
Comparative Interactome Profiling of Nonstructural Protein 3 Across SARS-CoV-2 Variants Emerged During the COVID-19 Pandemic

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Construct Design
2.2. Cell Culture and Transfection
2.3. FLAG IPs
2.4. Sample Preparation for TMT Label
2.5. MudPIT LC-MS/MS Analysis
2.6. Geneset Enrichment Analysis, Network Plots, and Comparative Heatmaps
3. Results
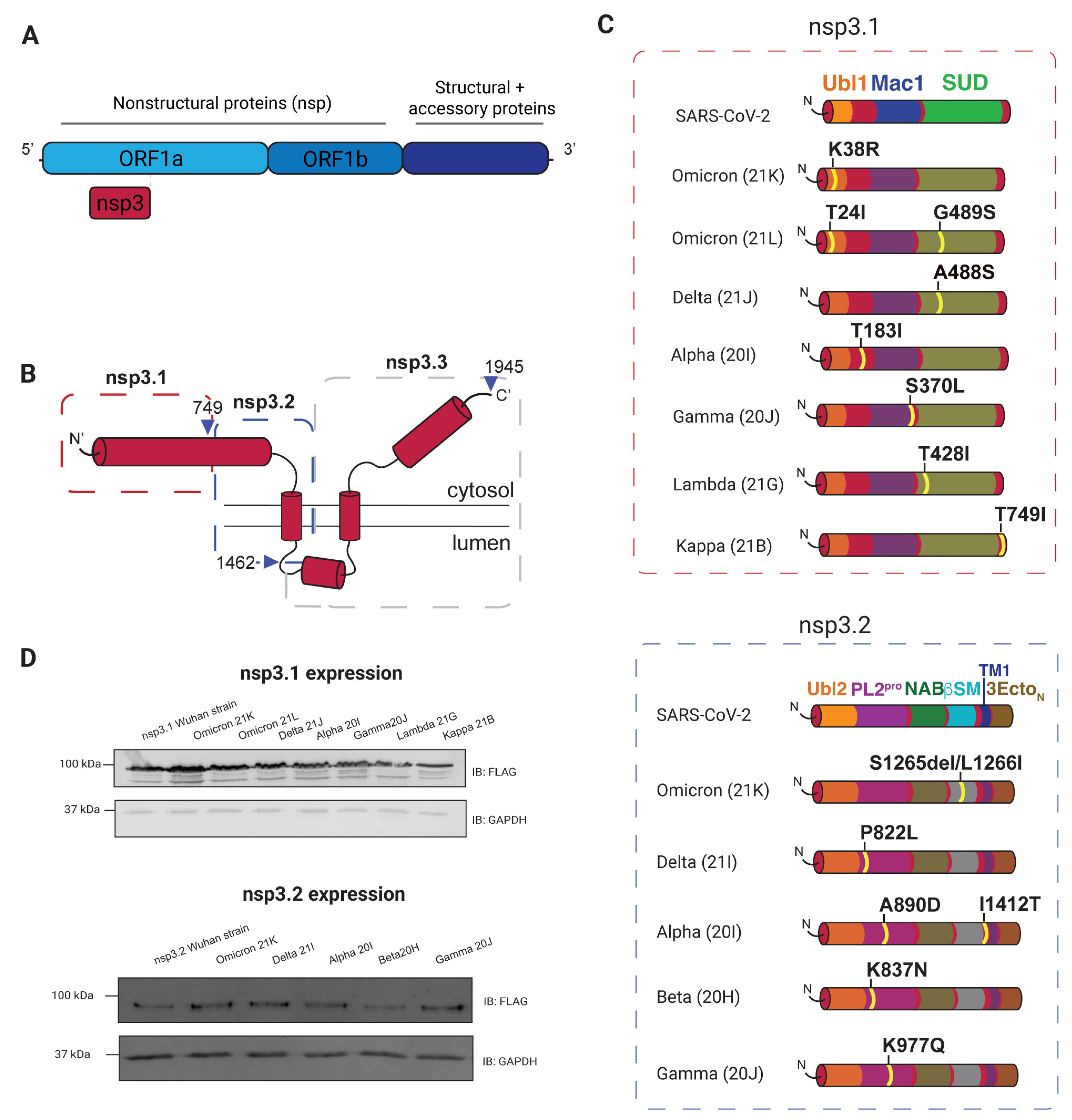
3.1. Selection and Expression of Variants
3.2. AP-MS of nsp3 Mutations
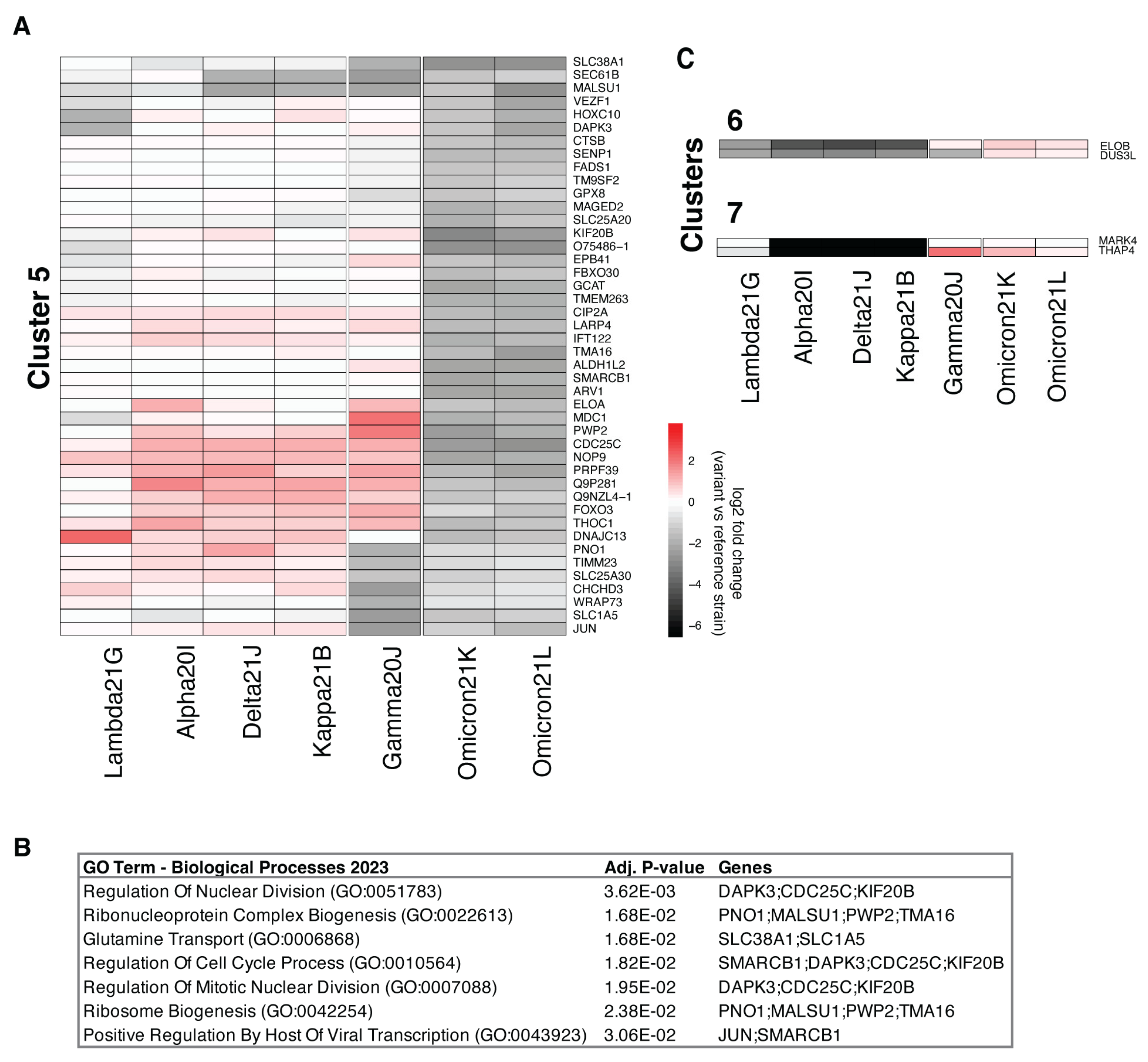
3.3. Comparison of nsp3.1 Interactors
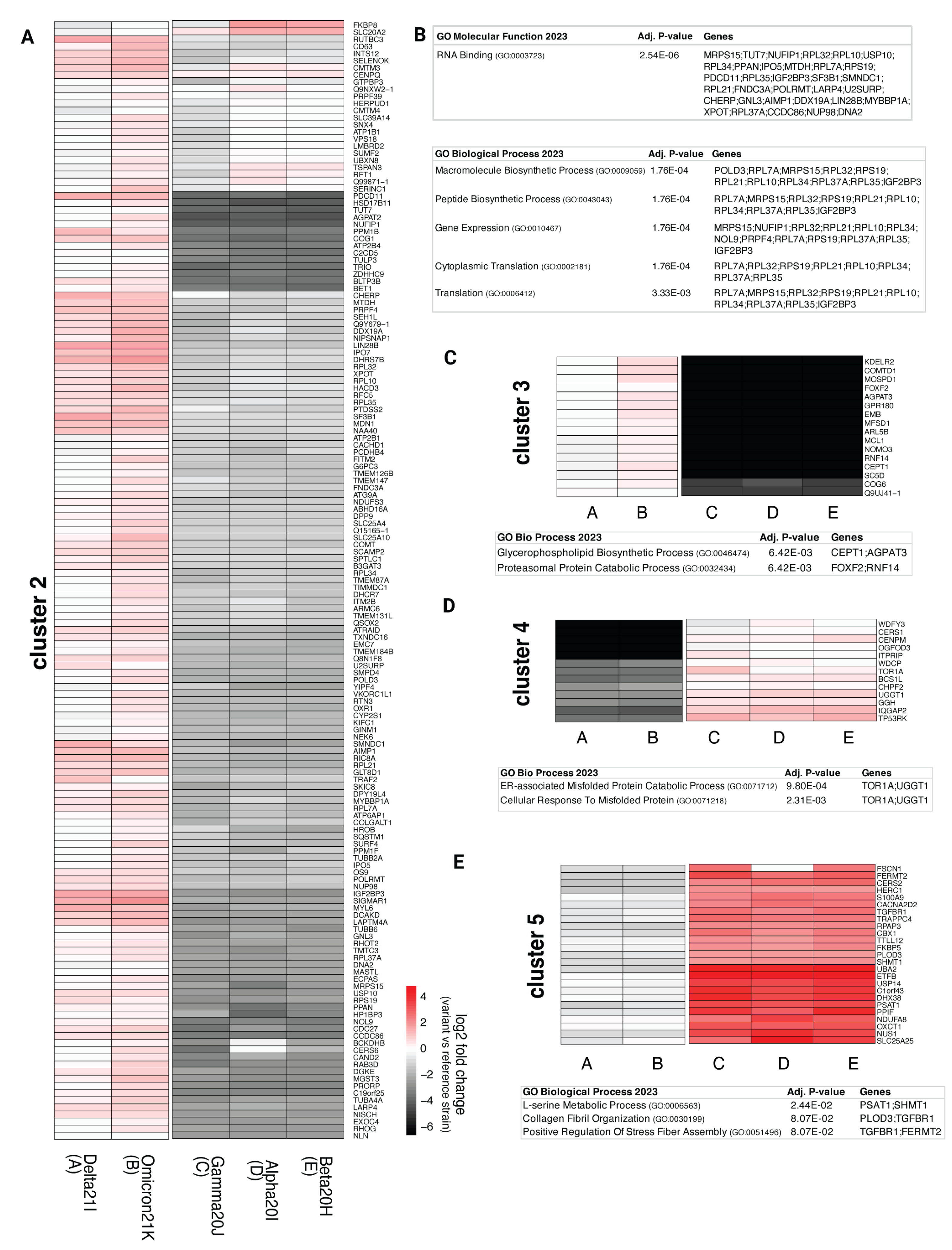
3.4. Comparison of nsp3.2 Interactors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Nsp3 | Nonstructural protein 3 |
| AP-MS | Affinity purification–mass spectrometry |
| IP | Immunoprecipitation |
| WHO | World Health Organization |
| TMTs | Tandem mass tags |
| Ubl1 | Ubiquitin-like domain |
| SUD | SARS-unique domain |
| Mac1 | Macrodomain |
| N protein | Nucleocapsid protein |
| PL2Pro | Papain-like Protease |
| βSM | Betacoronavirus-specific marker |
| FMRP | Fragile X-related protein |
| GO | Gene ontology |
| ERAD | Endoplasmic reticulum-associated degradation |
| MHV | Murine hepatitis virus |
| VOCs | Variants of concern |
| VOC-LUM | Variants of concern lineages under monitoring |
References
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced Sensitivity of SARS-CoV-2 Variant Delta to Antibody Neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.H.; Porrot, F.; Staropoli, I.; Lemoine, F.; et al. Considerable Escape of SARS-CoV-2 Omicron to Antibody Neutralization. Nature 2021, 602, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Krü, N.; Schulz, S.; Jä, H.-M.; Behrens, G.M.N.; Pö, S. The Omicron Variant Is Highly Resistant against Antibody-Mediated Neutralization: Implications for Control of the COVID-19 Pandemic. Cell 2022, 185, 447–456.e11. [Google Scholar] [CrossRef]
- Chatterjee, S.; Bhattacharya, M.; Nag, S.; Dhama, K.; Chakraborty, C. A Detailed Overview of SARS-CoV-2 Omicron: Its Sub-Variants, Mutations and Pathophysiology, Clinical Characteristics, Immunological Landscape, Immune Escape, and Therapies. Viruses 2023, 15, 167. [Google Scholar] [CrossRef]
