
Surveillance and Genomic Evolution of Infectious Precocity Virus (IPV) from 2011 to 2024

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. RNA Sequencing Library Collection
2.2. Sources of IPV Variants
2.3. Phylogenetic Analyses
2.4. Genetic Variance Analyses
2.5. Selection Pressure Analysis
3. Results
3.1. IPV Variant Dataset
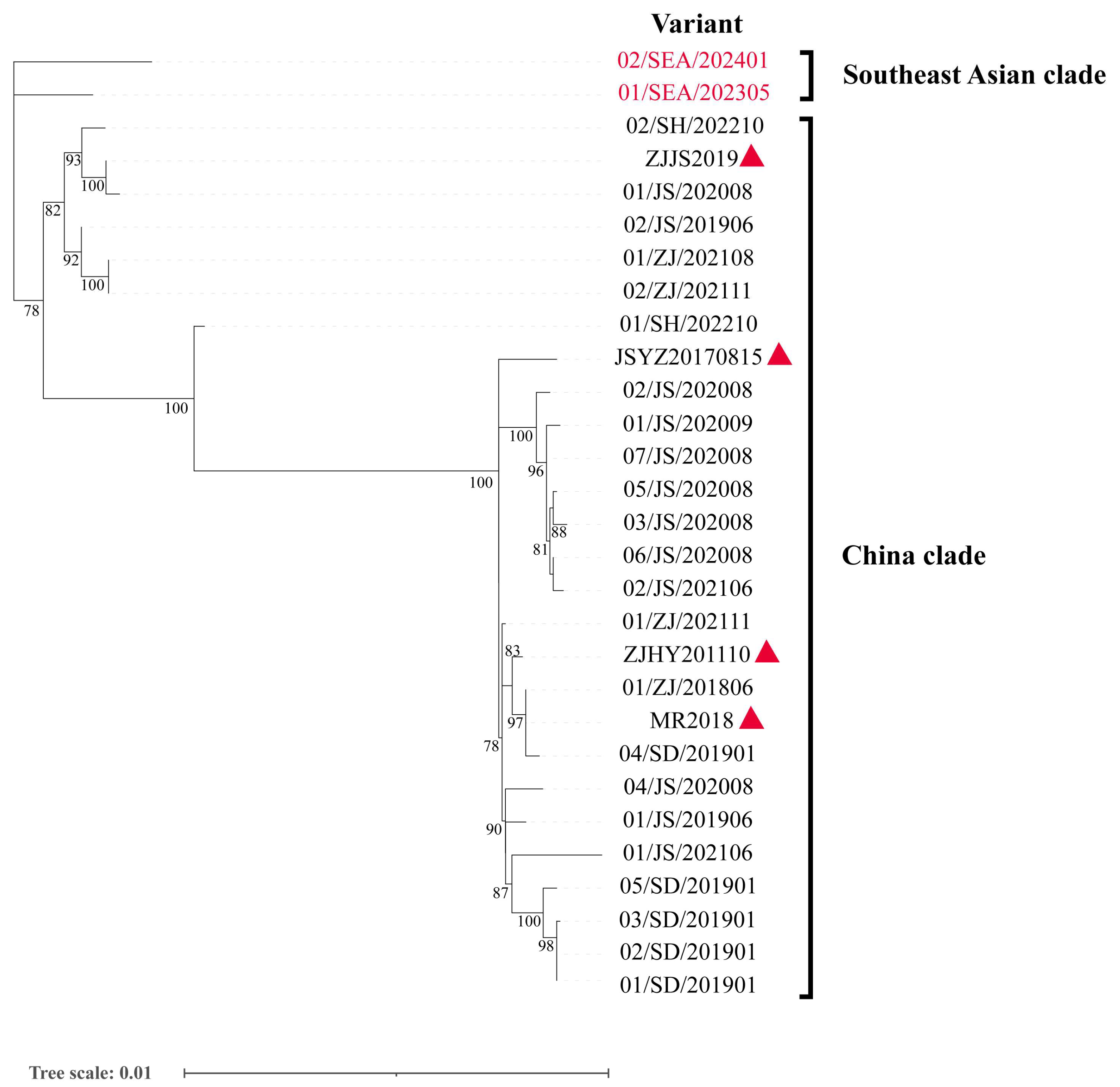
3.2. Phylogenetic Analyses in IPV Variants
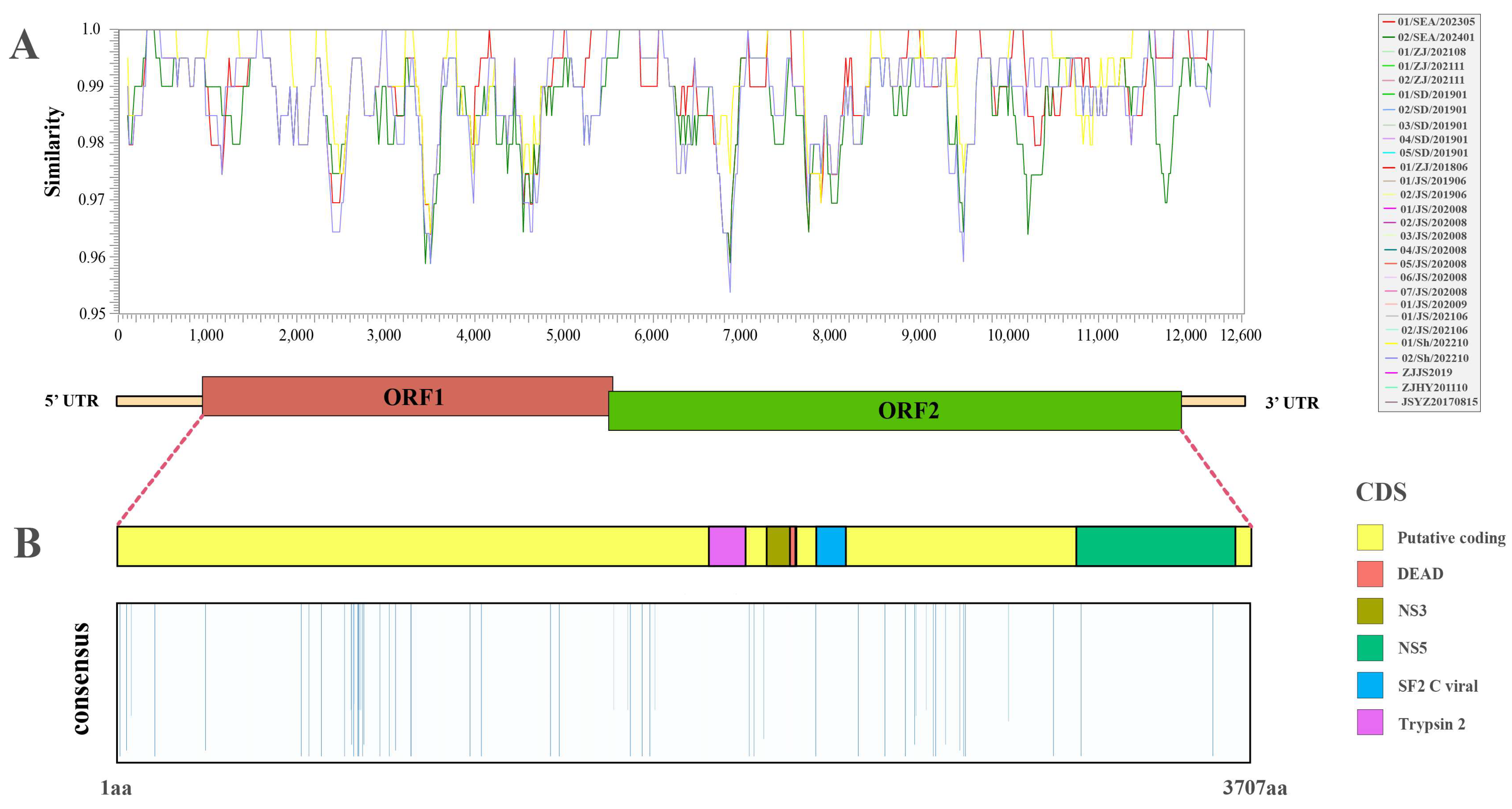
3.3. Genetic Variation in IPV Variants
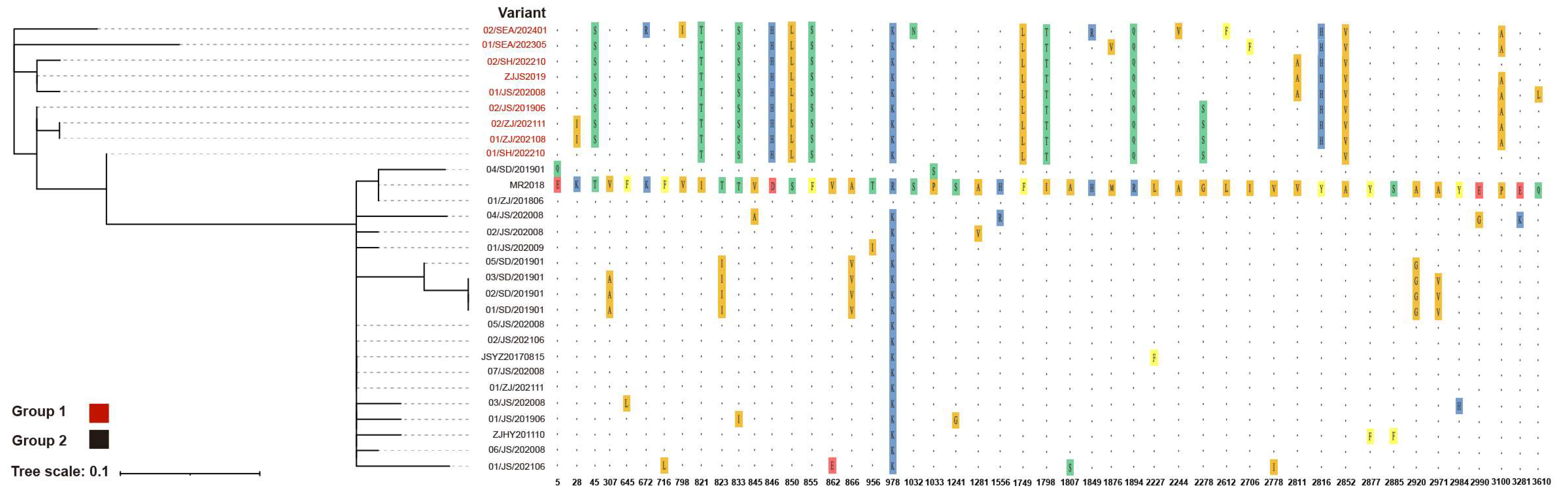
3.4. Amino Acid Polymorphism in IPV Variants
3.5. Selection Pressure in IPV Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Du, T.; Qi, H.; Lin, K.; Peng, X.; Gao, Q.; Yang, G.; Yi, S.; Tang, Q. Comprehensive evaluation of germplasm resources of nine Macrobrachium rosenbergii strains in China. Aquac. Rep. 2023, 33, 101755. [Google Scholar] [CrossRef]
- Alam, M.M.; Jørgensen, N.O.G.; Bass, D.; Santi, M.; Nielsen, M.; Rahman, M.A.; Hasan, N.A.; Bablee, A.L.; Bashar, A.; Hossain, M.I.; et al. Potential of integrated multitrophic aquaculture to make prawn farming sustainable in Bangladesh. Front. Sustain. Food Syst. 2024, 8, 1412919. [Google Scholar] [CrossRef]
- Ming, Y. Farming of giant freshwater prawn in China. World Aquac. 2014, 45, 48–51. [Google Scholar]
- Junming, Z.; Xilin, D.; Fei, J.; Fujiang, D. The preliminary analysis of the reasons for the poor growth of Macrobrachium rosenbergii in pond. J. Shanghai Ocean. Univ. 2017, 26, 853–861. [Google Scholar]
- Dong, X.; Wang, G.; Hu, T.; Li, J.; Li, C.; Cao, Z.; Shi, M.; Wang, Y.; Zou, P.; Song, J.; et al. A novel virus of flaviviridae associated with sexual precocity in Macrobrachium rosenbergii. mSystems 2021, 6, e0000321. [Google Scholar] [CrossRef]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV virus taxonomy profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef]
