
NAD+ Suppresses EV-D68 Infection by Enhancing Anti-Viral Effect of SIRT1

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells, Virus Strains, and Reagents
2.2. Virus Infection and TCID50
2.3. Plasmid Construction and siRNA
2.4. Western Blot
2.5. RNA Isolation and Quantitative Real-Time PCR
2.6. Dual-Luciferase Assays
2.7. Untargeted Metabolomics
2.8. Metabolomics Data Analysis
2.9. NAD+ Measurement
2.10. Immunofluorescence
2.11. CCK8
2.12. Mouse Pup Infection Model
2.13. H&E Stain
2.14. Statistical Analysis
3. Results
3.1. EV-D68 Infection Influences Cellular Metabolism
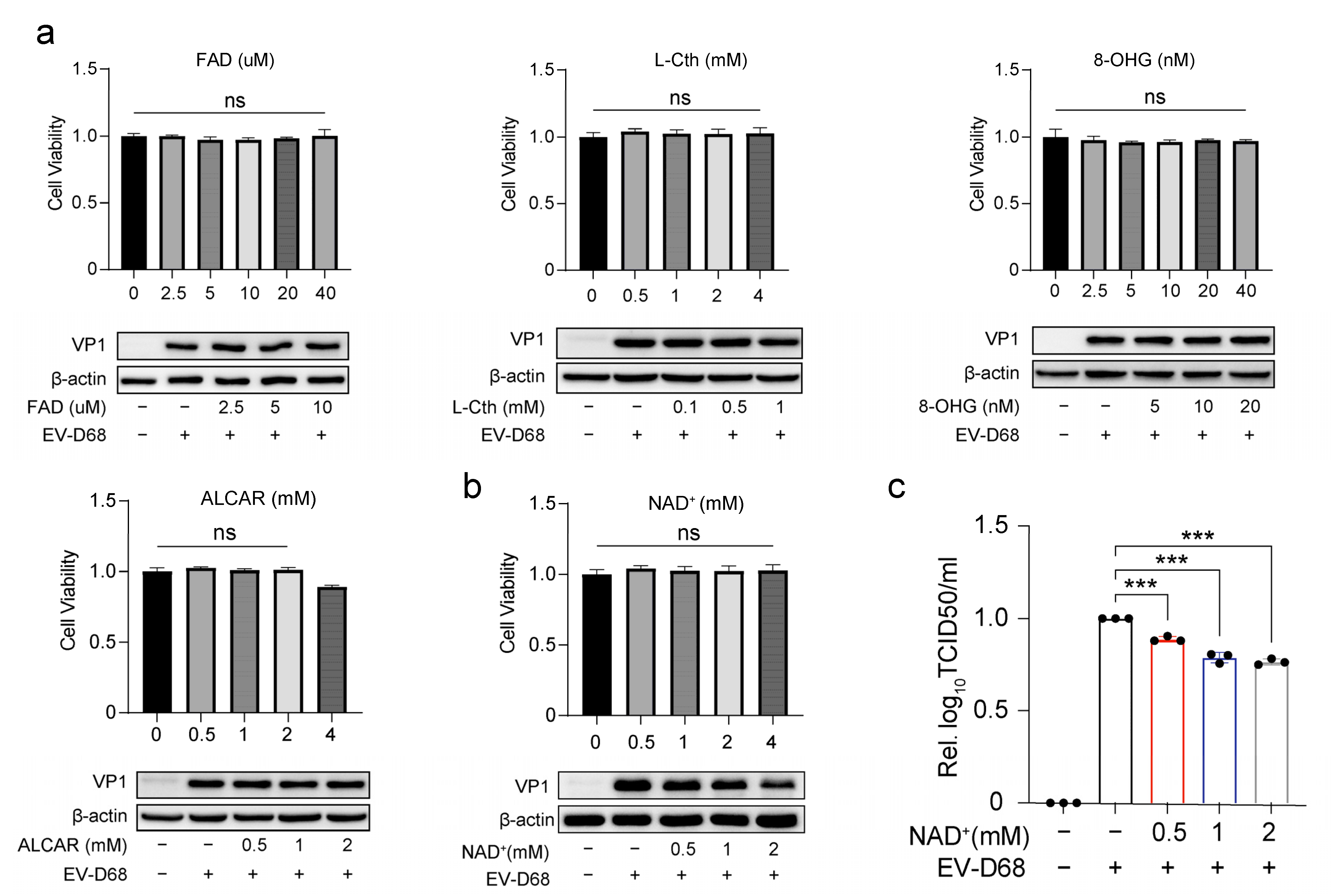
3.2. NAD+ Suppresses EV-D68 Proliferation In Vitro
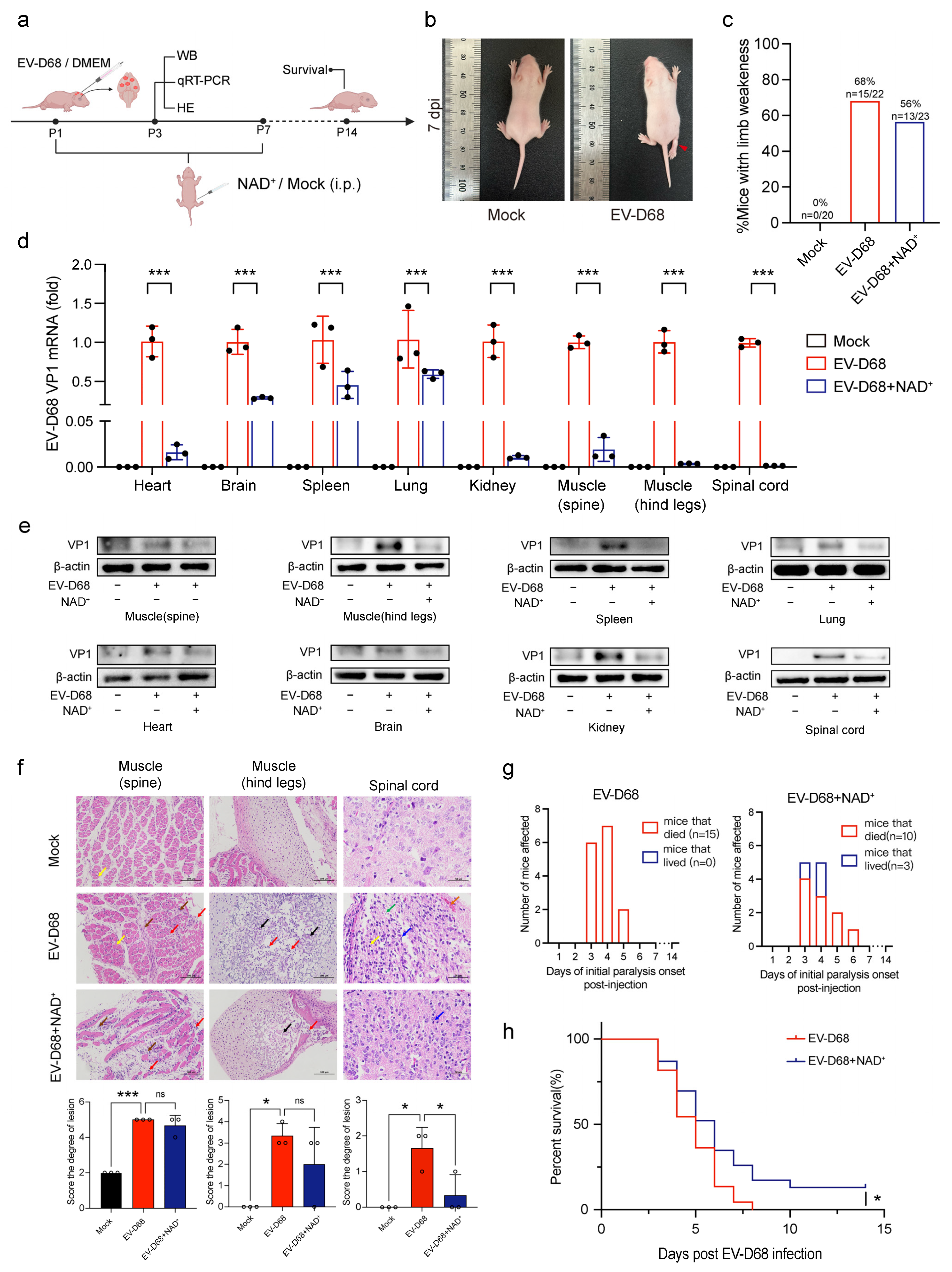
3.3. NAD+ Suppresses EV-D68 Proliferation In Vivo
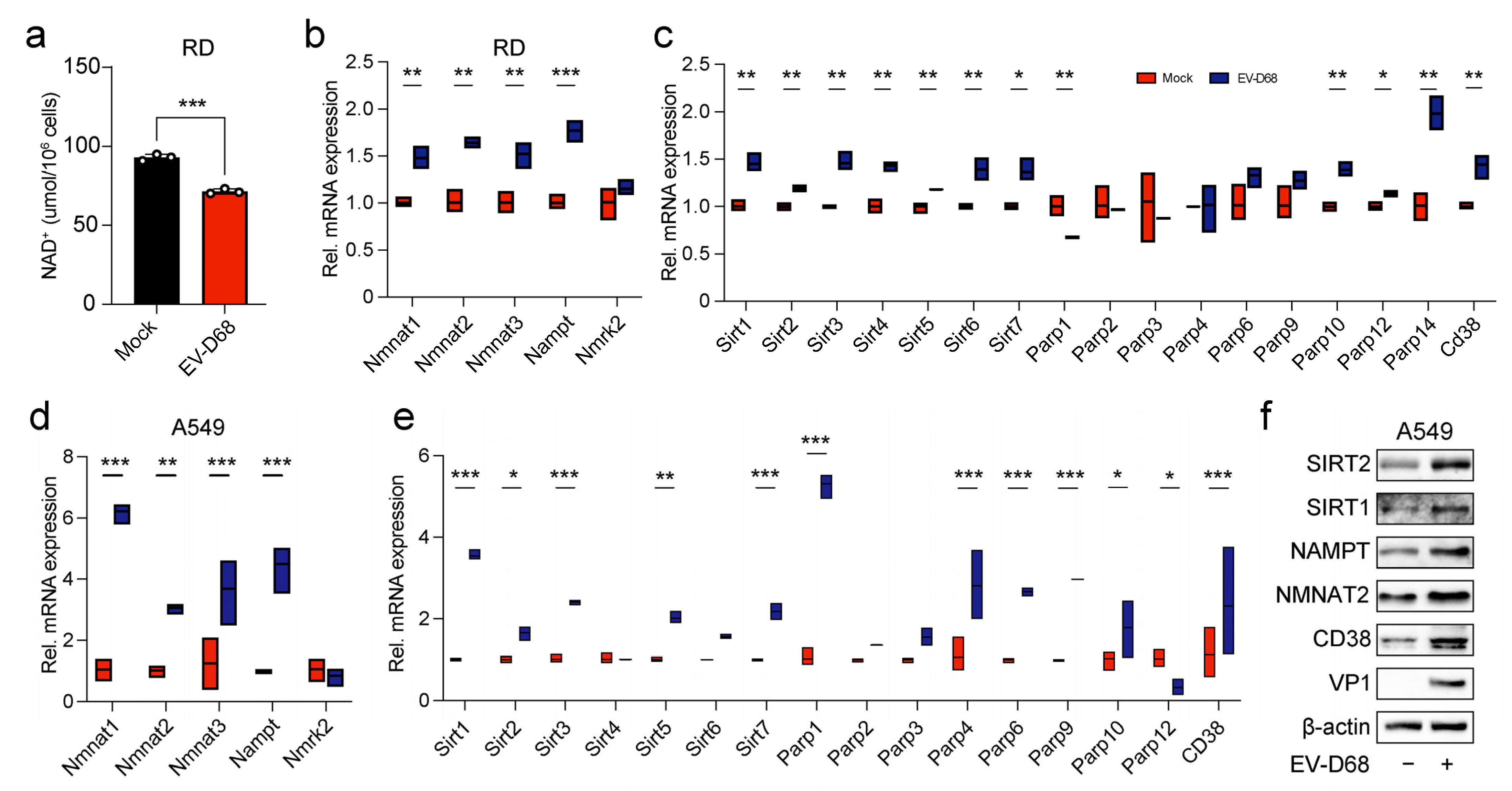
3.4. Viral Infection Regulated the Expression of Enzymes in the NAD+ Salvage Pathway, Leading to a Reduction in NAD+ Level
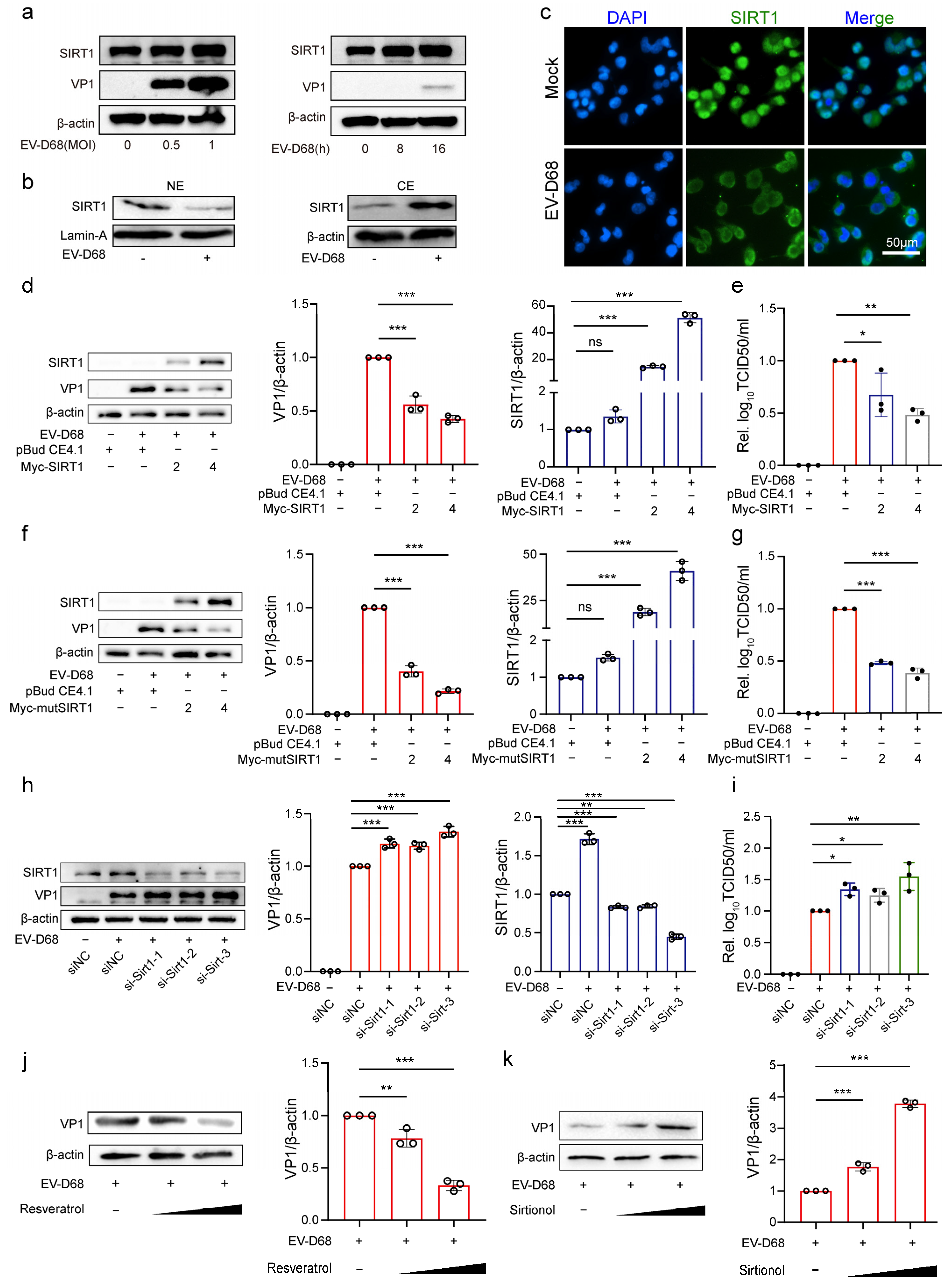
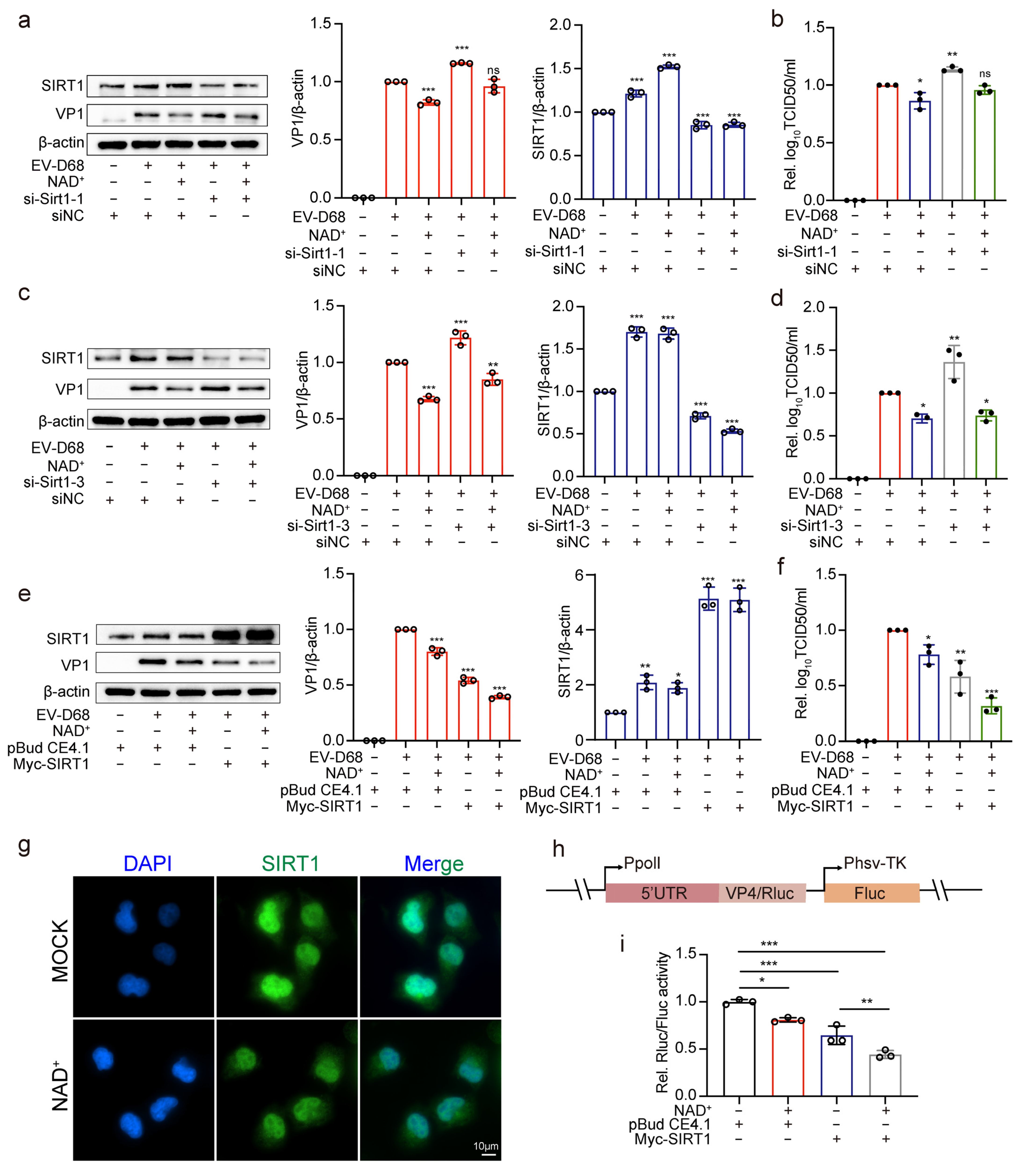
3.5. NAD+-Consuming Enzyme SIRT1 Inhibits EV-D68 Replication
3.6. NAD+ Augments the Antiviral Effect of SIRT1
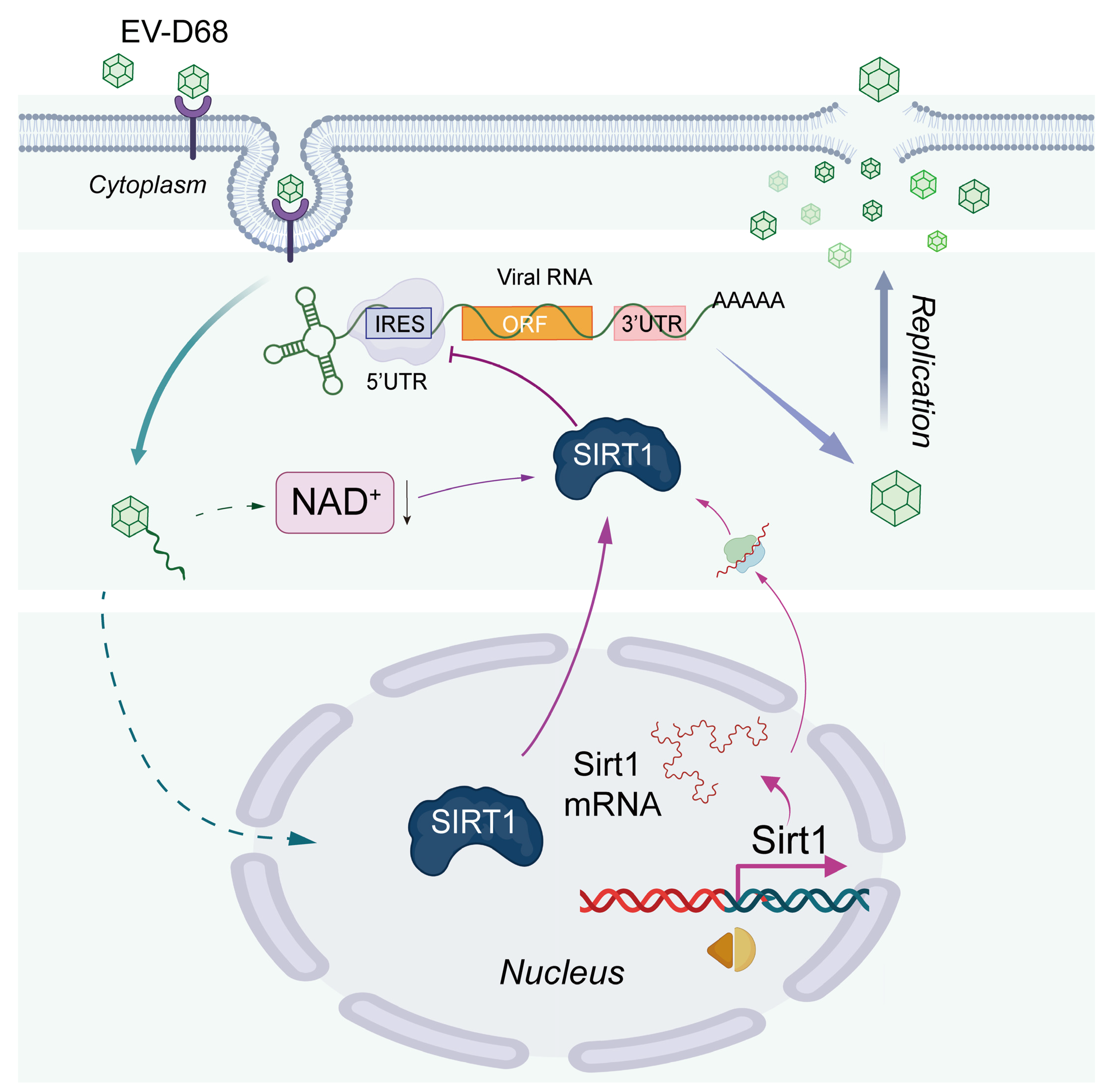
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Messacar, K.; Asturias, E.J.; Hixon, A.M.; Van Leer-Buter, C.; Niesters, H.G.M.; Tyler, K.L.; Abzug, M.J.; Dominguez, S.R. Enterovirus D68 and acute flaccid myelitis-evaluating the evidence for causality. Lancet Infect. Dis. 2018, 18, e239–e247. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Metabolomics for Investigating Physiological and Pathophysiological Processes. Physiol. Rev. 2019, 99, 1819–1875. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.L.; Lagunoff, M. Viral activation of cellular metabolism. Virology 2015, 479–480, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.F.; Hsieh, W.C.; Wu, H.C.; Lin, Y.J.; Tsai, H.W.; Huang, W.; Su, I.J. Hepatitis B Virus Pre-S2 Mutant Induces Aerobic Glycolysis through Mammalian Target of Rapamycin Signal Cascade. PLoS ONE 2015, 10, e0122373. [Google Scholar] [CrossRef] [PubMed]
- Vastag, L.; Koyuncu, E.; Grady, S.L.; Shenk, T.E.; Rabinowitz, J.D. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 2011, 7, e1002124. [Google Scholar] [CrossRef]
- Heaton, N.S.; Perera, R.; Berger, K.L.; Khadka, S.; Lacount, D.J.; Kuhn, R.J.; Randall, G. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 17345–17350. [Google Scholar] [CrossRef]
- Thaker, S.K.; Chapa, T.; Garcia, G., Jr.; Gong, D.; Schmid, E.W.; Arumugaswami, V.; Sun, R.; Christofk, H.R. Differential Metabolic Reprogramming by Zika Virus Promotes Cell Death in Human versus Mosquito Cells. Cell Metab. 2019, 29, 1206–1216.e4. [Google Scholar] [CrossRef]
- Mounce, B.C.; Cesaro, T.; Moratorio, G.; Hooikaas, P.J.; Yakovleva, A.; Werneke, S.W.; Smith, E.C.; Poirier, E.Z.; Simon-Loriere, E.; Prot, M.; et al. Inhibition of Polyamine Biosynthesis Is a Broad-Spectrum Strategy against RNA Viruses. J. Virol. 2016, 90, 9683–9692. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef]
