
Genetic Variations of Three Kazakhstan Strains of the SARS-CoV-2 Virus

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. RNA Extraction
2.3. cDNA Synthesis
2.4. Primer Design and Synthesis
2.5. Polymerase Chain Reaction (PCR) Setup
2.6. Determination of Nucleotide Sequences
2.7. Lineage Determination and Mutation Identification of the Studied Isolates
2.8. Analysis of Non-Synonymous Mutation Function
2.9. Phylogenetic Analysis of Nucleotide Sequences
3. Results
3.1. PCR Amplification of SARS-CoV-2 Virus Strains
3.2. Characteristics of the Genomes of the Studied SARS-CoV-2 Virus Strains
3.3. Impact of Mutations on Biological Function of Proteins in the Studied SARS-CoV-2 Samples
3.4. Phylogenetic Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shereen, M.A.; Khan, S.; Kazmi, A.; Bashir, N.; Siddique, R. COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020, 24, 91–98. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. COVID-19 Dashboard. Available online: https://covid19.who.int/ (accessed on 6 March 2025).
- Zhugunissov, K.; Zakarya, K.; Khairullin, B.; Orynbayev, M.; Abduraimov, Y.; Kassenov, M.; Sultankulova, K.; Kerimbayev, A.; Nurabayev, S.; Myrzakhmetova, B.; et al. Development of the Inactivated QazCovid-in Vaccine: Protective Efficacy of the Vaccine in Syrian Hamsters. Front. Microbiol. 2021, 12, 720437. [Google Scholar] [CrossRef] [PubMed]
- Usserbayev, B.; Zakarya, K.; Kutumbetov, L.; Orynbayev, M.; Sultankulova, K.; Abduraimov, Y.; Myrzakhmetova, B.; Zhugunissov, K.; Kerimbayev, A.; Melisbek, A.; et al. Near-complete genome sequence of a SARS-CoV-2 variant B. 1.1. 7 virus strain isolated in Kazakhstan. Microbiol. Resour. Announc. 2022, 11, e0061922. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Cosar, B.; Karagulleoglu, Z.Y.; Unal, S.; Ince, A.T.; Uncuoglu, D.B.; Tuncer, G.; Kilinc, B.R.; Ozkan, Y.E.; Ozkoc, H.C.; Demir, I.N.; et al. SARS-CoV-2 Mutations and their Viral Variants. Cytokine Growth Factor. Rev. 2022, 63, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Safari, I.; Elahi, E. Evolution of the SARS-CoV-2 genome and emergence of variants of concern. Arch. Virol. 2022, 167, 293–305. [Google Scholar] [CrossRef]
- Andre, M.; Lau, L.S.; Pokharel, M.D.; Ramelow, J.; Owens, F.; Souchak, J.; Akkaoui, J.; Ales, E.; Brown, H.; Shil, R.; et al. From alpha to omicron: How different variants of concern of the SARS-Coronavirus-2 impacted the world. Biology 2023, 12, 1267. [Google Scholar] [CrossRef]
- Bhardwaj, P.; Mishra, S.K.; Behera, S.P.; Zaman, K.; Kant, R.; Singh, R. Genomic evolution of the SARS-CoV-2 Variants of Concern: COVID-19 pandemic waves in India. EXCLI J. 2023, 22, 451–465. [Google Scholar]
- Vidanović, D.; Tešović, B.; Volkening, J.D.; Afonso, C.L.; Quick, J.; Šekler, M.; Knežević, A.; Janković, M.; Jovanović, T.; Petrović, T.; et al. First whole-genome analysis of the novel coronavirus (SARS-CoV-2) obtained from COVID-19 patients from five districts in Western Serbia. Epidemiol Infect 2021, 149, e246. [Google Scholar] [CrossRef]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef]
- LaTourrette, K.; Garcia-Ruiz, H. Determinants of Virus Variation, Evolution, and Host Adaptation. Pathogens 2022, 11, 1039. [Google Scholar] [CrossRef]
- Márquez, S.; Prado-Vivar, B.; Guadalupe, J.J.; Gutierrez, B.; Jibaja, M.; Tobar, M.; Mora, F.; Gaviria, J.; García, M.; Espinosa, F.; et al. Genome sequencing of the first SARS-CoV-2 reported from patients with COVID-19 in Ecuador. medRxiv 2020. [Google Scholar] [CrossRef]
- Mohammadi, E.; Shafiee, F.; Shahzamani, K.; Ranjbar, M.M.; Alibakhshi, A.; Ahangarzadeh, S.; Beikmohammadi, L.; Shariati, L.; Hooshmandi, S.; Ataei, B.; et al. Novel and emerging mutations of SARS-CoV-2: Biomedical implications. Biomed. Pharmacother. 2021, 139, 111599. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information (NCBI). SARS-CoV-2 Reference Genome. Available online: https://www.ncbi.nlm.nih.gov/nuccore/NC_045512.2 (accessed on 6 March 2025).
- National Center for Biotechnology Information (NCBI). BLAST: Basic Local Alignment Search Tool. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 6 March 2025).
- Burashev, Y. Primers for Whole Genome Sequencing of the Sars-Cov-2 Virus. Zenodo. 2022. Available online: https://zenodo.org/records/7264509 (accessed on 6 March 2025).