- Qin, X.C.; Shi, M.; Tian, J.H.; Lin, X.D.; Gao, D.Y.; He, J.R.; Wang, J.B.; Li, C.X.; Kang, Y.J.; Yu, B.; et al. A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors. Proc. Natl. Acad. Sci. USA 2014, 111, 6744–6749. [Google Scholar] [CrossRef]
- Wang, X.; Jing, X.; Shi, J.; Liu, Q.; Shen, S.; Cheung, P.P.-H.; Wu, J.; Deng, F.; Gong, P. A jingmenvirus RNA-dependent RNA polymerase structurally resembles the flavivirus counterpart but with different features at the initiation phase. Nucleic Acids Res. 2024, 52, 3278–3290. [Google Scholar] [CrossRef]
- Kobayashi, D.; Inoue, Y.; Suzuki, R.; Matsuda, M.; Shimoda, H.; Faizah, A.N.; Kaku, Y.; Ishijima, K.; Kuroda, Y.; Tatemoto, K.; et al. Identification and epidemiological study of an uncultured flavivirus from ticks using viral metagenomics and pseudoinfectious viral particles. Proc. Natl. Acad. Sci. USA 2024, 121, e2319400121. [Google Scholar] [CrossRef]
- Zhou, H.; Tian, R.R.; Wang, X.R.; Yang, J.X.; Wang, Y.X.; Zhao, M.L.; Zhang, X.D.; Ma, Y.H.; Lv, L.B.; Holmes, E.C.; et al. Identification of novel mammalian viruses in tree shrews (Tupaia belangeri chinensis). Zool. Res. 2024, 45, 429–438. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Vasilakis, N.; Tian, J.H.; Li, C.X.; Chen, L.J.; Eastwood, G.; Diao, X.N.; Chen, M.H.; Chen, X.; et al. Divergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the Flaviviridae and related viruses. J. Virol. 2016, 90, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Meng, F.; Zhou, C.; Li, J.; Hu, T.; Wang, Y.; Wang, G.; Luo, J.; Li, X.; Liu, S.; et al. Enormous diversity of RNA viruses in economic crustaceans. mSystems 2024, 9, e01016–e01024. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Y.; Dai, X. Transcriptomics-based analysis of Macrobrachium rosenbergii growth retardation. Comp. Biochem. Physiol. Part D Genom. Proteom. 2024, 52, 101298. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Li, X.; Sun, Y.; Hou, F.; Zhang, Y.; Li, F.; Gu, Z.; Liu, X. Insights into sexual precocity of female oriental river prawn Macrobrachium nipponense through transcriptome analysis. PLoS ONE 2016, 11, e0157173. [Google Scholar] [CrossRef]