- Pang, H.; Jiang, Y.; Li, J.; Wang, Y.; Nie, M.; Xiao, N.; Wang, S.; Song, Z.; Ji, F.; Chang, Y.; et al. Aberrant NAD(+) metabolism underlies Zika virus-induced microcephaly. Nat. Metab. 2021, 3, 1109–1124. [Google Scholar] [CrossRef]
- Sahoo, B.R.; Crook, A.A.; Pattnaik, A.; Torres-Gerena, A.D.; Khalimonchuk, O.; Powers, R.; Franco, R.; Pattnaik, A.K. Redox Regulation and Metabolic Dependency of Zika Virus Replication: Inhibition by Nrf2-Antioxidant Response and NAD(H) Antimetabolites. J. Virol. 2023, 97, e0136322. [Google Scholar] [CrossRef] [PubMed]
- Heer, C.D.; Sanderson, D.J.; Voth, L.S.; Alhammad, Y.M.O.; Schmidt, M.S.; Trammell, S.A.J.; Perlman, S.; Cohen, M.S.; Fehr, A.R.; Brenner, C. Coronavirus infection and PARP expression dysregulate the NAD metabolome: An actionable component of innate immunity. J. Biol. Chem. 2020, 295, 17986–17996. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Ott, M. The ups and downs of SIRT1. Trends Biochem. Sci. 2008, 33, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zang, W.; Wang, J.; Huang, Y.; He, Y.; Yan, L.; Liu, J.; Zheng, W. The chemical biology of sirtuins. Chem. Soc. Rev. 2015, 44, 5246–5264. [Google Scholar] [CrossRef]
- Jiang, T.; Qin, T.; Gao, P.; Tao, Z.; Wang, X.; Wu, M.; Gu, J.; Chu, B.; Zheng, Z.; Yi, J.; et al. SIRT1 attenuates blood-spinal cord barrier disruption after spinal cord injury by deacetylating p66Shc. Redox Biol. 2023, 60, 102615. [Google Scholar] [CrossRef]
