes (Bubalus bubalis) in India. Arch. Virol. 2024, 170, 11. [Google Scholar] [CrossRef]
- Sprygin, A.; Mazloum, A.; Van Schalkwyk, A.; Krotova, A.; Bydovskaya, O.; Prokhvatilova, L.; Chvala, I. Development and application of a real-time polymerase chain reaction assay to detect lumpy skin disease virus belonging to the Kenyan sheep and goat pox group. BMC Res. Notes 2023, 16, 247. [Google Scholar] [CrossRef]
- Haegeman, A.; De Leeuw, I.; Philips, W.; De Regge, N. Development and Validation of a New DIVA Real-Time PCR Allowing to Differentiate Wild-Type Lumpy Skin Disease Virus Strains, Including the Asian Recombinant Strains, from Neethling-Based Vaccine Strains. Viruses 2023, 15, 870. [Google Scholar] [CrossRef] [PubMed]
- Philips, W.; Haegeman, A.; Krešić, N.; Mostin, L.; De Regge, N. Neethling Strain-Based Homologous Live Attenuated LSDV Vaccines Provide Protection Against Infection with a Clade 2.5 Recombinant LSDV Strain. Vaccines 2024, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Q.; Zhou, X.; Du, L.; Yang, N.; Lou, Y.; Liu, J.; Zhai, S. A Real-Time Recombinase Polymerase Amplification Assay for Specific Detection of Lumpy Skin Disease Virus. Vet. Sci. 2023, 10, 625. [Google Scholar] [CrossRef] [PubMed]