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 genome annotator. Available online: http://giorgilab.unibo.it/coronannotator/ (accessed on 6 March 2025).
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA 11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef]
- Burashev, Y.; Usserbayev, B.; Kutumbetov, L.; Abduraimov, Y.; Kassenov, M.; Kerimbayev, A.; Myrzakhmetova, B.; Melisbek, A.; Shirinbekov, M.; Khaidarov, S.; et al. Coding Complete Genome Sequence of the SARS-CoV-2 Virus Strain, Variant B.1.1, Sampled from Kazakhstan. Microbiol. Resour. Announc. 2022, 11, e0111422. [Google Scholar] [CrossRef]
- Usserbayev, B.; Abduraimov, Y.; Kozhabergenov, N.; Melisbek, A.; Shirinbekov, M.; Smagul, M.; Nusupbayeva, G.; Nakhanov, A.; Burashev, Y. Complete Coding Sequence of a Lineage AY.122 SARS-CoV-2 Virus Strain Detected in Kazakhstan. Microbiol. Resour. Announc. 2023, 12, e0030123. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.A.K.; Medina, P.M.B. Temporal changes in the accessory protein mutations of SARS-CoV-2 variants and their predicted structural and functional effects. J. Med. Virol. 2022, 94, 5189–5200. [Google Scholar] [CrossRef] [PubMed]
- Cleaveland, S.; Laurenson, M.K.; Taylor, L.H. Diseases of humans and their domestic mammals: Pathogen characteristics, host range and the risk of emergence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Padhan, K.; Parvez, M.K.; Al-Dosari, M.S. Comparative sequence analysis of SARS-CoV-2 suggests its high transmissibility and pathogenicity. Future Virol. 2021, 16, 245–254. [Google Scholar] [CrossRef]
- Parvez, M.K.; Parveen, S. Evolution and Emergence of Pathogenic Viruses: Past, Present, and Future. Intervirology 2017, 60, 1–7. [Google Scholar] [CrossRef]
- Lee, S.H. A Routine Sanger Sequencing Target Specific Mutation Assay for SARS-CoV-2 Variants of Concern and Interest. Viruses 2021, 13, 2386. [Google Scholar] [CrossRef]
- Li, X.; Wang, W.; Zhao, X.; Zai, J.; Zhao, Q.; Li, Y.; Chaillon, A. Transmission dynamics and evolutionary history of 2019-nCoV. J Med Virol 2020, 92, 501–511. [Google Scholar] [CrossRef]
- Li, Y.; Lai, D.Y.; Zhang, H.N.; Jiang, H.W.; Tian, X.; Ma, M.L.; Qi, H.; Meng, Q.F.; Guo, S.J.; Wu, Y.; et al. Linear epitopes of SARS-CoV-2 spike protein elicit neutralizing antibodies in COVID-19 patients. Cell Mol. Immunol. 2020, 17, 1095–1097. [Google Scholar] [CrossRef]
- Abbasian, M.H.; Mahmanzar, M.; Rahimian, K.; Mahdavi, B.; Tokhanbigli, S.; Moradi, B.; Sisakht, M.M.; Deng, Y. Global landscape of SARS-CoV-2 mutations and conserved regions. J. Transl. Med. 2023, 21, 152. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Esman, A.; Dubodelov, D.; Khafizov, K.; Kotov, I.; Roev, G.; Golubeva, A.; Gasanov, G.; Korabelnikova, M.; Turashev, A.; Cherkashin, E.; et al. Development and Application of Real-Time PCR-Based Screening for Identification of Omicron SARS-CoV-2 Variant Sublineages. Genes 2023, 14, 1218. [Google Scholar] [CrossRef]
- Singh, L.; San, J.E.; Tegally, H.; Brzoska, P.M.; Anyaneji, U.J.; Wilkinson, E.; Clark, L.; Giandhari, J.; Pillay, S.; Lessells, R.J.; et al. Targeted Sanger sequencing to recover key mutations in SARS-CoV-2 variant genome assemblies produced by next-generation sequencing. Microb. Genom. 2022, 8, 000774. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Genomic Sequencing of SARS-CoV-2: A Guide to Implementation for Maximum Impact on Public Health; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Supasa, P.; Zhou, D.; Dejnirattisai, W.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Nutalai, R.; Tuekprakhon, A.; et al. Reduced neutralization of SARS-CoV-2 B.1.1.7 variant by convalescent and vaccine sera. Cell 2021, 184, 2201–2211.e7. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, L.; Chen, S.; Ji, W.; Li, C.; Ren, L. Recent progress on the mutations of SARS-CoV-2 spike protein and suggestions for prevention and controlling of the pandemic. Infect. Genet. Evol. 2021, 93, 104971. [Google Scholar] [CrossRef]
- Majumdar, P.; Niyogi, S. SARS-CoV-2 mutations: The biological trackway towards viral fitness. Epidemiol. Infect. 2021, 149, e110. [Google Scholar] [CrossRef]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.T.M.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef]
- Weng, S.; Zhou, H.; Ji, C.; Li, L.; Han, N.; Yang, R.; Shang, J.; Wu, A. Conserved Pattern and Potential Role of Recurrent Deletions in SARS-CoV-2 Evolution. Microbiol. Spectr. 2022, 10, e0219121. [Google Scholar] [CrossRef]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science 2021, 371, 1139–1142. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.; Pearson, C.A.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and severity of novel SARS-CoV-2 variant of concern 202012/01 in England. medRxiv. 2020. [Google Scholar] [CrossRef]
- Liu, H.; Yuan, M.; Huang, D.; Bangaru, S.; Zhao, F.; Lee, C.D.; Peng, L.; Barman, S.; Zhu, X.; Nemazee, D.; et al. A combination of cross-neutralizing antibodies synergizes to prevent SARS-CoV-2 and SARS-CoV pseudovirus infection. Cell Host Microbe 2021, 29, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Khetran, S.R.; Mustafa, R. Mutations of SARS-CoV-2 Structural Proteins in the Alpha, Beta, Gamma, and Delta Variants: Bioinformatics Analysis. JMIR Bioinform. Biotech. 2023, 4, e43906. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Lubinski, B.; Fernandes, M.H.V.; Frazier, L.; Tang, T.; Daniel, S.; Diel, D.G.; Jaimes, J.A.; Whittaker, G.R. Functional evaluation of the P681H mutation on the proteolytic activation the SARS-CoV-2 variant B.1.1.7 (Alpha) spike. bioRxiv 2021. [Google Scholar] [CrossRef]
- SARS-CoV-2 Lineage Tree. Available online: https://observablehq.com/embed/6475ff63fc3ebfb3 (accessed on 6 March 2025).