- Qin, Z.; Fang, X.; Sun, W.; Ma, Z.; Dai, T.; Wang, S.; Zong, Z.; Huang, H.; Ru, H.; Lu, H.; et al. Deactylation by SIRT1 enables liquid-liquid phase separation of IRF3/IRF7 in innate antiviral immunity. Nat. Immunol. 2022, 23, 1193–1207. [Google Scholar] [CrossRef]
- Tang, H.; Yang, C.; Kang, Y.; TIAN, Y.-B.; HE, Y.-L.; XU, L.; ZHANG, G.-Y.; TANG, X.; LU, N. Effect of EV-D68 virus 5′ UTR spacer region on downstream gene expression. Microbiol. China 2020, 47, 4196–4204. [Google Scholar] [CrossRef]
- Mann, P. International Harmonization of Nomenclature and Diagnostic Criteria for Lesions in Rats and Mice (INHAND); Yang, F., Translator; China Agriculture Press: Beijing, China, 2019. [Google Scholar]
- Zheng, M.; Schultz, M.B.; Sinclair, D.A. NAD(+) in COVID-19 and viral infections. Trends Immunol. 2022, 43, 283–295. [Google Scholar] [CrossRef]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD(+) homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef]
- Chiarugi, A.; Dölle, C.; Felici, R.; Ziegler, M. The NAD metabolome--a key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef]
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Imai, S.I. NAD+ biosynthesis, aging, and disease. F1000Research 2018, 7, 132. [Google Scholar] [CrossRef] [PubMed]
- Moreira, J.D.; Hamraz, M.; Abolhassani, M.; Bigan, E.; Pérès, S.; Paulevé, L.; Nogueira, M.L.; Steyaert, J.M.; Schwartz, L. The Redox Status of Cancer Cells Supports Mechanisms behind the Warburg Effect. Metabolites 