- Xu, H.; Ren, J.; Xu, X.; Lou, B.; Zhang, D. Gut mcirobiomeis associated with hepatopancreatic and gonadal transcriptomes and together they impact on growth traits in M. rosenbergii. Preprints 2023. [Google Scholar] [CrossRef]
- Phonsiri, K.; Mavichak, R.; Panserat, S.; Boonanuntanasarn, S. Differential responses of hepatopancreas transcriptome between fast and slow growth in giant freshwater prawns (Macrobrachium rosenbergii) fed a plant-based diet. Sci. Rep. 2024, 14, 4957. [Google Scholar] [CrossRef]
- Suwansa-ard, S.; Thongbuakaew, T.; Wang, T.; Zhao, M.; Elizur, A.; Hanna, P.J.; Sretarugsa, P.; Cummins, S.F.; Sobhon, P. In silico neuropeptidome of female Macrobrachium rosenbergii based on transcriptome and peptide mining of eyestalk, central nervous system and ovary. PLoS ONE 2015, 10, e0123848. [Google Scholar] [CrossRef]
- Baliarsingh, S.; Chung, J.M.; Sahoo, S.; Sarkar, A.; Mohanty, J.; Han, Y.; Lee, Y.S.; Patnaik, B.B. Transcriptome analysis of Macrobrachium rosenbergii hepatopancreas in response to vibrio harveyi infection. Aquac. Res. 2020, 52, 1855–1875. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Zhou, L.; Feng, T.; Xu, S.; Gao, F.; Lam, T.T.; Wang, Q.; Wu, T.; Huang, H.; Zhan, L.; Li, L.; et al. ggmsa: A visual exploration tool for multiple sequence alignment and associated data. Brief. Bioinform. 2022, 23, bbac222. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer International Publishing: Basingstoke, UK, 2016. [Google Scholar]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef]
- Zhou, D.; Liu, S.; Guo, G.; He, X.; Xing, C.; Miao, Q.; Chen, G.; Chen, X.; Yan, H.; Zeng, J.; et al. Virome analysis of normal and growth retardation disease-affected Macrobrachium rosenbergii. Microbiol. Spectr. 2022, 10, e0146222. [Google Scholar] [CrossRef]