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; Du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- O’Toole, Á.; Pybus, O.G.; Abram, M.E.; Kelly, E.J.; Rambaut, A. Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences. BMC Genom. 2022, 23, 121. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, M.; Sharma, A.; Priyanka Thakur, N.; Rajkhowa, T.K.; Choudhary, O.P. Delta variant (B.1.617.2) of SARS-CoV-2: Mutations, impact, challenges and possible solutions. Hum. Vaccin. Immunother. 2022, 18, 2068883. [Google Scholar] [CrossRef]
- Rahman, F.I.; Ether, S.A.; Islam, M.R. The “Delta Plus” COVID-19 variant has evolved to become the next potential variant of concern: Mutation history and measures of prevention. J. Basic. Clin. Physiol. Pharmacol. 2021, 33, 109–112. [Google Scholar] [CrossRef]
- Kumar, S.; Thambiraja, T.S.; Karuppanan, K.; Subramaniam, G. Omicron and Delta variant of SARS-CoV-2: A comparative computational study of spike protein. J. Med. Virol. 2022, 94, 1641–1649. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Mlcochova, P.; Kemp, S.A.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.A.T.M.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S.; et al. SARS-CoV-2 B.1.617.2 delta variant replication and immune evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Abavisani, M.; Rahimian, K.; Mahdavi, B.; Tokhanbigli, S.; Mollapour Siasakht, M.; Farhadi, A.; Kodori, M.; Mahmanzar, M.; Meshkat, Z. Mutations in SARS-CoV-2 structural proteins: A global analysis. Virol. J. 2022, 19, 220. [Google Scholar] [CrossRef]
- Periwal, N.; Rathod, S.B.; Sarma, S.; Johar, G.S.; Jain, A.; Barnwal, R.P.; Srivastava, K.R.; Kaur, B.; Arora, P.; Sood, V. Time Series Analysis of SARS-CoV-2 Genomes and Correlations among Highly Prevalent Mutations. Microbiol. Spectr. 2022, 10, e0121922. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]
- Hossain, M.S.; Pathan, A.Q.M.S.U.; Islam, M.N.; Tonmoy, M.I.Q.; Rakib, M.I.; Munim, M.A.; Saha, O.; Fariha, A.; Reza, H.A.; Roy, M.; et al. Genome-wide identification and prediction of SARS-CoV-2 mutations show an abundance of variants: Integrated study of bioinformatics and deep neural learning. Inform. Med. Unlocked 2021, 27, 100798. [Google Scholar] [CrossRef]
- Subissi, L.; Imbert, I.; Ferron, F.; Collet, A.; Coutard, B.; Decroly, E.; Canard, B. SARS-CoV ORF1b-encoded nonstructural proteins 12-16: Replicative enzymes as antiviral targets. Antiviral Res. 2014, 101, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Haddad, D.; John, S.E.; Mohammad, A.; Hammad, M.M.; Hebbar, P.; Channanath, A.; Nizam, R.; Al-Qabandi, S.; Al Madhoun, A.; Alshukry, A.; et al. SARS-CoV-2: Possible recombination and emergence of potentially more virulent strains. PLoS ONE 2021, 16, e0251368. [Google Scholar] [CrossRef]
- Archana, A.; Long, C.; Chandran, K. Analysis of SARS-CoV-2 amino acid mutations in New York City Metropolitan wastewater (2020–2022) reveals multiple traits with human health implications across the genome and environment-specific distinctions. medRxiv 2022. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef]
- Ferreira, I.A.T.M.; Kemp, S.A.; Datir, R.; Saito, A.; Meng, B.; Rakshit, P.; Takaori-Kondo, A.; Kosugi, Y.; Uriu, K.; Kimura, I.; et al. SARS-CoV-2 B.1.617 Mutations L452R and E484Q Are Not Synergistic for Antibody Evasion. J. Infect. Dis. 2021, 224, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M. New ‘Delta Plus’ Variant of SARS-CoV-2 Identified; Here’s What We Know So Far. India Toda. 2021. Available online: https://www.indiatoday.in/coronavirus-outbreak/story/delta-plus-variant-covid-corona-coronavirus-sarscov2-1814768-2021-06-14 (accessed on 6 March 2025).