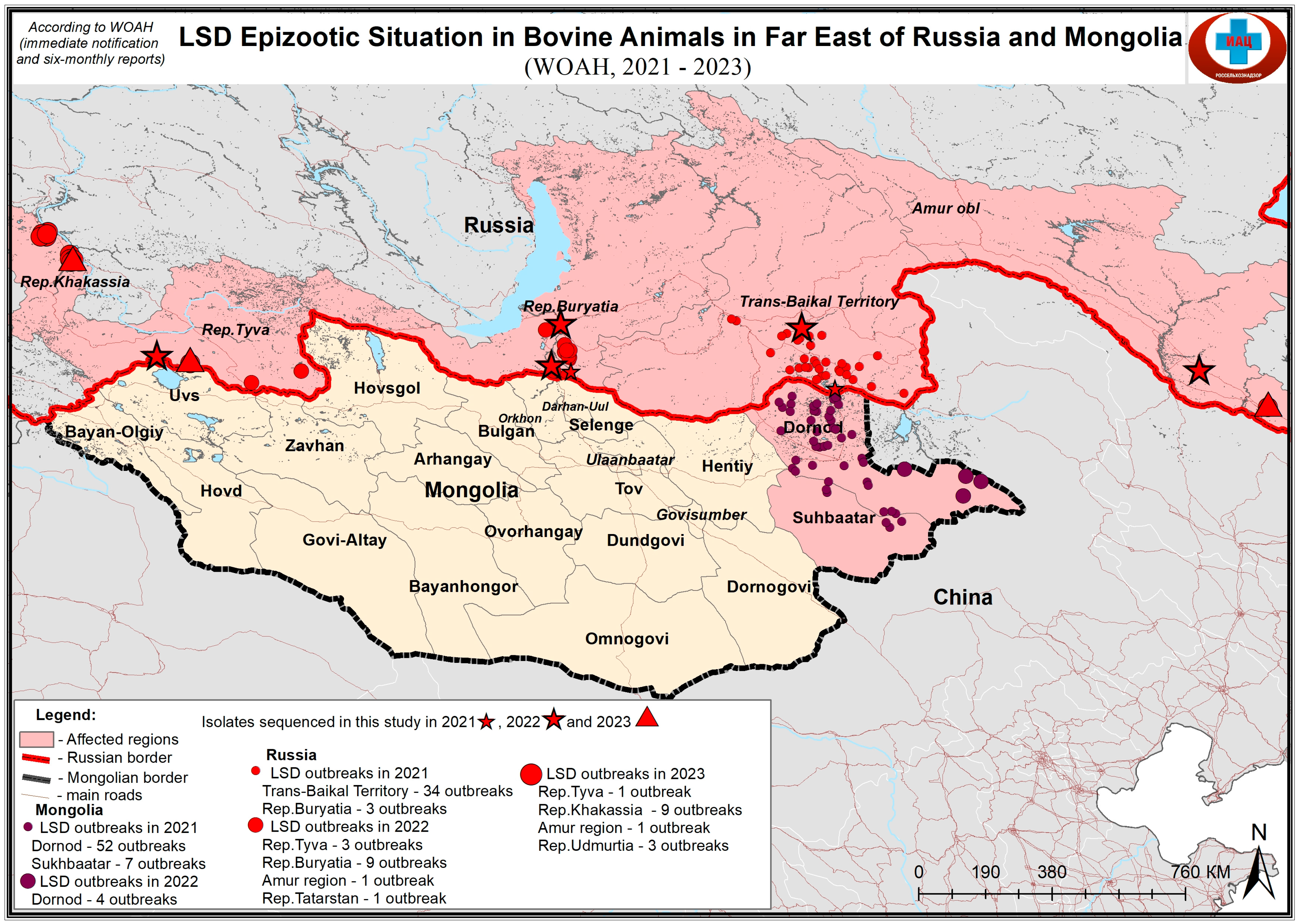
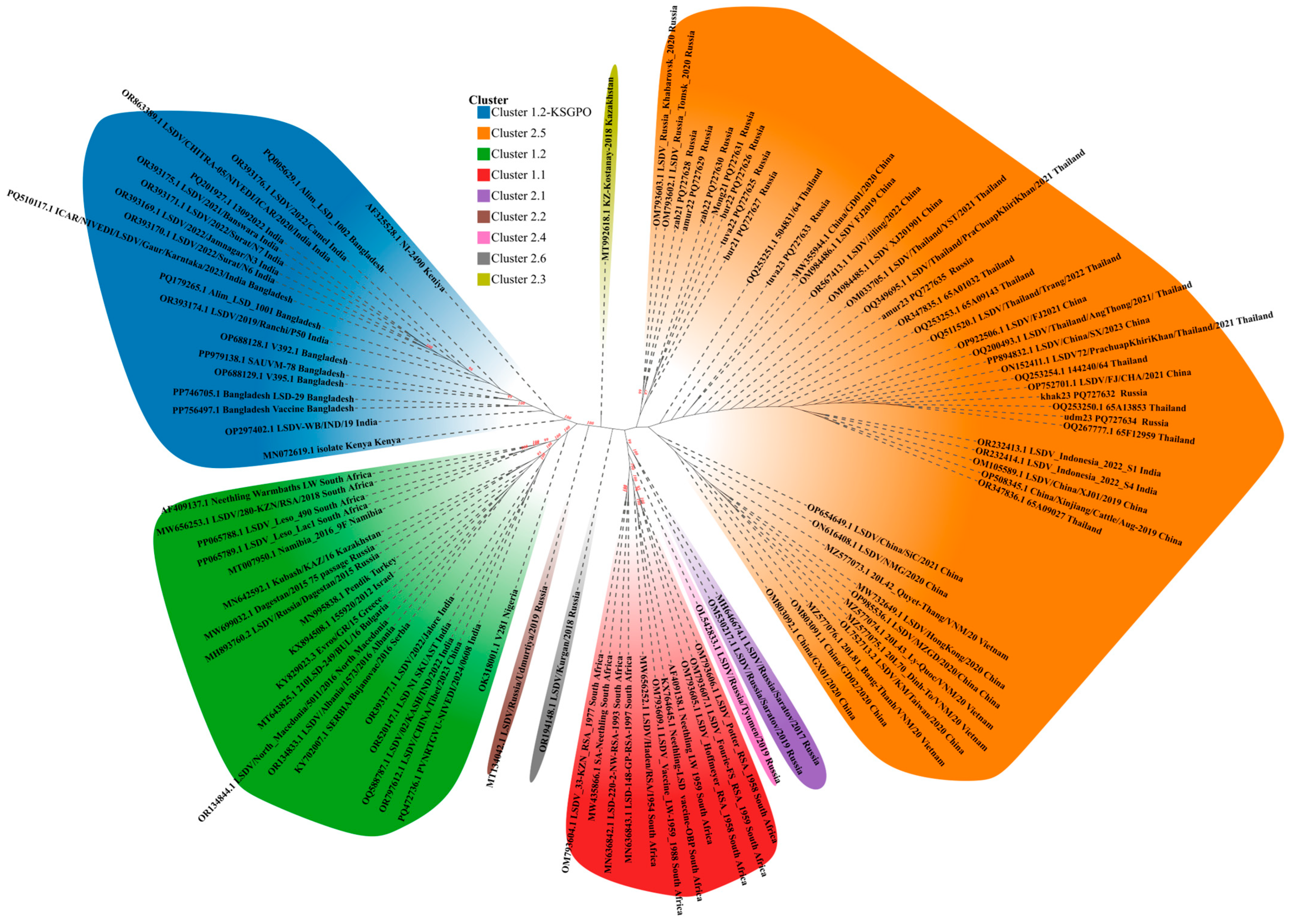

{kind=link}
{kind=link}
| Sample Name | Location | Year of Origin | Sample Type |
|---|---|---|---|
| tuva22 | Tuva republic, Russia | 2022 | Skin lesion |
| bur22 | Buryatiya republic, Russia | 2022 | Skin lesion |
| bur21 | Buryatiya republic, Russia | 2021 | Skin lesion |
| zab21 | Transbaykal territory, Russia | 2021 | Skin lesion |
| amur22 | Amur oblast, Russia | 2022 | Skin lesion |
| zab22 | Buryatiya republic, Russia | 2022 | Skin lesion |
| khak23 | Khakassia republic, Russia | 2023 | Skin lesion |
| tuva23 | Tuva republic, Russia | 2023 | Skin lesion |
| udm23 | Udmurtiya republic, Russia | 2023 | Skin lesion |
| amur23 | Amur oblast, Russia | 2023 | Skin lesion |
| Mong21 | Mongolia | 2021 | Skin lesion |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sprygin, A.; Krotova, A.; Jun, M.; Byadovskaya, O.; Kirpichenko, V.; Chen, J.; Sainnokhoi, T.; Chvala, I. Whole Genome Sequencing of Lumpy Skin Disease Virus from 2021–2023 in Eastern Eurasia Reveals No More Recombination Signals in the Circulating Pool of Strains. Viruses 2025, 17, 468. https://doi.org/10.3390/v17040468
