
A T-Cell-Derived 3-Gene Signature Distinguishes SARS-CoV-2 from Common Respiratory Viruses

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
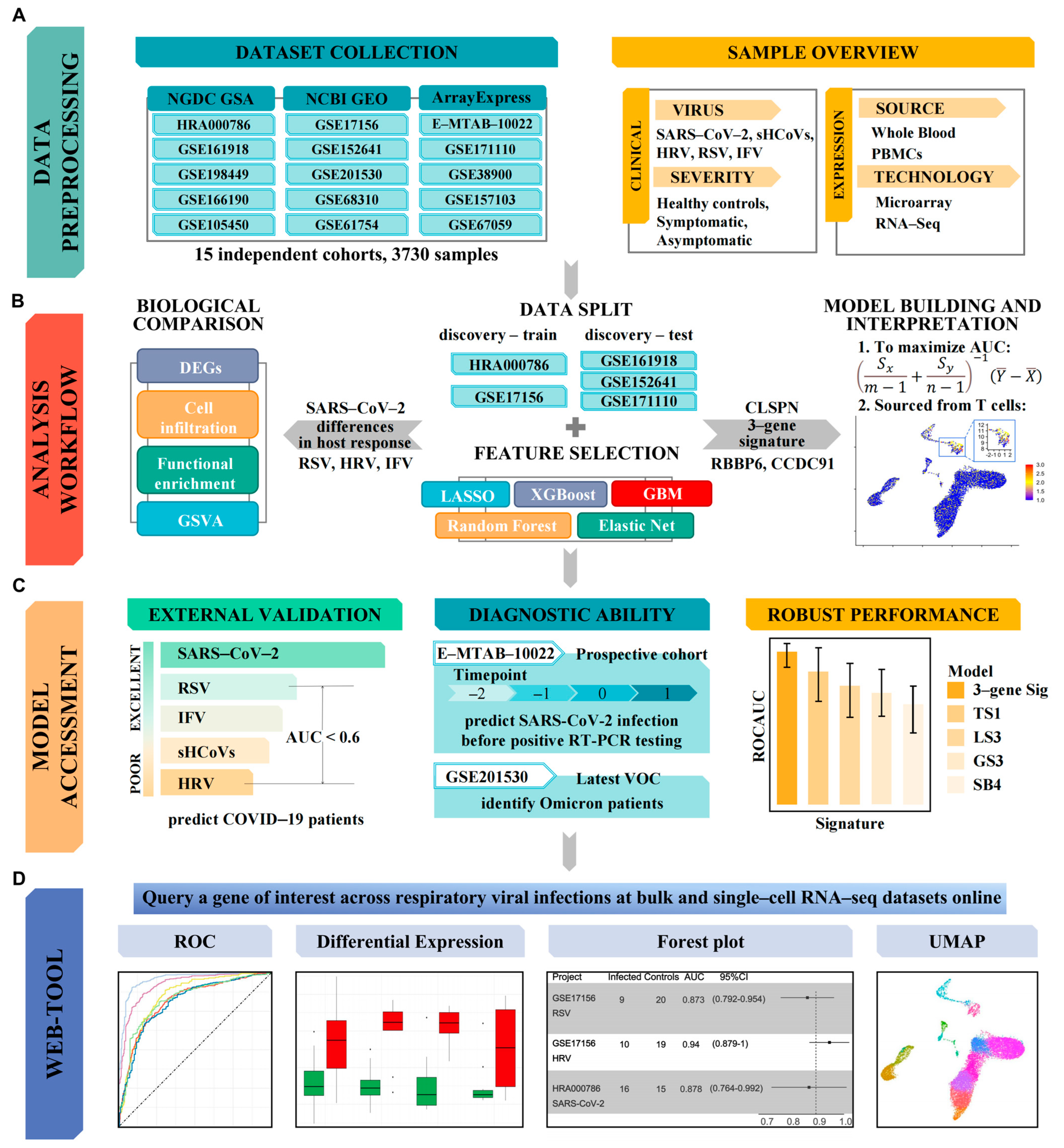
2. Materials and Methods
2.1. Dataset Collection
2.2. Data Preprocessing