- Hu, F.H.; Jia, Y.J.; Zhao, D.Y.; Fu, X.L.; Zhang, W.Q.; Tang, W.; Hu, S.Q.; Wu, H.; Ge, M.W.; Du, W.; et al. Clinical Outcomes of the Severe Acute Respiratory Syndrome Coronavirus 2 Omicron and Delta Variant: Systematic Review and Meta-Analysis of 33 Studies Covering 6 037 144 Coronavirus Disease 2019–Positive Patients. Clin. Microbiol. Infect. 2023, 29, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.R.; Graham, R.L.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses. RNA Biol. 2011, 120, 215–218. [Google Scholar] [CrossRef]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 79, 710. [Google Scholar] [CrossRef]
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R. The Coronavirus Proofreading Exoribonuclease Mediates Extensive Viral Recombination. PLoS Pathog. 2021, 17, e1009226. [Google Scholar] [CrossRef]
- Yi, H. 2019 Novel Coronavirus Is Undergoing Active Recombination. Clin. Infect. Dis. 2020, 71, 884–887. [Google Scholar] [CrossRef]
- Paraskevis, D.; Kostaki, E.G.; Magiorkinis, G.; Panayiotakopoulos, G.; Sourvinos, G.; Tsiodras, S. Full-Genome Evolutionary Analysis of the Novel Corona Virus (2019-NCoV) Rejects the Hypothesis of Emergence as a Result of a Recent Recombination Event. Infect. Genet. Evol. 2020, 79, 104212. [Google Scholar] [CrossRef] [PubMed]
- Perlman, S.; Zhao, J. Human Coronavirus EMC Is Not the Same as Severe Acute Respiratory Syndrome Coronavirus. MBio 2013, 4, e00002-13. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional Assessment of Cell Entry and Receptor Usage for SARS-CoV-2 and Other Lineage B Betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Snijder, E.J.; Bredenbeek, P.J.; Dobbe, J.C.; Thiel, V.; Ziebuhr, J.; Poon, L.L.M.; Guan, Y.; Rozanov, M.; Spaan, W.J.M.; Gorbalenya, A.E. Unique and Conserved Features of Genome and Proteome of SARS-Coronavirus, an Early Split-off From the Coronavirus Group 2 Lineage. J. Mol. Biol. 2003, 331, 991–1004. [Google Scholar] [CrossRef]
- Arya, R.; Kumari, S.; Pandey, B.; Mistry, H.; Bihani, S.C.; Das, A.; Prashar, V.; Gupta, G.D.; Panicker, L.; Kumar, M. Structural Insights into SARS-CoV-2 Proteins. J. Mol. Biol. 2021, 433, 166725. [Google Scholar] [CrossRef]