- Wang, G.; Guo, X.; Huang, X.; Wang, D.; Chen, Y.; Qin, J.; Yang, G.; Tang, K.F.J.; Dong, X.; Huang, J. The quantitative characteristics of infection with infectious precocity virus (IPV) revealed with a new TaqMan probe-based real-time RT-PCR method. Aquaculture 2023, 566, 739179. [Google Scholar] [CrossRef]
- Zhao, C.; Miu, Q.; Liu, S.; Zhou, D.; He, X.; Pang, J.; Weng, S.; He, J. Detection methods, epidemiological investigation, and host ranges of infectious precocity virus (IPV). Aquaculture 2023, 562, 738818. [Google Scholar] [CrossRef]
- Chiang, Y.-R.; Lu, Y.-Y.; Lin, H.-Y. Quantitative analysis of infectious precocity virus load and stunted growth syndrome via an immunohistochemical assay and reverse transcriptase quantitative real-time PCR in Macrobrachium rosenbergii. Aquaculture 2024, 583, 740577. [Google Scholar] [CrossRef]
- Xu, W.f.; Zhao, C.; Pan, X.; He, X.; Liu, X.; Hou, J.; Pang, J. Transmission routes and a new freshwater crustacean host for infectious precocity virus (IPV). Transbound. Emerg. Dis. 2023, 2023, 3950107. [Google Scholar] [CrossRef]
- Wang, G.; Guo, X.; Zhou, C.; Lou, H.; Li, X.; Yao, J.; Dong, X.; Yang, G.; Huang, J. Coinfection with infectious precocity virus and decapod iridescent virus 1 in farmed giant freshwater prawn (Macrobrachium rosenbergii). Aquaculture 2024, 586, 740830. [Google Scholar] [CrossRef]
- Ying, N.; Wang, Y.; Song, X.; Qin, B.; Wu, Y.; Yang, L.; Fang, W. Transcriptome analysis of Macrobrachium rosenbergii: Identification of precocious puberty and slow-growing information. J. Invertebr. Pathol. 2022, 190, 107752. [Google Scholar] [CrossRef]
- Li, X.-L.; Shen, P.-J.; Jiang, W.-P.; Meng, J.-L.; Cheng, H.-H.; Gao, Q. Metabonomic analysis of Macrobrachium rosenbergii with iron prawn syndrome (IPS). Fishes 2023, 8, 196. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Variant | GenBank Accession | Length | Virus Reads | FPKM | Library ID | Time | Location | Group |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Infectious precocity virus 01/SEA/202305 | PQ786402 | 12,630 | 633 | 9.57 × 102 | 20230518002 | 202305 | Southeast Asia | Previously Collected Samples |