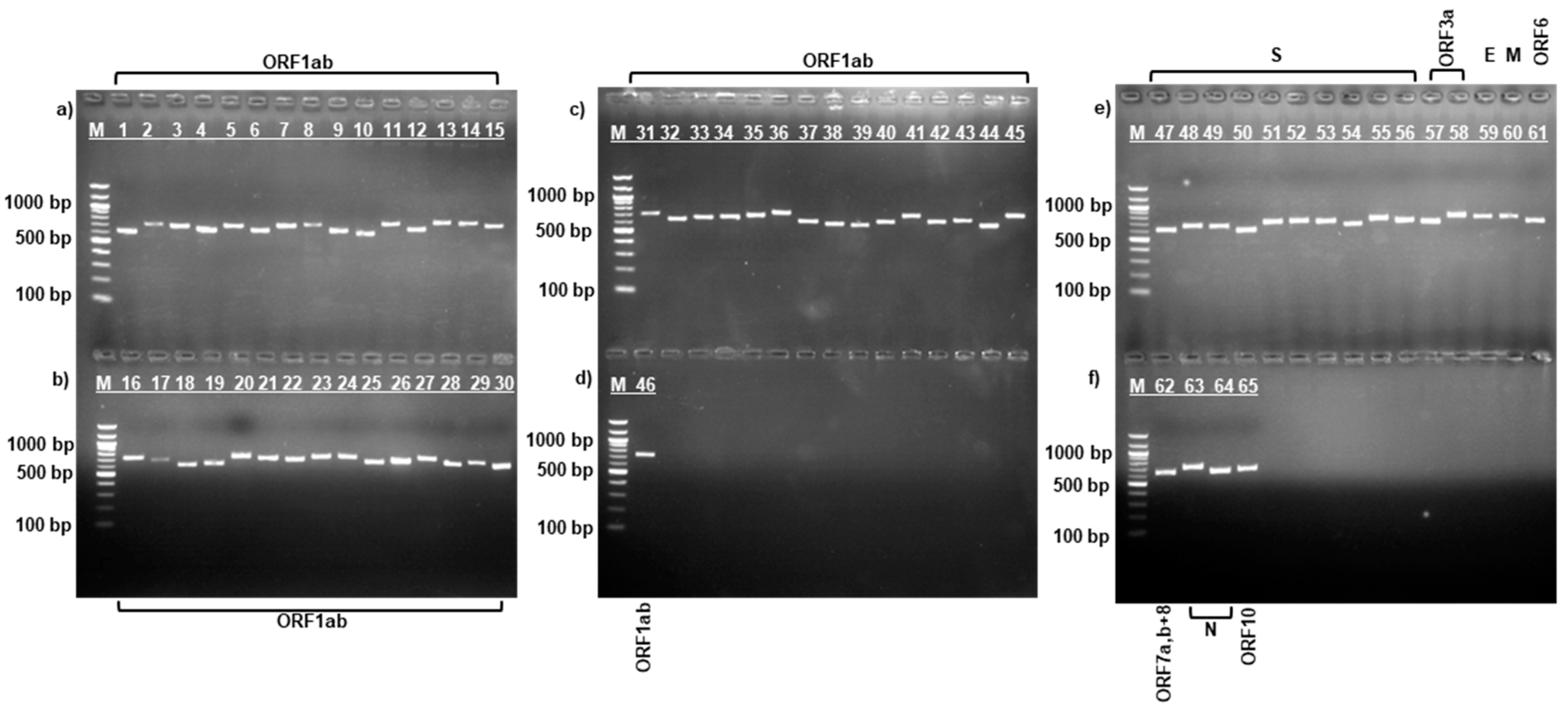

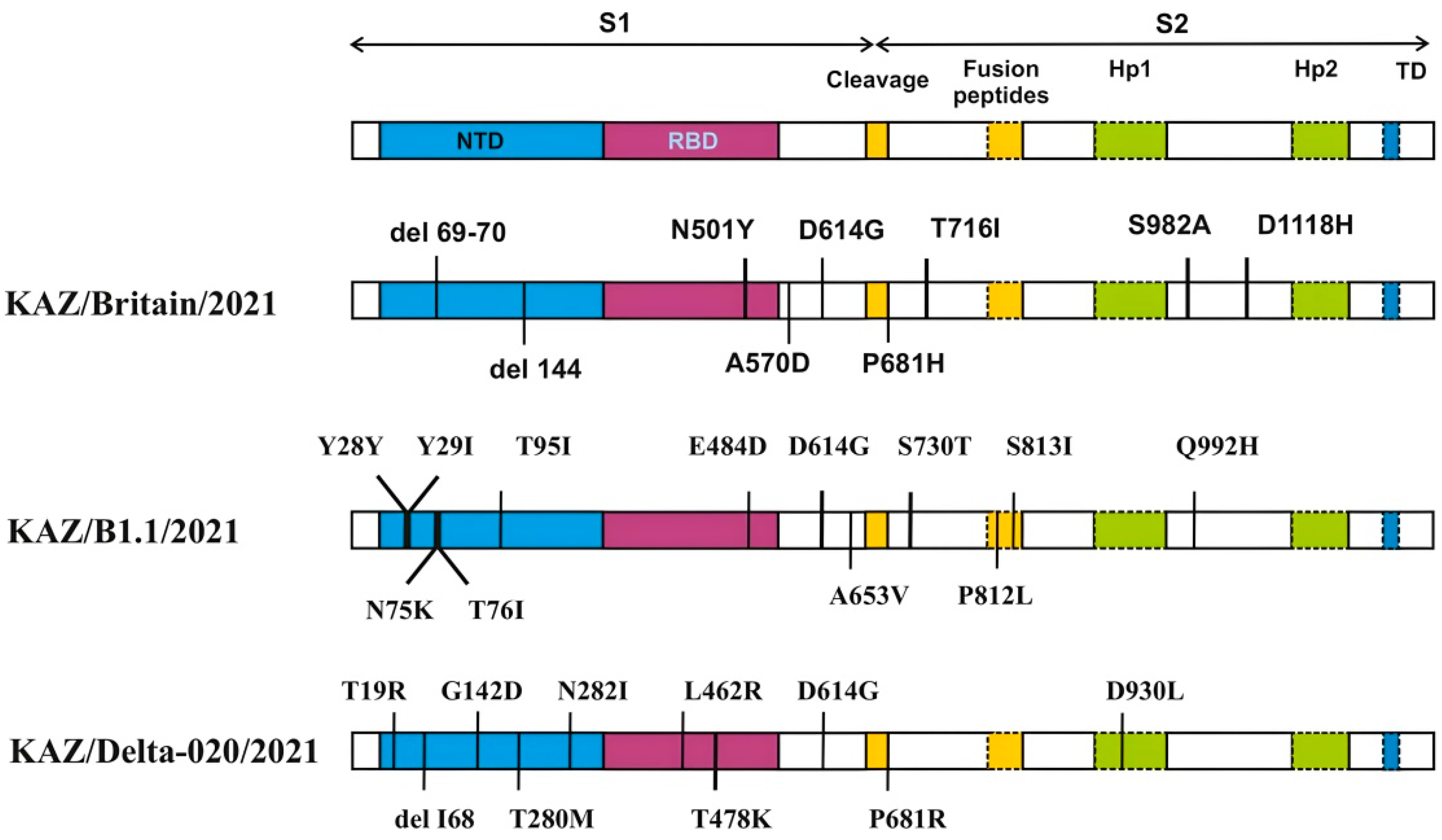


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Data for Strain: | Amino Acid Change | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Wuhan-Hu-1 a | KAZ/Britain/2021 | KAZ/B1.1/2021 | KAZ/Delta020/2021 | Type of Mutation | ||||||
| Position | Variant | Variant | Position | Variant | Position | Variant | Position | |||
| 5′ UTR b | 106 | C | – | – | T | 29 | – | – | – | 106 |
| 210 | G | – | – | – | – | T | 184 | – | 210 | |
| 241 | C | T | 215 | T | 164 | T | 215 | – | 241 | |
| ORF1ab | 344 | C | - | - | T | 267 | – | – | SNP | L27F |
| 913 | C | T | 887 | – | – | – | – | SNP_silent | S36S | |
| 1048 | G | – | – | – | – | T | 1022 | SNP | K81N c | |
| 1688 | A | C | 1662 | – | – | – | – | SNP | I295L | |
| 1899 | G | – | – | – | – | T | 1873 | SNP | R365L | |
| 2110 | C | T | 2084 | – | – | – | – | SNP_silent | N435N | |
| 2530 | A | – | – | G | 2453 | – | – | SNP_silent | E575E | |
| 3037 | C | T | 3011 | T | 2960 | T | 3011 | SNP_silent | F106F | |
| 3267 | C | T | 3241 | – | – | – | – | SNP | T183I | |
| 4181 | G | – | – | – | – | T | 4155 | SNP | A488S | |
| 4449 | C | – | – | A | 4372 | – | – | SNP | T577N | |
| 4455 | C | – | – | T | 4378 | – | – | SNP | A579V | |
| 4475 | C | – | – | T | 4398 | – | – | SNP | R586C | |
| 5388 | C | A | 5362 | – | – | – | – | SNP | A890D | |
| 5829 | A | – | – | C | 5752 | – | – | SNP | K1037T | |
| 5986 | C | T | 5960 | – | – | – | – | SNP_silent | F1089F | |
| 6402 | C | – | – | – | – | T | 6376 | SNP | P1228L | |
| 6954 | T | C | 6928 | – | – | – | – | SNP | I1412T | |
| 7042 | G | T | 7016 | – | – | – | – | SNP | M1441I | |
| 7124 | C | – | – | – | – | T | 7098 | SNP | P1469S | |
| 8986 | C | – | – | – | – | T | 8960 | SNP_silent | D144D | |
| 9053 | G | – | – | – | – | T | 9027 | SNP | V167L | |
| 9749 | A | – | – | G | 9672 | – | – | SNP | K399E | |
| 9867 | T | – | – | G | 9790 | – | – | SNP | L438R | |
| 10,029 | C | – | – | – | – | T | 10,003 | SNP | T492I | |
| 10,198 | C | – | – | T | 10,121 | – | – | SNP_silent | D48D | |
| 11,195 | C | T | 11,169 | – | – | – | – | SNP | L75F | |
| 11,201 | A | – | – | – | – | G | 11,175 | SNP | T77A | |
| 11,288 | TCTGGTTTT | del | 11,261 | del | 11,210 | – | – | SNP_stop | S106 | |
| 11,332 | A | – | – | G | 11,306 | SNP_silent | V120V | |||
| 14,120 | C | T | 14,085 | – | – | SNP | P218L | |||
| 14,408 | C | T | 14,373 | T | 14,322 | T | 14,382 | SNP | P314L | |
| 14,676 | C | T | 14,641 | – | – | – | – | SNP_silent | P403P | |