- Davies, J.P.; Almasy, K.M.; McDonald, E.F.; Plate, L. Comparative Multiplexed Interactomics of SARS-CoV-2 and Homologous Coronavirus Nonstructural Proteins Identifies Unique and Shared Host-Cell Dependencies. ACS Infect. Dis. 2020, 6, 3174–3189. [Google Scholar] [CrossRef]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel Proteomics Reveals Host Perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Gordon, D.E.; Hiatt, J.; Bouhaddou, M.; Rezelj, V.V.; Ulferts, S.; Braberg, H.; Jureka, A.S.; Obernier, K.; Guo, J.Z.; Batra, J.; et al. Comparative Host-Coronavirus Protein Interaction Networks Reveal Pan-Viral Disease Mechanisms. Science 2020, 370, eabe9403. [Google Scholar] [CrossRef]
- Shah, P.S.; Link, N.; Jang, G.M.; Sharp, P.P.; Zhu, T.; Swaney, D.L.; Johnson, J.R.; Von Dollen, J.; Ramage, H.R.; Satkamp, L.; et al. Comparative Flavivirus-Host Protein Interaction Mapping Reveals Mechanisms of Dengue and Zika Virus Pathogenesis. Cell 2018, 175, 1931–1945.e18. [Google Scholar] [CrossRef]
- Jäger, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K.; et al. Global Landscape of HIV–Human Protein Complexes. Nature 2011, 481, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Kandasamy, K.; Alvisi, G.; Mulhern, O.; Sacco, R.; Habjan, M.; Binder, M.; Stefanovic, A.; Eberle, C.A.; Goncalves, A.; et al. Viral Immune Modulators Perturb the Human Molecular Network by Common and Unique Strategies. Nature 2012, 487, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Almasy, K.M.; Davies, J.P.; Plate, L. Comparative Host Interactomes of the SARS-CoV-2 Nonstructural Protein 3 and Human Coronavirus Homologs. Mol. Cell. Proteom. 2021, 20, 100120. [Google Scholar] [CrossRef]
- Samavarchi-Tehrani, P.; Abdouni, H.; Knight, J.D.R.; Astori, A.; Samson, R.; Lin, Z.-Y.; Kim, D.-K.; Knapp, J.J.; St-Germain, J.; Go, C.D.; et al. A SARS-CoV-2—Host Proximity Interactome. bioRxiv 2020. [Google Scholar] [CrossRef]
- May, D.G.; Martin-Sancho, L.; Anschau, V.; Liu, S.; Chrisopulos, R.J.; Scott, K.L.; Halfmann, C.T.; Peña, R.D.; Pratt, D.; Campos, A.R.; et al. A BioID-Derived Proximity Interactome for SARS-CoV-2 Proteins. Viruses 2022, 14, 611. [Google Scholar] [CrossRef]
- Lindner, H.A.; Fotouhi-Ardakani, N.; Lytvyn, V.; Lachance, P.; Sulea, T.; Ménard, R. The Papain-Like Protease from the Severe Acute Respiratory Syndrome Coronavirus Is a Deubiquitinating Enzyme. J. Virol. 2005, 79, 15199–15208. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. Coronaviruses 2015, 1282, 1. [Google Scholar] [CrossRef]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of Coronaviruses: Structures and Functions of a Large Multidomain Protein. Antiviral. Res. 2018, 149, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Deng, J.; Han, L.; Zhuang, M.W.; Xu, Y.; Zhang, J.; Nan, M.L.; Xiao, Y.; Zhan, P.; Liu, X.; et al. SARS-CoV-2 NSP5 and N Protein Counteract the RIG-I Signaling Pathway by Suppressing the Formation of Stress Granules. Signal Transduct. Target. Ther. 2022, 7, 22. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like Protease Regulates SARS-CoV-2 Viral Spread and Innate Immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Ratia, K.; Kilianski, A.; Baez-Santos, Y.M.; Baker, S.C.; Mesecar, A. Structural Basis for the Ubiquitin-Linkage Specificity and DeISGylating Activity of SARS-CoV Papain-Like Protease. PLoS Pathog. 2014, 10, e1004113. [Google Scholar] [CrossRef]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar, A.D.; Baker, S.C. The Papain-like Protease of Severe Acute Respiratory Syndrome Coronavirus Has Deubiquitinating Activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef]
- Osipiuk, J.; Azizi, S.A.; Dvorkin, S.; Endres, M.; Jedrzejczak, R.; Jones, K.A.; Kang, S.; Kathayat, R.S.; Kim, Y.; Lisnyak, V.G.; et al. Structure of Papain-like Protease from SARS-CoV-2 and Its Complexes with Non-Covalent Inhibitors. Nat. Commun. 2021, 12, 743. [Google Scholar] [CrossRef]
- Wydorski, P.M.; Osipiuk, J.; Lanham, B.T.; Tesar, C.; Endres, M.; Engle, E.; Jedrzejczak, R.; Mullapudi, V.; Michalska, K.; Fidelis, K.; et al. Dual Domain Recognition Determines SARS-CoV-2 PLpro Selectivity for Human ISG15 and K48-Linked Di-Ubiquitin. Nat. Commun. 2023, 14, 2366. [Google Scholar] [CrossRef] [PubMed]
- Kusov, Y.; Tan, J.; Alvarez, E.; Enjuanes, L.; Hilgenfeld, R. A G-Quadruplex-Binding Macrodomain Within the “SARS-Unique Domain” Is Essential for the Activity of the SARS-Coronavirus Replication–Transcription Complex. Virology 2015, 484, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Kusov, Y.; Mutschall, D.; Tech, S.; Nagarajan, K.; Hilgenfeld, R.; Schmidt, C.L. The “SARS-Unique Domain” (SUD) of SARS Coronavirus Is an Oligo(G)-Binding Protein. Biochem. Biophys. Res. Commun. 2007, 364, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Vonrhein, C.; Smart, O.S.; Bricogne, G.; Bollati, M.; Kusov, Y.; Hansen, G.; Mesters, J.R.; Schmidt, C.L.; Hilgenfeld, R. The SARS-Unique Domain (SUD) of SARS Coronavirus Contains Two Macrodomains That Bind G-Quadruplexes. PLoS Pathog. 2009, 5, e1000428. [Google Scholar] [CrossRef]