| 2 | Infectious precocity virus 02/SEA/202401 | PQ786403 | 12,630 | 113,059 | 3.00 × 105 | 20240131030 | 202401 | Southeast Asia | Previously Collected Samples |
| 3 | Infectious precocity virus 01/ZJ/202108 | PP215336 | 12,519 | 10,280 | 1.14 × 104 | 20210828001 | 202108 | Zhejiang Province, China | Previously Collected Samples |
| 4 | Infectious precocity virus 01/ZJ/202111 | PP215337 | 12,608 | 1,798,970 | 3.87 × 106 | 20211103001 | 202108 | Zhejiang Province, China | Previously Collected Samples |
| 5 | Infectious precocity virus 02/ZJ/202111 | PP215338 | 12,519 | 1,112,410 | 2.30 × 106 | 20211103002 | 202108 | Zhejiang Province, China | Previously Collected Samples |
| 6 | Infectious precocity virus 01/SD/201901 | PP215321 | 12,550 | 24,730 | 3.97 × 104 | 20190123003-X | 201901 | Shandong Province, China | Previously Collected Samples |
| 7 | Infectious precocity virus 02/SD/201901 | PP215322 | 12,559 | 24,078 | 4.74 × 101 | 20190123006-O | 201901 | Shandong Province, China | Previously Collected Samples |
| 8 | Infectious precocity virus 03/SD/201901 | PP215323 | 12,528 | 3788 | 6.40 × 103 | 20190123006-X | 201901 | Shandong Province, China | Previously Collected Samples |
| 9 | Infectious precocity virus 04/SD/201901 | PP215324 | 12,556 | 14,978 | 2.86 × 104 | 20190123009-O | 201901 | Shandong Province, China | Previously Collected Samples |
| 10 | Infectious precocity virus 05/SD/201901 | PP215325 | 12,535 | 285,626 | 6.26 × 105 | 20190123011-O | 201901 | Shandong Province, China | Previously Collected Samples |
| 11 | Infectious precocity virus 01/ZJ/201806 | PP054173 | 12,288 | 649,714 | 4.09 × 106 | A | 201806 | Jiangsu Province, China | Previously Collected Samples |
| 12 | Infectious precocity virus 01/JS/201906 | PP215326 | 12,585 | 436,014 | 8.40 × 105 | 20190602H1H2H3 | 201906 | Jiangsu Province, China | Previously Collected Samples |
