
The Emergence of Coxsackievirus A16 Subgenotype B1c: A Key Driver of the Hand, Foot, and Mouth Disease Epidemic in Guangdong, China

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. HFMD Surveillance and Sample Collection
2.2. Virus Isolation and VP1 Sequencing
2.3. Phylogenetic Analysis
3. Results
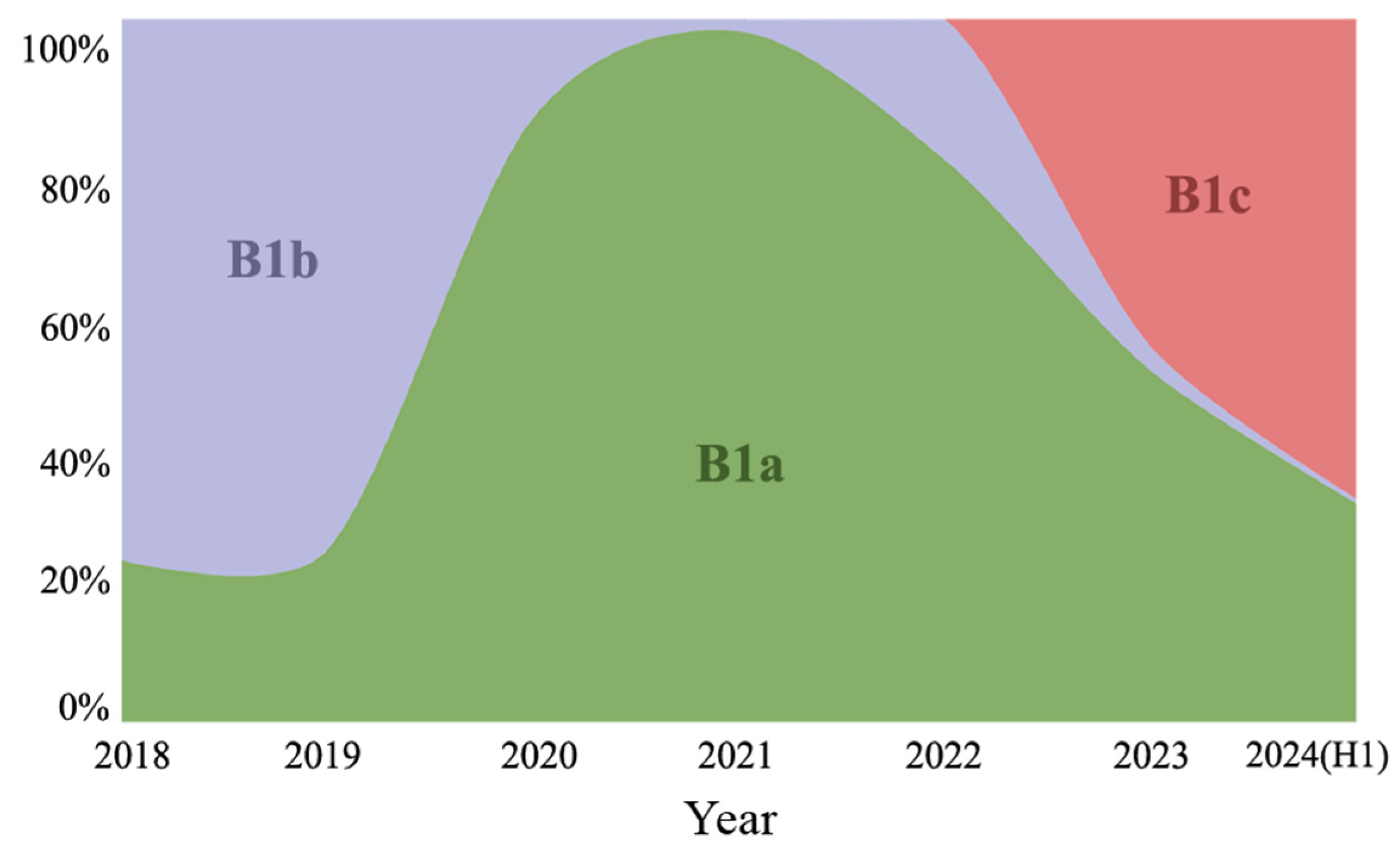
3.1. Incidence of HFMD Infection from 2018 to 2024
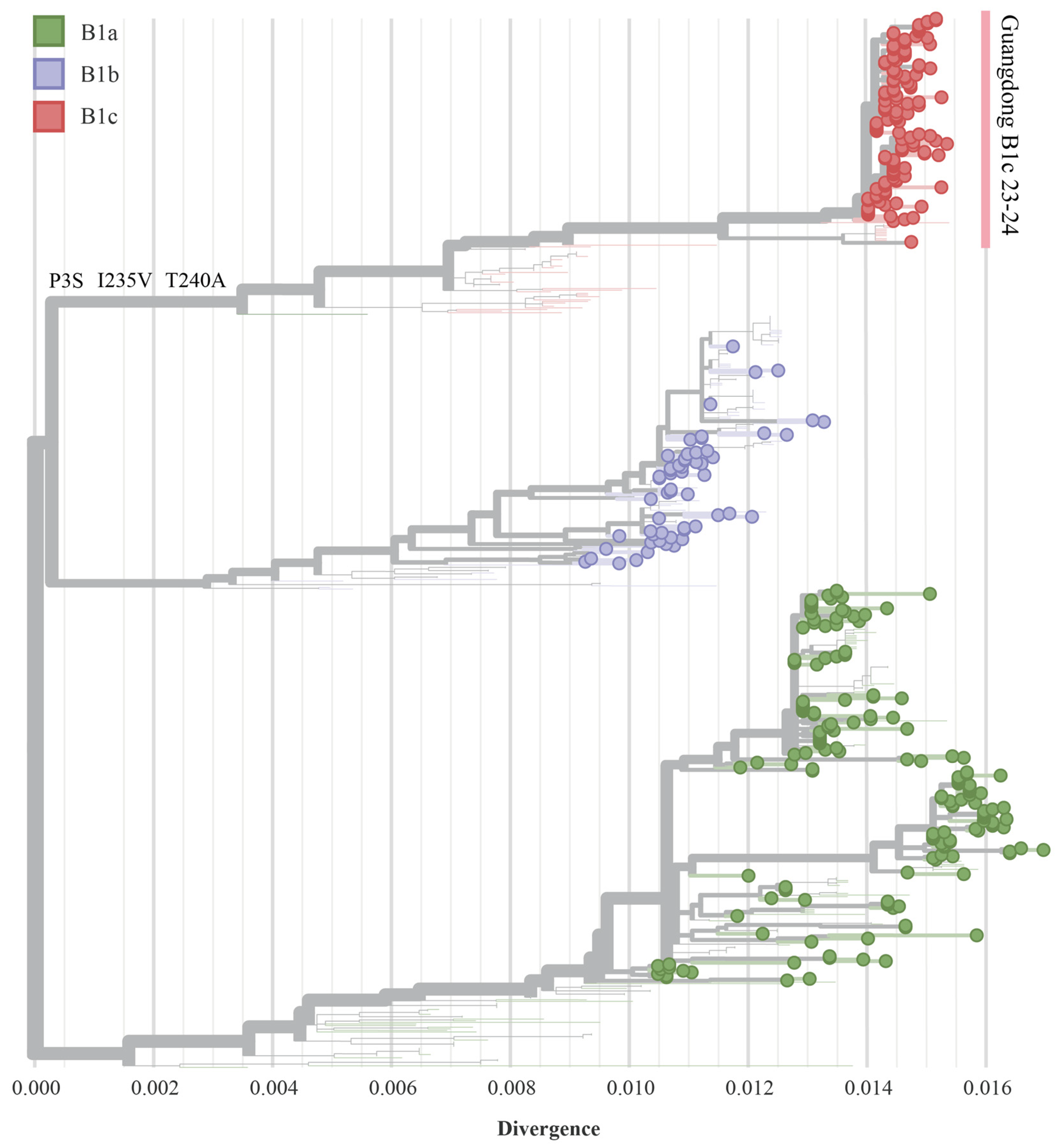
3.2. Phylogenetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lian, H.; Yi, L.; Qiu, M.; Li, B.; Sun, L.; Zeng, H.; Zeng, B.; Yang, F.; Yang, H.; Yang, M.; et al. Genomic epidemiology of CVA10 in Guangdong, China, 2013–2021. Virol. J. 2024, 21, 122. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Xie, Y.; Ji, F.; Zhao, F.; Song, X.; Lu, S.; Li, Z.; Geng, J.; Yang, H.; Long, J.; et al. Effectiveness of EV-A71 Vaccine and Its Impact on the Incidence of Hand, Foot and Mouth Disease: A Systematic Review. Vaccines 2024, 12, 1028. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Shah, P.; Ma, Z.; Wang, Y.; Xing, L. Genotyping and phylogeographic dynamics of coxsackievirus A16. Heliyon 2024, 10, e38248. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-F.; Jia, L.-P.; Yu, F.-Y.; Liu, L.-Y.; Song, Q.-W.; Dong, H.-J.; Deng, J.; Qian, Y.; Zhao, L.-Q.; Deng, L.; et al. Molecular epidemiology of coxsackievirus A16 circulating in children in Beijing, China from 2010 to 2019. World J. Pediatr. 2021, 17, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Lin, C.; Huo, D.; Yang, Y.; Feng, Z.; Cui, S.; Wu, D.; Ren, Z.; Li, D.; Jia, L.; et al. First detection of multiple cases related to CV-A16 strain of B1c clade in Beijing in 2022. J. Med. Virol. 