| 15,017 | C | – | – | T | 14,931 | – | – | SNP | A517V | |
| 15,279 | C | T | 15,244 | – | – | – | – | SNP_silent | H604H | |
| 15,451 | G | – | - | – | – | A | 15,425 | SNP | G662S | |
| 16,176 | T | C | 16,141 | – | – | – | – | SNP | T903T | |
| 16,466 | C | – | – | – | – | T | 16,440 | SNP | P77L | |
| 18,271 | G | – | – | – | – | A | 18,245 | SNP | E78K | |
| 18,337 | G | – | – | – | – | T | 18,311 | SNP | A100S | |
| 19,220 | C | – | – | – | – | T | 19,194 | SNP | A394V | |
| 20,405 | C | T | 20,370 | – | – | – | – | SNP | P262L | |
| 20,759 | C | – | – | T | 20,673 | – | – | SNP | A34V | |
| 21,080 | A | – | – | G | 20,994 | – | – | SNP | K141R | |
| 21,215 | A | G | 21,180 | – | – | – | – | SNP | H186R | |
| 21,446 | A | – | – | G | 21,360 | – | – | SNP | K263R | |
| 3′ UTR | 27,389 | C | – | – | T | 27,303 | – | – | – | 27,389 |
| 29,733 | – | – | – | TA | 29,648 | – | – | – | 29,733 | |
| 29,742 | G | – | – | – | – | T | 29,716 | – | 29,742 | |
| 29,755 | – | – | – | C | 29,672 | – | – | – | 29,755 | |
| 29,790 | – | – | – | T | 29,708 | – | – | – | 29,790 | |
| Protein | Data for Strain: | |||||||
|---|---|---|---|---|---|---|---|---|
| Wuhan-Hu-1 a | KAZ/Britain/2021 | KAZ/B1.1/2021 | KAZ/Delta020/2021 | |||||
| Position | Variant | Variant | Position | Variant | Position | Variant | Position | |
| S | 21,618 | C | – | – | – | – | G | 21592 |
| 21,646 | C | – | – | T | 21,560 | – | – | |
| 21,648 | C | – | – | T | 21,562 | – | – | |
| 21,765 | TACATG | del | 21,729 | – | – | – | – | |
| 21,766 | A | – | – | – | – | del | 21,739 | |
| 21,784 | T | – | – | A | 21,698 | – | – | |
| 21,789 | C | – | – | T | 21,703 | – | – | |
| 21,846 | C | – | – | T | 21,760 | – | – | |
| 21,987 | G | – | – | – | – | A | 21,961 | |
| 21,993 | ATT | del | 21,951 | – | – | – | – | |
| 22,185 | C | – | – | – | – | T | 22,159 | |
| 22,407 | A | – | – | – | – | T | 22,381 | |
| 22,917 | T | – | – | – | – | G | 22,891 | |
| 22,995 | C | – | – | – | – | A | 22,969 | |
| 23,014 | A | – | – | C | 22,928 | – | – | |
| 23,063 | A | T | 23,019 | – | – | – | – | |
| 23,271 | C | A | 32,227 | – | – | – | – | |
| 23,403 | A | G | 23,359 | G | 23,317 | G | 23,377 | |
| 23,520 | C | - | – | T | 23,434 | – | – | |
| 23,604 | C | A | 23,560 | – | – | G | 23,578 | |
| 23,709 | C | T | 23,665 | – | – | – | – | |
| 23,751 | C | – | – | T | 23,665 | – | – | |
| 23,997 | C | – | – | T | 23,911 | – | – | |
| 24,000 | G | – | – | T | 23,914 | – | – | |
| 24,410 | G | – | – | – | – | A | 24,384 | |
| 24,506 | T | G | 24,462 | – | – | – | – | |
| 24,538 | A | – | – | T | 24,452 | – | – | |