2.3. Re-Standardized Group
2.4. Differentially Expressed Genes Screening
2.5. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Analyses
2.6. Multiple-Cohort Integration and Co-normalization
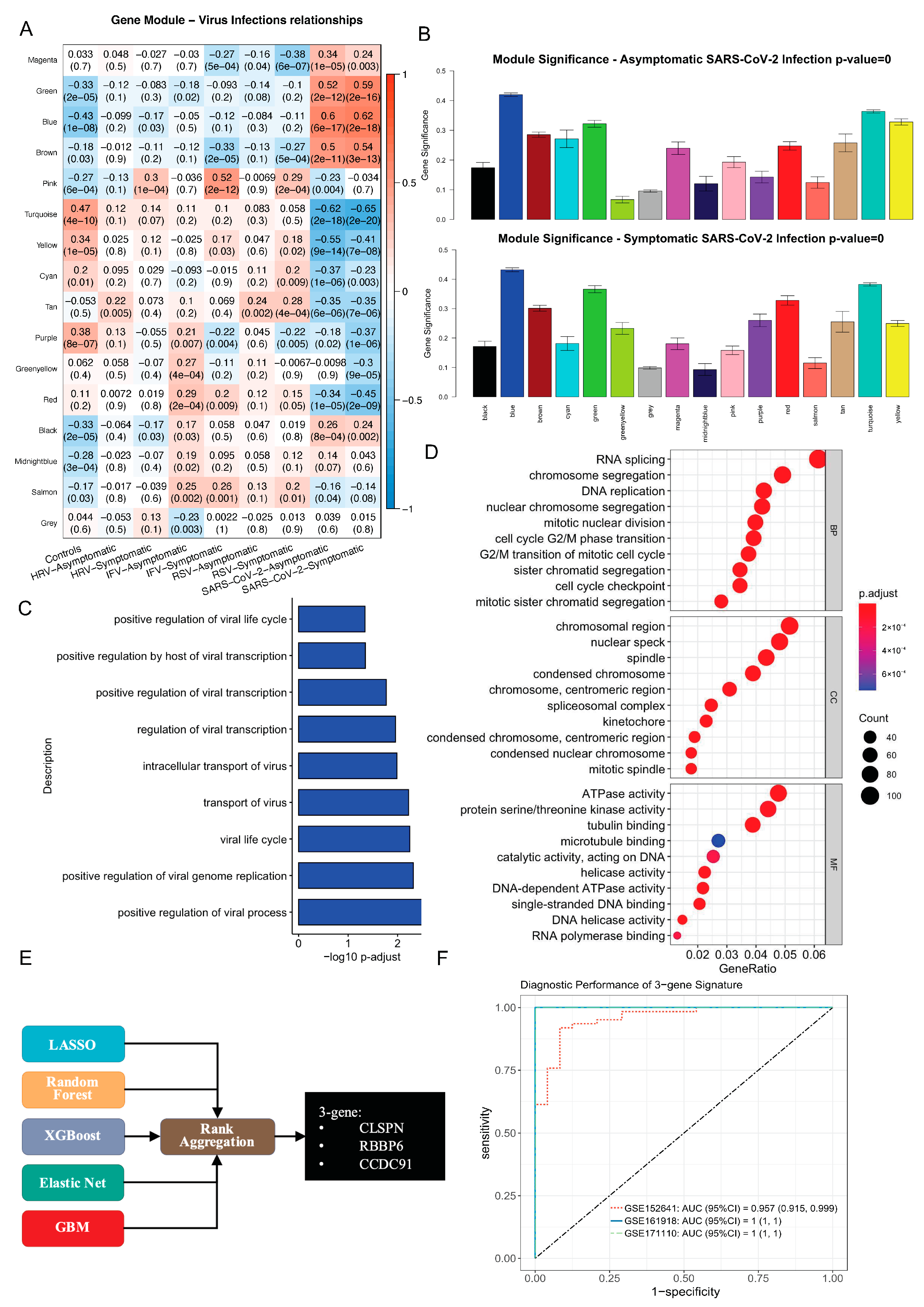
2.7. Co-Expression Network Construction