2024, 96, e29796. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Zeng, H.; Zheng, H.; Peng, J.; Guo, X.; Liu, L.; Xiong, Q.; Sun, L.; Tan, X.; He, J.; et al. Molecular surveillance of coxsackievirus A16 in southern China, 2008–2019. Arch. Virol. 2021, 166, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zheng, H.; Yi, L.; Guo, X.; Liu, L.; Sun, L.; Tan, X.; Li, H.; Ke, C.; Lin, J. Hand, foot and mouth disease in Guangdong, China, in 2013: New trends in the continuing epidemic. Clin. Microbiol. Infect. 2014, 20, O442–O445. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tan, X.; Li, J.; Jin, Y.; Gong, L.; Hong, M.; Shi, Y.; Zhang, B.; Zhang, S.; Zhang, Y.; et al. Molecular Epidemiology of Coxsackievirus A16: Intratype and Prevalent Intertype Recombination Identified. PLoS ONE 2013, 8, e82861. [Google Scholar] [CrossRef] [PubMed]
- Ganorkar, N.N.; Patil, P.R.; Tikute, S.S.; Gopalkrishna, V. Genetic characterization of enterovirus strains identified in Hand, Foot and Mouth Disease (HFMD): Emergence of B1c, C1 subgenotypes, E2 sublineage of CVA16, EV71 and CVA6 strains in India. Infect. Genet. Evol. 2017, 54, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Teng, Z.; Cui, X.; Li, C.; Pan, H.; Zheng, Y.; Mao, S.; Yang, Y.; Wu, L.; Guo, X.; et al. Epidemiological and serological surveillance of hand-foot-and-mouth disease in Shanghai, China, 2012–2016. Emerg. Microbes Infect. 2018, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Yang, X.