| 24,914 | G | C | 24,870 | – | – | – | – | |
| Protein | Data for Strain: | Amino Acid Change | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Wuhan-Hu-1 a | KAZ/Britain/2021 | KAZ/B1.1/2021 | KAZ/Delta020/2021 | Type of Mutation | ||||||
| Position | Variant | Variant | Position | Variant | Position | Variant | Position | |||
| ORF3a | 25,459 | C | – | – | – | – | T | 25,443 | SNP | S26L b |
| 25,688 | C | – | – | T | 25,602 | – | – | SNP | A99V | |
| 25,838 | G | T | 25,794 | – | – | – | – | SNP | W149L | |
| 26,110 | C | – | – | T | 26,024 | – | – | SNP | P240S | |
| M | 26,767 | T | – | – | – | – | C | 26,741 | SNP | I82T |
| 26,895 | C | – | – | T | 26,809 | – | – | SNP | H125Y | |
| 27,008 | G | – | – | T | 26,922 | – | – | SNP | K162N | |
| ORF6 | 27,281 | GG | AA | 27,237 | – | – | – | – | SNP_stop | W27 |
| 27,285 | TC | AT | 27,241 | – | – | – | – | SNP | NL28KF | |
| ORF7a | 27,527 | C | – | – | – | – | T | 27,501 | SNP | P45L |
| 27,638 | T | – | – | – | – | C | 27,612 | SNP | V82A | |
| 27,630 | C | – | – | T | 27,544 | – | – | SNP_silent | A79A | |
| 27,667 | G | – | – | A | 27,581 | – | – | SNP | E92K | |
| 27,739 | C | – | – | T | 27,653 | – | – | SNP | L116F | |
| 27,752 | C | – | – | – | T | 27,726 | SNP | T120I | ||
| ORF7b | 27,874 | C | – | – | – | – | T | 27,848 | SNP | T40I |
| ORF8 | 27,919 | T | – | – | – | – | C | 27,893 | SNP | I9T |
| 27,972 | C | T | 27,928 | – | – | – | – | SNP_stop | Q27 | |
| 28,048 | G | T | 28,004 | – | – | – | – | SNP | R52I | |
| 28,095 | A | T | 28,051 | – | – | – | – | SNP_stop | K68 | |
| 28,111 | A | G | 28,067 | – | – | – | – | SNP | Y73C | |
| 28,251 | T | – | – | – | – | C | 28,225 | SNP | F120L | |
| 28,253 | C | – | – | – | – | A | 28,227 | SNP | F120L | |
| 28,255 | T | – | – | – | – | A | 28,229 | SNP | I121N | |
| 28,258 | A | – | – | – | – | G | 28,232 | SNP_silent | 122 * | |
| N | 28,280 | GAT | CTA | 28,236 | – | – | – | – | SNP | D3L |
| 28,461 | A | – | – | – | G | 28,435 | SNP | D63G | ||
| 28,881 | GGG | AAC | 28,837 | AAC | 28,837 | – | – | SNP | RG203KR | |
| 28,881 | G | – | – | – | – | T | 28,855 | SNP | R203M | |
| 28,916 | G | – | – | – | – | T | 28,890 | SNP | G215C | |
| 28,977 | C | T | 28,933 | – | – | – | – | SNP | S235F | |
| 29,236 | C | – | – | – | – | T | 29,210 | SNP_silent | G312G | |
| 29,402 | G | – | – | – | – | T | 29,376 | SNP | D377Y | |
| 29,436 | A | – | – | T | 29,350 | – | – | SNP | K388I | |