2016, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.J.; Li, H.Y.; Chen, Z.H.; Zhou, W.J.; Li, J.J.; Zhang, J.Y.; Wang, J.; Luo, X.Y.; Zeng, T.; Shi, Z.; et al. NAMPT promotes hepatitis B virus replication and liver cancer cell proliferation through the regulation of aerobic glycolysis. Oncol. Lett. 2021, 21, 390. [Google Scholar] [CrossRef]
- Langsfeld, E.S.; Bodily, J.M.; Laimins, L.A. The Deacetylase Sirtuin 1 Regulates Human Papillomavirus Replication by Modulating Histone Acetylation and Recruitment of DNA Damage Factors NBS1 and Rad51 to Viral Genomes. PLoS Pathog. 2015, 11, e1005181. [Google Scholar] [CrossRef]
- Cheng, S.T.; Ren, J.H.; Cai, X.F.; Jiang, H.; Chen, J. HBx-elevated SIRT2 promotes HBV replication and hepatocarcinogenesis. Biochem. Biophys. Res. Commun. 2018, 496, 904–910. [Google Scholar] [CrossRef]
- Lins, L.; Farias, É.; Brites-Alves, C.; Torres, A.; Netto, E.M.; Brites, C. Increased expression of CD38 and HLADR in HIV-infected patients with oral lesion. J. Med. Virol. 2017, 89, 1782–1787. [Google Scholar] [CrossRef]
- Jiang, Y.; Deng, Y.; Pang, H.; Ma, T.; Ye, Q.; Chen, Q.; Chen, H.; Hu, Z.; Qin, C.F.; Xu, Z. Treatment of SARS-CoV-2-induced pneumonia with NAD(+) and NMN in two mouse models. Cell Discov. 2022, 8, 38. [Google Scholar] [CrossRef]
- Shang, J.; Smith, M.R.; Anmangandla, A.; Lin, H. NAD+-consuming enzymes in immune defense against viral infection. Biochem. J. 2021, 478, 4071–4092. [Google Scholar] [CrossRef]