-H.; Hu, S.-Q.; Zhan, W.-L.; Zhang, C.-B.; Liu, H.; Zhao, H.-Y.; Chai, H.-Y.; Chen, K.-Y.; Du, Q.-Y.; et al. Co-circulation of coxsackieviruses A-6, A-10, and A-16 causes hand, foot, and mouth disease in Guangzhou city, China. BMC Infect. Dis. 2020, 20, 271. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, H.; Zeng, B.; Yi, L.; Qu, L.; Cao, J.; Yang, F.; Yang, H.; Xie, C.; Yan, Y.; Deng, W.; et al. The Emergence of Coxsackievirus A16 Subgenotype B1c: A Key Driver of the Hand, Foot, and Mouth Disease Epidemic in Guangdong, China. Viruses 2025, 17, 219. https://doi.org/10.3390/v17020219
Zeng H, Zeng B, Yi L, Qu L, Cao J, Yang F, Yang H, Xie C, Yan Y, Deng W, et al. The Emergence of Coxsackievirus A16 Subgenotype B1c: A Key Driver of the Hand, Foot, and Mouth Disease Epidemic in Guangdong, China. Viruses. 2025; 17(2):219. https://doi.org/10.3390/v17020219
Chicago/Turabian StyleZeng, Huiling, Biao Zeng, Lina Yi, Lin Qu, Jiadian Cao, Fen Yang, Haiyi Yang, Chunyan Xie, Yuxi Yan, Wenwen Deng, and et al. 2025. "The Emergence of Coxsackievirus A16 Subgenotype B1c: A Key Driver of the Hand, Foot, and Mouth Disease Epidemic in Guangdong, China" Viruses 17, no. 2: 219. https://doi.org/10.3390/v17020219
APA StyleZeng, H., Zeng, B., Yi, L., Qu, L., Cao, J., Yang, F., Yang, H., Xie, C., Yan, Y., Deng, W., Li, S., Zhang, Y., Li, B., Lu, J., & Zeng, H. (2025). The Emergence of Coxsackievirus A16 Subgenotype B1c: A Key Driver of the Hand, Foot, and Mouth Disease Epidemic in Guangdong, China. Viruses, 17(2), 219. https://doi.org/10.3390/v17020219