| Protein | KAZ/Britain/2021 | KAZ/B1.1/2021 | KAZ/Delta020/2021 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Amino Acid Change | PROVEAN Assessment | The Effect of Variation on Protein | Amino Acid Change | PROVEAN Assessment | The Effect of Variation on Protein | Amino Acid Change | PROVEAN Assessment | The Effect of Variation on Protein | |
| ORF1ab | I295L | 0.232 | Neutral | L27F | −0.047 | Neutral | K81N | −0.070 | Neutral |
| T183I | 0.216 | Neutral | A579V | 0.011 | Neutral | R365L | −0.939 | Neutral | |
| T577N | 0.240 | Neutral | R586C | −0.727 | Neutral | A488S | −0.061 | Neutral | |
| A890D | −1.749 | Neutral | K1037T | −1.196 | Neutral | P1228L | −1.038 | Neutral | |
| I1412T | −0.370 | Neutral | K399E | −1.877 | Neutral | P1469S | 0.338 | Neutral | |
| M1441I | 0.263 | Neutral | L438R | 0.659 | Neutral | V167L | −0.696 | Neutral | |
| L75F | −2.290 | Neutral | P314L | −0.446 | Neutral | T492I | 1.435 | Neutral | |
| P218L | −5.021 | Deleterious | A517V | −1.291 | Neutral | T77A | −0.878 | Neutral | |
| P314L | −0.446 | Neutral | A34V | 1.158 | Neutral | P314L | −0.446 | Neutral | |
| P262L | −0.014 | Neutral | K141R | −0.221 | Neutral | G662S | −2.475 | Neutral | |
| H186R | −0.267 | Neutral | K263R | −1.344 | Neutral | P77L | −6.845 | Deleterious | |
| – | – | – | E78K | −1.123 | Neutral | ||||
| – | – | – | – | – | – | A100S | 1.338 | Neutral | |
| – | – | – | – | – | – | A394V | −1.523 | Neutral | |
| S | H69del | 0.260 | Neutral | Y28Y | 0.000 | Neutral | T19R | −0.839 | Neutral |
| Y145del | 0.853 | Neutral | T29I | −1.538 | Neutral | I68del | −0.821 | Neutral | |
| N501Y | −0.090 | Neutral | N74K | −1.309 | Neutral | G142D | −0.277 | Neutral | |
| A570D | −0.682 | Neutral | T76I | −0.115 | Neutral | T208M | −0.314 | Neutral | |
| D614G | 0.598 | Neutral | T95I | −1.214 | Neutral | N282I | −3.717 | Deleterious | |
| P681H | 0.060 | Neutral | E484D | −0.210 | Neutral | L452R | 0.559 | Neutral | |
| T716I | −3.293 | Deleterious | D614G | 0.598 | Neutral | T478K | −0.524 | Neutral | |
| S982A | −1.505 | Neutral | A653V | −0.715 | Neutral | D614G | 0.598 | Neutral | |
| D1118H | −1.142 | Neutral | S730T | −0.040 | Neutral | P681R | 0.741 | Neutral | |
| – | – | – | P812L | −0.868 | Neutral | D950N | −1.631 | Neutral | |
| – | – | – | S813I | −2.867 | Deleterious | – | – | – | |
| – | – | – | Q992H | −4.059 | Deleterious | – | – | – | |
| ORF3a | – | – | – | – | – | – | S26L | −2.314 | Neutral |
| A99V | −1.962 | Neutral | – | – | – | ||||
| W149L | −9.419 | Deleterious | – | – | – | – | – | ||
| – | – | – | P240S | −1.495 | Neutra | – | – | – | |
| M | – | – | – | I82T | −3.853 | Deleterious | |||