| 13 | Infectious precocity virus 02/JS/201906 | PP215328 | 12,589 | 64,182 | 2.39 × 105 | 20190602S3S4 | 201906 | Jiangsu Province, China | Previously Collected Samples |
| 14 | Infectious precocity virus 01/JS/202008 | PP215327 | 12,589 | 267,378 | 8.55 × 105 | 20200821007 | 202008 | Jiangsu Province, China | Previously Collected Samples |
| 15 | Infectious precocity virus 02/JS/202008 | PP215329 | 12,554 | 71,914 | 1.60 × 105 | 20200821008 | 202008 | Jiangsu Province, China | Previously Collected Samples |
| 16 | Infectious precocity virus 03/JS/202008 | PP215330 | 12,592 | 519,744 | 1.17 × 106 | 20200821010 | 202008 | Jiangsu Province, China | Previously Collected Samples |
| 17 | Infectious precocity virus 04/JS/202008 | PP215331 | 12,588 | 162,836 | 3.75 × 105 | 20200821011 | 202008 | Jiangsu Province, China | Previously Collected Samples |
| 18 | Infectious precocity virus 05/JS/202008 | PP215332 | 12,537 | 69,390 | 1.39 × 105 | 20200821-tie10 | 202008 | Jiangsu Province, China | Previously Collected Samples |
| 19 | Infectious precocity virus 06/JS/202008 | PP054190 | 12,522 | 1872 | 3.93 × 103 | 20200828005 | 202008 | Jiangsu Province, China | Previously Collected Samples |
| 20 | Infectious precocity virus 07/JS/202008 | PP054191 | 12,565 | 89,740 | 2.40 × 105 | 20200828006 | 202008 | Jiangsu Province, China | Previously Collected Samples |
| 21 | Infectious precocity virus 01/JS/202009 | PP215333 | 12,544 | 3678 | 8.59 × 103 | 20200929004 | 202009 | Jiangsu Province, China | Previously Collected Samples |
| 22 | Infectious precocity virus 01/JS/202106 | PP215334 | 12,582 | 272,778 | 6.12 × 105 | 20210626-T1 | 202106 | Jiangsu Province, China | Previously Collected Samples |
| 23 | Infectious precocity virus 02/JS/202106 | PP215335 | 12,598 | 501,334 | 1.16 × 106 | 20210626-T2 | 202106 | Jiangsu Province, China | Previously Collected Samples |
| 24 | Infectious precocity virus 01/SH/202210 | PQ786404 | 12,630 | 237 | 4.18 × 102 | J5_fastqc | 202210 | Shanghai, China | Public Database Samples |