- Silva, R.F.E.; Bassi, G.; Câmara, N.O.S.; Moretti, N.S. Sirtuins: Key pieces in the host response to pathogens’ puzzle. Mol. Immunol. 2023, 160, 150–160. [Google Scholar] [CrossRef]
- Yamai, T.; Hikita, H.; Fukuoka, M.; Fukutomi, K.; Murai, K.; Nakabori, T.; Yamada, R.; Miyakawa, K.; Watashi, K.; Ryo, A.; et al. SIRT1 enhances hepatitis virus B transcription independent of hepatic autophagy. Biochem. Biophys. Res. Commun. 2020, 527, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.H.; Tao, Y.; Zhang, Z.Z.; Chen, W.X.; Cai, X.F.; Chen, K.; Ko, B.C.; Song, C.L.; Ran, L.K.; Li, W.Y.; et al. Sirtuin 1 regulates hepatitis B virus transcription and replication by targeting transcription factor AP-1. J. Virol. 2014, 88, 2442–2451. [Google Scholar] [CrossRef] [PubMed]
- Piracha, Z.Z.; Kwon, H.; Saeed, U.; Kim, J.; Jung, J.; Chwae, Y.J.; Park, S.; Shin, H.J.; Kim, K. Sirtuin 2 Isoform 1 Enhances Hepatitis B Virus RNA Transcription and DNA Synthesis through the AKT/GSK-3β/β-Catenin Signaling Pathway. J. Virol. 2018, 92, e00955-18. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.; Chen, I.P.; Vallejo-Gracia, A.; Kim, I.J.; Bielska, O.; Lam, V.L.; Hayashi, J.M.; Cruz, A.; Shah, S.; Soveg, F.W.; et al. SIRT5 is a proviral factor that interacts with SARS-CoV-2 Nsp14 protein. PLoS Pathog. 2022, 18, e1010811. [Google Scholar] [CrossRef]