- Wolff, G.; Limpens, R.W.A.L.; Zevenhoven-Dobbe, J.C.; Laugks, U.; Zheng, S.; de Jong, A.W.M.; Koning, R.I.; Agard, D.A.; Grünewald, K.; Koster, A.J.; et al. A Molecular Pore Spans the Double Membrane of the Coronavirus Replication Organelle. Science 2020, 369, 1395–1398. [Google Scholar] [CrossRef]
- Taha, T.Y.; Suryawanshi, R.K.; Chen, I.P.; Correy, G.J.; McCavitt-Malvido, M.; O’Leary, P.C.; Jogalekar, M.P.; Diolaiti, M.E.; Kimmerly, G.R.; Tsou, C.L.; et al. A Single Inactivating Amino Acid Change in the SARS-CoV-2 NSP3 Mac1 Domain Attenuates Viral Replication in Vivo. PLoS Pathog. 2023, 19, e1011614. [Google Scholar] [CrossRef]
- Alhammad, Y.M.; Parthasarathy, S.; Ghimire, R.; Kerr, C.M.; O’Connor, J.J.; Pfannenstiel, J.J.; Chanda, D.; Miller, C.A.; Baumlin, N.; Salathe, M.; et al. SARS-CoV-2 Mac1 Is Required for IFN Antagonism and Efficient Virus Replication in Cell Culture and in Mice. Proc. Natl. Acad. Sci. USA 2023, 120, e2302083120. [Google Scholar] [CrossRef]
- Emma, B. Hodcroft. CoVariants: SARS-CoV-2 Mutations and Variants of Interest. Available online: https://covariants.org/ (accessed on 19 February 2025).
- Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 19 February 2025).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations-SARS-CoV-2 Coronavirus/nCoV-2019 Genomic Epidemiology—Virological. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 13 February 2025).
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 Variant of Concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Mascola, J.R.; Graham, B.S.; Fauci, A.S. SARS-CoV-2 Viral Variants—Tackling a Moving Target. JAMA 2021, 325, 1261–1262. [Google Scholar] [CrossRef]
- Ong, S.W.X.; Chiew, C.J.; Ang, L.W.; Mak, T.M.; Cui, L.; Toh, M.P.H.S.; Lim, Y.D.; Lee, P.H.; Lee, T.H.; Chia, P.Y.; et al. Clinical and Virological Features of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Variants of Concern: A Retrospective Cohort Study Comparing B.1.1.7 (Alpha), B.1.351 (Beta), and B.1.617.2 (Delta). Clin. Infect. Dis. 2021, 75, e1128–e1136. [Google Scholar] [CrossRef]
- Classification of Omicron (B.1.1.529): SARS-CoV-2 Variant of Concern. Available online: https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern (accessed on 13 February 2025).