| 25 | Infectious precocity virus 02/SH/202210 | PQ786405 | 12,630 | 758 | 1.20 × 103 | J4_fastqc | 202210 | Shanghai, China | Public Database Samples |
| 26 | Infectious precocity virus ZJJS2019 | ON382579.1 | 12,594 | NA | NA | NA | 201908 | Zhejiang Province, China | Public Database Samples |
| 27 | Macrobrachium flavivirus 1 ZJHY201110 | MT648663.1 | 12,628 | NA | NA | NA | 201110 | Zhejiang Province, China | Public Database Samples |
| 28 | Macrobrachium flavivirus 1 JSYZ20170815 | MT648664.1 | 12,628 | NA | NA | NA | 201708 | Jiangsu Province, China | Public Database Samples |
| 29 | Infectious precocity virus MR2018 | MT084113.1 | 12,630 | NA | NA | NA | 2018 | China | Public Database Samples |
| 30 | Infectious precocity virus 01/JS/201806 | PP210859 | 8789 | 608 | 8.03 × 10−1 | T4 | 201806 | Jiangsu Province, China | Previously Collected Samples |
| 31 | Infectious precocity virus 02/JS/201806 | PP210860 | 3515 | 572 | 8.85 × 10−1 | T3 | 201806 | Jiangsu Province, China | Previously Collected Samples |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, C.; Wang, G.; Zhou, Q.; Meng, F.; Liu, S.; Huang, J.; Dong, X. Surveillance and Genomic Evolution of Infectious Precocity Virus (IPV) from 2011 to 2024. Viruses 2025, 17, 425. https://doi.org/10.3390/v17030425
Zhou C, Wang G, Zhou Q, Meng F, Liu S, Huang J, Dong X. Surveillance and Genomic Evolution of Infectious Precocity Virus (IPV) from 2011 to 2024. Viruses. 2025; 17(3):425. https://doi.org/10.3390/v17030425
Chicago/Turabian StyleZhou, Chengyan, Guohao Wang, Qingqing Zhou, Fanzeng Meng, Shufang Liu, Jie Huang, and Xuan Dong. 2025. "Surveillance and Genomic Evolution of Infectious Precocity Virus (IPV) from 2011 to 2024" Viruses 17, no. 3: 425. https://doi.org/10.3390/v17030425
APA StyleZhou, C., Wang, G., Zhou, Q., Meng, F., Liu, S., Huang, J., & Dong, X. (2025). Surveillance and Genomic Evolution of Infectious Precocity Virus (IPV) from 2011 to 2024. Viruses, 17(3), 425. https://doi.org/10.3390/v17030425