- Owczarczyk, A.B.; Schaller, M.A.; Reed, M.; Rasky, A.J.; Lombard, D.B.; Lukacs, N.W. Sirtuin 1 Regulates Dendritic Cell Activation and Autophagy during Respiratory Syncytial Virus-Induced Immune Responses. J. Immunol. 2015, 195, 1637–1646. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, C.; Zha, H.; Tang, J.; Rong, F.; Chen, X.; Fan, S.; Xu, C.; Du, J.; Zhu, J.; et al. SIRT5 impairs aggregation and activation of the signaling adaptor MAVS through catalyzing lysine desuccinylation. EMBO J. 2020, 39, e103285. [Google Scholar] [CrossRef]
- Shi, J.; Jia, X.; He, Y.; Ma, X.; Qi, X.; Li, W.; Gao, S.J.; Yan, Q.; Lu, C. Immune evasion strategy involving propionylation by the KSHV interferon regulatory factor 1 (vIRF1). PLoS Pathog. 2023, 19, e1011324. [Google Scholar] [CrossRef]
- Han, Y.; Wang, L.; Cui, J.; Song, Y.; Luo, Z.; Chen, J.; Xiong, Y.; Zhang, Q.; Liu, F.; Ho, W.; et al. SIRT1 inhibits EV71 genome replication and RNA translation by interfering with the viral polymerase and 5’UTR RNA. J. Cell Sci. 2016, 129, 4534–4547. [Google Scholar] [CrossRef]
- Kane, A.E.; Sinclair, D.A. Sirtuins and NAD(+) in the Development and Treatment of Metabolic and Cardiovascular Diseases. Circ. Res. 2018, 123, 868–885. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Y.; Wang, Y.; Chao, Y.; Zhang, J.; Jia, Y.; Tie, J.; Hu, D. Regulation of SIRT1 and Its Roles in Inflammation. Front. Immunol. 2022, 13, 831168. [Google Scholar] [CrossRef]
- Pinnoji, R.C.; Bedadala, G.R.; George, B.; Holland, T.C.; Hill, J.M.; Hsia, S.C. Repressor element-1 silencing transcription factor/neuronal restrictive silencer factor (REST/NRSF) can regulate HSV-1 immediate-early transcription via histone modification. Virol. J. 2007, 4, 56. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.J.; Zhang, T.N.; Chen, H.H.; Yu, X.F.; Lv, J.L.; Liu, Y.Y.; Liu, Y.S.; Zheng, G.; Zhao, J.Q.; Wei, Y.F.; et al. The sirtuin family in health and disease. Signal Transduct. Target. Ther. 2022, 7, 402. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Dong, W. SIRT1-Related Signaling Pathways and Their Association With Bronchopulmonary Dysplasia. Front. Med. 2021, 8, 595634. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Li, H.; Huang, X.; Huang, Y.; Lv, M.; Tang, H.; Han, X.; Liu, J.; Liang, Y.; Zang, G.; et al. NAD+ Suppresses EV-D68 Infection by Enhancing Anti-Viral Effect of SIRT1. Viruses 2025, 17, 175. https://doi.org/10.3390/v17020175
Wang Y, Li H, Huang X, Huang Y, Lv M, Tang H, Han X, Liu J, Liang Y, Zang G, et al. NAD+ Suppresses EV-D68 Infection by Enhancing Anti-Viral Effect of SIRT1. Viruses. 2025; 17(2):175. https://doi.org/10.3390/v17020175
Chicago/Turabian StyleWang, Yue, Haiyu Li, Xia Huang, Yan Huang, Mingqi Lv, Hong Tang, Xinyue Han, Juntong Liu, Yan Liang, Guangchao Zang, and et al. 2025. "NAD+ Suppresses EV-D68 Infection by Enhancing Anti-Viral Effect of SIRT1" Viruses 17, no. 2: 175. https://doi.org/10.3390/v17020175
APA StyleWang, Y., Li, H., Huang, X., Huang, Y., Lv, M., Tang, H., Han, X., Liu, J., Liang, Y., Zang, G., Lu, N., & Zhang, G. (2025). NAD+ Suppresses EV-D68 Infection by Enhancing Anti-Viral Effect of SIRT1. Viruses, 17(2), 175. https://doi.org/10.3390/v17020175