- Shuai, H.; Chan, J.F.W.; Hu, B.; Chai, Y.; Yuen, T.T.T.; Yin, F.; Huang, X.; Yoon, C.; Hu, J.C.; Liu, H.; et al. Attenuated Replication and Pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature 2022, 603, 693–699. [Google Scholar] [CrossRef]
- Sigal, A.; Milo, R.; Jassat, W. Estimating Disease Severity of Omicron and Delta SARS-CoV-2 Infections. Nat. Rev. Immunol. 2022, 22, 267–269. [Google Scholar] [CrossRef]
- Jiang, Y.; Tong, K.; Yao, R.; Zhou, Y.; Lin, H.; Du, L.; Jin, Y.; Cao, L.; Tan, J.; Zhang, X.D.; et al. Genome-Wide Analysis of Protein–Protein Interactions and Involvement of Viral Proteins in SARS-CoV-2 Replication. Cell Biosci 2021, 11, 140. [Google Scholar] [CrossRef]
- Fonslow, B.R.; Niessen, S.M.; Singh, M.; Wong, C.C.L.; Xu, T.; Carvalho, P.C.; Choi, J.; Park, S.K.; Yates, J.R. Single-Step Inline Hydroxyapatite Enrichment Facilitates Identification and Quantitation of Phosphopeptides from Mass-Limited Proteomes with MudPIT. J. Proteome. Res. 2012, 11, 2697–2709. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Serrano, P.; Johnson, M.A.; Almeida, M.S.; Horst, R.; Herrmann, T.; Joseph, J.S.; Neuman, B.W.; Subramanian, V.; Saikatendu, K.S.; Buchmeier, M.J.; et al. Nuclear Magnetic Resonance Structure of the N-Terminal Domain of Nonstructural Protein 3 from the Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2007, 81, 12049–12060. [Google Scholar] [CrossRef] [PubMed]
- Hurst, K.R.; Ye, R.; Goebel, S.J.; Jayaraman, P.; Masters, P.S. An Interaction between the Nucleocapsid Protein and a Component of the Replicase-Transcriptase Complex Is Crucial for the Infectivity of Coronavirus Genomic RNA. J. Virol. 2010, 84, 10276–10288. [Google Scholar] [CrossRef] [PubMed]
- Hurst, K.R.; Koetzner, C.A.; Masters, P.S. Characterization of a Critical Interaction between the Coronavirus Nucleocapsid Protein and Nonstructural Protein 3 of the Viral Replicase-Transcriptase Complex. J. Virol. 2013, 87, 9159–9172. [Google Scholar] [CrossRef]
- Eriksson, K.K.; Cervantes-Barragán, L.; Ludewig, B.; Thiel, V. Mouse Hepatitis Virus Liver Pathology Is Dependent on ADP-Ribose-1″-Phosphatase, a Viral Function Conserved in the Alpha-like Supergroup. J. Virol. 2008, 82, 12325–12334. [Google Scholar] [CrossRef] [PubMed]
- Kuri, T.; Eriksson, K.K.; Putics, A.; Züst, R.; Snijder, E.J.; Davidson, A.D.; Siddell, S.G.; Thiel, V.; Ziebuhr, J.; Weber, F. The ADP-Ribose-1″-Monophosphatase Domains of Severe Acute Respiratory Syndrome Coronavirus and Human Coronavirus 229E Mediate Resistance to Antiviral Interferon Responses. J. Gen. Virol. 2011, 92, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Channappanavar, R.; Jankevicius, G.; Fett, C.; Zhao, J.; Athmer, J.; Meyerholz, D.K.; Ahel, I.; Perlman, S. The Conserved Coronavirus Macrodomain Promotes Virulence and Suppresses the Innate Immune Response during Severe Acute Respiratory Syndrome Coronavirus Infection. MBio 2016, 7, e01721-16. [Google Scholar] [CrossRef]
- Alhammad, Y.M.O.; Kashipathy, M.M.; Roy, A.; Gagné, J.-P.; McDonald, P.; Gao, P.; Nonfoux, L.; Battaile, K.P.; Johnson, D.K.; Holmstrom, E.D.; et al. The SARS-CoV-2 Conserved Macrodomain Is a Mono-ADP-Ribosylhydrolase. J. Virol. 2021, 95, e01969-20. [Google Scholar] [CrossRef]
- Lei, J.; Ma-Lauer, Y.; Han, Y.; Thoms, M.; Buschauer, R.; Jores, J.; Thiel, V.; Beckmann, R.; Deng, W.; Leonhardt, H.; et al. The SARS-unique Domain (SUD) of SARS-CoV and SARS-CoV-2 Interacts with Human Paip1 to Enhance Viral RNA Translation. EMBO J. 2021, 40, e102277. [Google Scholar] [CrossRef]
- Qin, B.; Li, Z.; Tang, K.; Wang, T.; Xie, Y.; Aumonier, S.; Wang, M.; Yuan, S.; Cui, S. Identification of the SARS-Unique Domain of SARS-CoV-2 as an Antiviral Target. Nat. Commun. 2023, 14, 3999. [Google Scholar] [CrossRef]