Sprygin A, Krotova A, Jun M, Byadovskaya O, Kirpichenko V, Chen J, Sainnokhoi T, Chvala I. Whole Genome Sequencing of Lumpy Skin Disease Virus from 2021–2023 in Eastern Eurasia Reveals No More Recombination Signals in the Circulating Pool of Strains. Viruses. 2025; 17(4):468. https://doi.org/10.3390/v17040468
Chicago/Turabian StyleSprygin, Alexander, Alena Krotova, Ma Jun, Olga Byadovskaya, Vladimir Kirpichenko, Jinchao Chen, Tserenchimed Sainnokhoi, and Ilya Chvala. 2025. "Whole Genome Sequencing of Lumpy Skin Disease Virus from 2021–2023 in Eastern Eurasia Reveals No More Recombination Signals in the Circulating Pool of Strains" Viruses 17, no. 4: 468. https://doi.org/10.3390/v17040468
APA StyleSprygin, A., Krotova, A., Jun, M., Byadovskaya, O., Kirpichenko, V., Chen, J., Sainnokhoi, T., & Chvala, I. (2025). Whole Genome Sequencing of Lumpy Skin Disease Virus from 2021–2023 in Eastern Eurasia Reveals No More Recombination Signals in the Circulating Pool of Strains. Viruses, 17(4), 468. https://doi.org/10.3390/v17040468