| – | – | – | H125Y | 0.799 | Neutral | – | – | – | |
| – | – | – | K162N | 0.501 | Neutral | – | – | – | |
| ORF7a | – | – | – | – | – | – | P45L | −10.000 | Deleterious |
| – | – | – | – | – | – | V82A | −2.667 | Deleterious | |
| – | – | – | A79A | 0.000 | Neutral | – | – | – | |
| – | – | – | E92K | −1.842 | Neutral | – | – | – | |
| – | – | – | L116F | −1.263 | Neutral | – | – | – | |
| – | – | – | – | – | – | T120I | −1.789 | Neutral | |
| ORF7b | – | – | – | – | – | – | T40I | −2.000 | Neutral |
| ORF8 | – | – | – | I9T | −1.333 | Neutral | |||
| R52I | −6.417 | Deleterious | – | – | – | – | – | – | |
| Y73C | −4.500 | Deleterious | – | – | – | – | – | – | |
| – | – | – | – | – | – | F120L | −2.667 | Deleterious | |
| – | – | – | – | – | – | F120L | −2.667 | Deleterious | |
| – | – | – | – | – | – | I121N | −0.667 | Neutral | |
| N | D3L | −0.230 | Neutral | – | – | – | |||
| – | – | – | – | – | – | D63G | −0.929 | Neutral | |
| – | – | – | – | – | – | R203M | −3.304 | Deleterious | |
| – | – | – | – | – | – | G215C | −0.953 | Neutral | |
| – | – | – | – | – | – | S235F | −1.738 | Neutral | |
| – | – | – | – | – | – | D377Y | −1.779 | Neutral | |
| – | – | – | K388I | −1.204 | Neutral | – | – | – | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usserbayev, B.; Sultankulova, K.T.; Burashev, Y.; Melisbek, A.; Shirinbekov, M.; Myrzakhmetova, B.S.; Zhunushov, A.; Smekenov, I.; Kerimbaev, A.; Nurabaev, S.; et al. Genetic Variations of Three Kazakhstan Strains of the SARS-CoV-2 Virus. Viruses 2025, 17, 415. https://doi.org/10.3390/v17030415
Usserbayev B, Sultankulova KT, Burashev Y, Melisbek A, Shirinbekov M, Myrzakhmetova BS, Zhunushov A, Smekenov I, Kerimbaev A, Nurabaev S, et al. Genetic Variations of Three Kazakhstan Strains of the SARS-CoV-2 Virus. Viruses. 2025; 17(3):415. https://doi.org/10.3390/v17030415
Chicago/Turabian StyleUsserbayev, Bekbolat, Kulyaisan T. Sultankulova, Yerbol Burashev, Aibarys Melisbek, Meirzhan Shirinbekov, Balzhan S. Myrzakhmetova, Asankadir Zhunushov, Izat Smekenov, Aslan Kerimbaev, Sergazy Nurabaev, and et al. 2025. "Genetic Variations of Three Kazakhstan Strains of the SARS-CoV-2 Virus" Viruses 17, no. 3: 415. https://doi.org/10.3390/v17030415
APA StyleUsserbayev, B., Sultankulova, K. T., Burashev, Y., Melisbek, A., Shirinbekov, M., Myrzakhmetova, B. S., Zhunushov, A., Smekenov, I., Kerimbaev, A., Nurabaev, S., Chervyakova, O., Kozhabergenov, N., & Kutumbetov, L. B. (2025). Genetic Variations of Three Kazakhstan Strains of the SARS-CoV-2 Virus. Viruses, 17(3), 415. https://doi.org/10.3390/v17030415