- Harcourt, B.H.; Jukneliene, D.; Kanjanahaluethai, A.; Bechill, J.; Severson, K.M.; Smith, C.M.; Rota, P.A.; Baker, S.C. Identification of Severe Acute Respiratory Syndrome Coronavirus Replicase Products and Characterization of Papain-Like Protease Activity. J. Virol. 2004, 78, 13600–13612. [Google Scholar] [CrossRef]
- Bouhaddou, M.; Reuschl, A.K.; Polacco, B.J.; Thorne, L.G.; Ummadi, M.R.; Ye, C.; Rosales, R.; Pelin, A.; Batra, J.; Jang, G.M.; et al. SARS-CoV-2 Variants Evolve Convergent Strategies to Remodel the Host Response. Cell 2023, 186, 4597–4614.e26. [Google Scholar] [CrossRef]
- Watson, J.; Smith, M.; Francavilla, C.; Schwartz, J.M. SubcellulaRVis: A Web-Based Tool to Simplify and Visualise Subcellular Compartment Enrichment. Nucleic. Acids. Res. 2022, 50, W718–W725. [Google Scholar] [CrossRef]
- Siomi, M.C.; Zhang, Y.; Siomi, H.; Dreyfuss, G. Specific Sequences in the Fragile X Syndrome Protein FMR1 and the FXR Proteins Mediate Their Binding to 60S Ribosomal Subunits and the Interactions among Them. Mol. Cell Biol. 1996, 16, 3825–3832. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Wu, M.; He, Y.; Jiang, B.; He, M.L. Metabolic Alterations upon SARS-CoV-2 Infection and Potential Therapeutic Targets against Coronavirus Infection. Signal Transduct. Target. Ther. 2023, 8, 237. [Google Scholar] [CrossRef]
- Guizani, I.; Fourti, N.; Zidi, W.; Feki, M.; Allal-Elasmi, M. SARS-CoV-2 and Pathological Matrix Remodeling Mediators. Inflamm. Res. 2021, 70, 847–858. [Google Scholar] [CrossRef]
- Swain, J.; Merida, P.; Rubio, K.; Bracquemond, D.; Neyret, A.; Aguilar-Ordoñez, I.; Günther, S.; Barreto, G.; Muriaux, D. F-Actin Nanostructures Rearrangements and Regulation Are Essential for SARS-CoV-2 Particle Production in Host Pulmonary Cells. iScience 2023, 26, 107384. [Google Scholar] [CrossRef]
- Butt, A.A.; Dargham, S.R.; Tang, P.; Chemaitelly, H.; Hasan, M.R.; Coyle, P.V.; Kaleeckal, A.H.; Latif, A.N.; Loka, S.; Shaik, R.M.; et al. COVID-19 Disease Severity in Persons Infected with the Omicron Variant Compared with the Delta Variant in Qatar. J. Glob. Health 2022, 12, 05032. [Google Scholar] [CrossRef]
- Ao, D.; He, X.; Hong, W.; Wei, X. The Rapid Rise of SARS-CoV-2 Omicron Subvariants with Immune Evasion Properties: XBB.1.5 and BQ.1.1 Subvariants. MedComm 2023, 4, e239. [Google Scholar] [CrossRef]
- Zhu, K.; Sah, M.; Mahimainathan, L.; Liu, Y.; Xing, C.; Roush, K.; Clark, A.; SoRelle, J. Prospective Clinical Performance of CoVarScan in Identifying SARS-CoV-2 Omicron Subvariants. Microbiol. Spectr. 2025, 13, e01385-24. [Google Scholar] [CrossRef] [PubMed]
- Barrera, A.; Martínez-Valdebenito, C.; Angulo, J.; Palma, C.; Hormazábal, J.; Vial, C.; Aguilera, X.; Castillo-Torres, P.; Pardo-Roa, C.; Balcells, M.E.; et al. SARS-CoV-2 Infectivity and Antigenic Evasion: Spotlight on Isolated Omicron Sub-Lineages. Front Med. 2024, 11, 1414331. [Google Scholar] [CrossRef] [PubMed]
- Voth, L.S.; O’Connor, J.J.; Kerr, C.M.; Doerger, E.; Schwarting, N.; Sperstad, P.; Johnson, D.K.; Fehr, A.R. Unique Mutations in the Murine Hepatitis Virus Macrodomain Differentially Attenuate Virus Replication, Indicating Multiple Roles for the Macrodomain in Coronavirus Replication. J. Virol. 2021, 95, 766–787. [Google Scholar] [CrossRef]
- Schmidt, N.; Lareau, C.A.; Keshishian, H.; Ganskih, S.; Schneider, C.; Hennig, T.; Melanson, R.; Werner, S.; Wei, Y.; Zimmer, M.; et al. The SARS-CoV-2 RNA–Protein Interactome in Infected Human Cells. Nat. Microbiol. 2020, 6, 339–353. [Google Scholar] [CrossRef]
- Zhao, H.; Cai, Z.; Rao, J.; Wu, D.; Ji, L.; Ye, R.; Wang, D.; Chen, J.; Cao, C.; Hu, N.; et al. SARS-CoV-2 RNA Stabilizes Host MRNAs to Elicit Immunopathogenesis. Mol. Cell 2024, 84, 490–505.e9. [Google Scholar] [CrossRef]
- Nikolic, J.; Le Bars, R.; Lama, Z.; Scrima, N.; Lagaudrière-Gesbert, C.; Gaudin, Y.; Blondel, D. Negri Bodies Are Viral Factories with Properties of Liquid Organelles. Nat. Commun. 2017, 8, 58. [Google Scholar] [CrossRef]
- Peng, Q.; Wang, L.; Qin, Z.; Wang, J.; Zheng, X.; Wei, L.; Zhang, X.; Zhang, X.; Liu, C.; Li, Z.; et al. Phase Separation of Epstein-Barr Virus EBNA2 and Its Coactivator EBNALP Controls Gene Expression. J. Virol. 2020, 94, e01771-19. [Google Scholar] [CrossRef]
- Henninger, J.E.; Oksuz, O.; Shrinivas, K.; Sagi, I.; LeRoy, G.; Zheng, M.M.; Andrews, J.O.; Zamudio, A.V.; Lazaris, C.; Hannett, N.M.; et al. RNA-Mediated Feedback Control of Transcriptional Condensates. Cell 2021, 184, 207–225.e24. [Google Scholar] [CrossRef]
- Carlson, C.R.; Asfaha, J.B.; Ghent, C.M.; Howard, C.J.; Hartooni, N.; Safari, M.; Frankel, A.D.; Morgan, D.O. Phosphoregulation of Phase Separation by the SARS-CoV-2 N Protein Suggests a Biophysical Basis for Its Dual Functions. Mol. Cell 2020, 80, 1092–1103.e4. [Google Scholar] [CrossRef]
- Ke, Z.; Zhang, H.; Wang, Y.; Wang, J.; Peng, F.; Wang, J.; Liu, X.; Hu, H.; Li, Y. N Terminus of SARS-CoV-2 Nonstructural Protein 3 Interrupts RNA-Driven Phase Separation of N Protein by Displacing RNA. J. Biol. Chem. 2024, 300, 107828. [Google Scholar] [CrossRef]
- Garvanska, D.H.; Alvarado, R.E.; Mundt, F.O.; Lindqvist, R.; Duel, J.K.; Coscia, F.; Nilsson, E.; Lokugamage, K.; Johnson, B.A.; Plante, J.A.; et al. The NSP3 Protein of SARS-CoV-2 Binds Fragile X Mental Retardation Proteins to Disrupt UBAP2L Interactions. EMBO Rep. 2024, 25, 902–926. [Google Scholar] [CrossRef]
- Gessert, S.; Bugner, V.; Tecza, A.; Pinker, M.; Kühl, M. FMR1/FXR1 and the MiRNA Pathway Are Required for Eye and Neural Crest Development. Dev. Biol. 2010, 341, 222–235. [Google Scholar] [CrossRef]
- Qin, M.; Fan, W.; Li, L.; Xu, T.; Zhang, H.; Chen, F.; Man, J.; Kombe, A.J.K.; Zhang, J.; Shi, Y.; et al. PRMT1 and TDRD3 Promote Stress Granule Assembly by Rebuilding the Protein-RNA Interaction Network. Int. J. Biol. Macromol. 2024, 277, 134411. [Google Scholar] [CrossRef]
- Davies, J.P.; Plate, L. The Glycoprotein Quality Control Factor Malectin Promotes Coronavirus Replication and Viral Protein Biogenesis. Elife 2024, 13, RP100834. [Google Scholar] [CrossRef]
- Shi, R.; Feng, Z.; Zhang, X. Integrative Multi-Omics Landscape of Nonstructural Protein 3 of Severe Acute Respiratory Syndrome Coronaviruses. Genom. Proteom. Bioinform. 2021, 19, 707–726. [Google Scholar] [CrossRef]
- Wang, W.; Lusvarghi, S.; Subramanian, R.; Epsi, N.J.; Wang, R.; Goguet, E.; Fries, A.C.; Echegaray, F.; Vassell, R.; Coggins, S.A.; et al. Antigenic Cartography of Well-Characterized Human Sera Shows SARS-CoV-2 Neutralization Differences Based on Infection and Vaccination History. Cell Host Microbe 2022, 30, 1745–1758.e7. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia Lopez, V.; Plate, L. Comparative Interactome Profiling of Nonstructural Protein 3 Across SARS-CoV-2 Variants Emerged During the COVID-19 Pandemic. Viruses 2025, 17, 447. https://doi.org/10.3390/v17030447
Garcia Lopez V, Plate L. Comparative Interactome Profiling of Nonstructural Protein 3 Across SARS-CoV-2 Variants Emerged During the COVID-19 Pandemic. Viruses. 2025; 17(3):447. https://doi.org/10.3390/v17030447
Chicago/Turabian StyleGarcia Lopez, Valeria, and Lars Plate. 2025. "Comparative Interactome Profiling of Nonstructural Protein 3 Across SARS-CoV-2 Variants Emerged During the COVID-19 Pandemic" Viruses 17, no. 3: 447. https://doi.org/10.3390/v17030447
APA StyleGarcia Lopez, V., & Plate, L. (2025). Comparative Interactome Profiling of Nonstructural Protein 3 Across SARS-CoV-2 Variants Emerged During the COVID-19 Pandemic. Viruses, 17(3), 447. https://doi.org/10.3390/v17030447