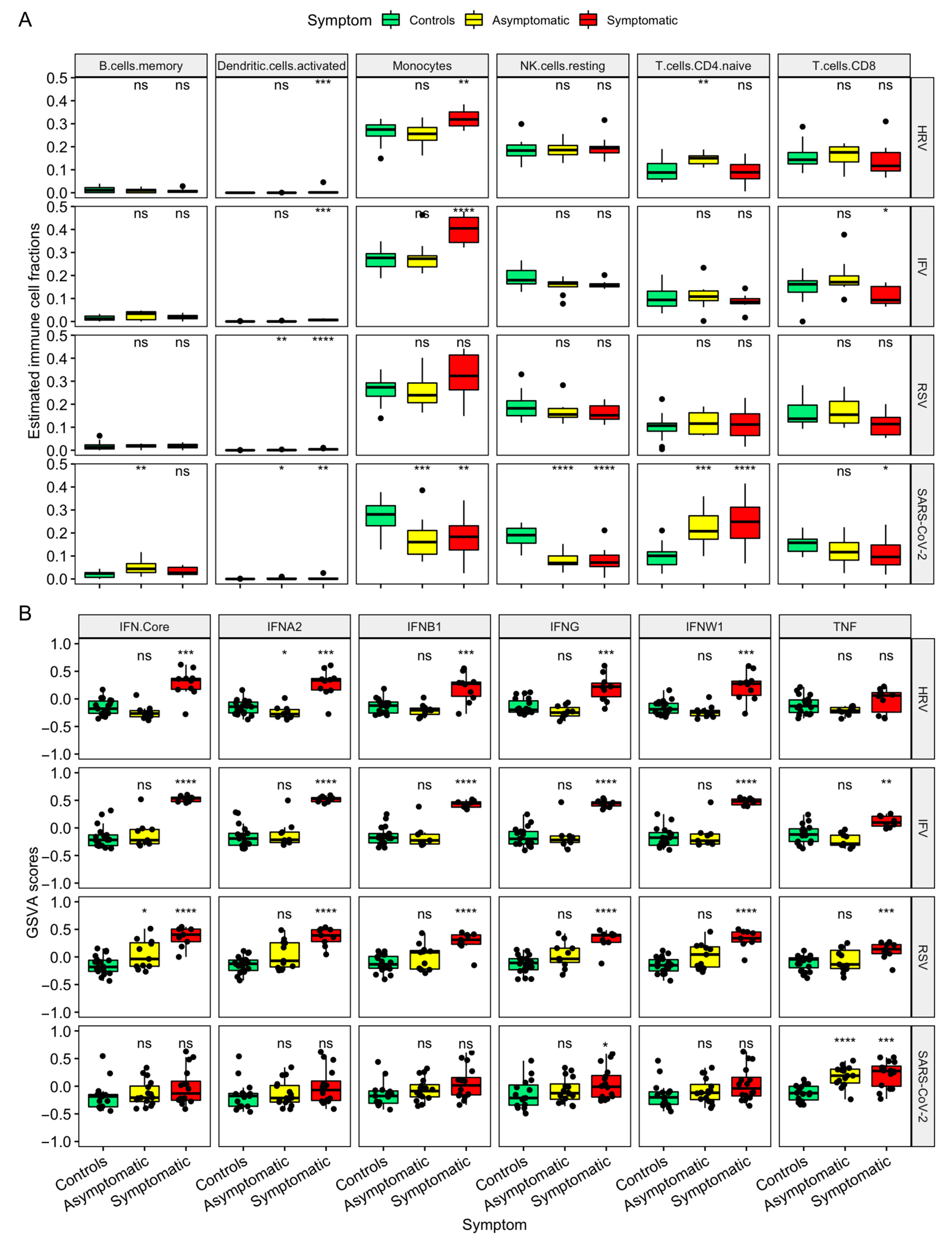
2.8. Estimation of Immune Cell Type Abundances
2.9. Gene Set Variation Analysis (GSVA) with Pre-Defined Interferon Gene Sets
2.10. Feature Selection with Bioinformatics and Machine Learning
2.11. External Validation
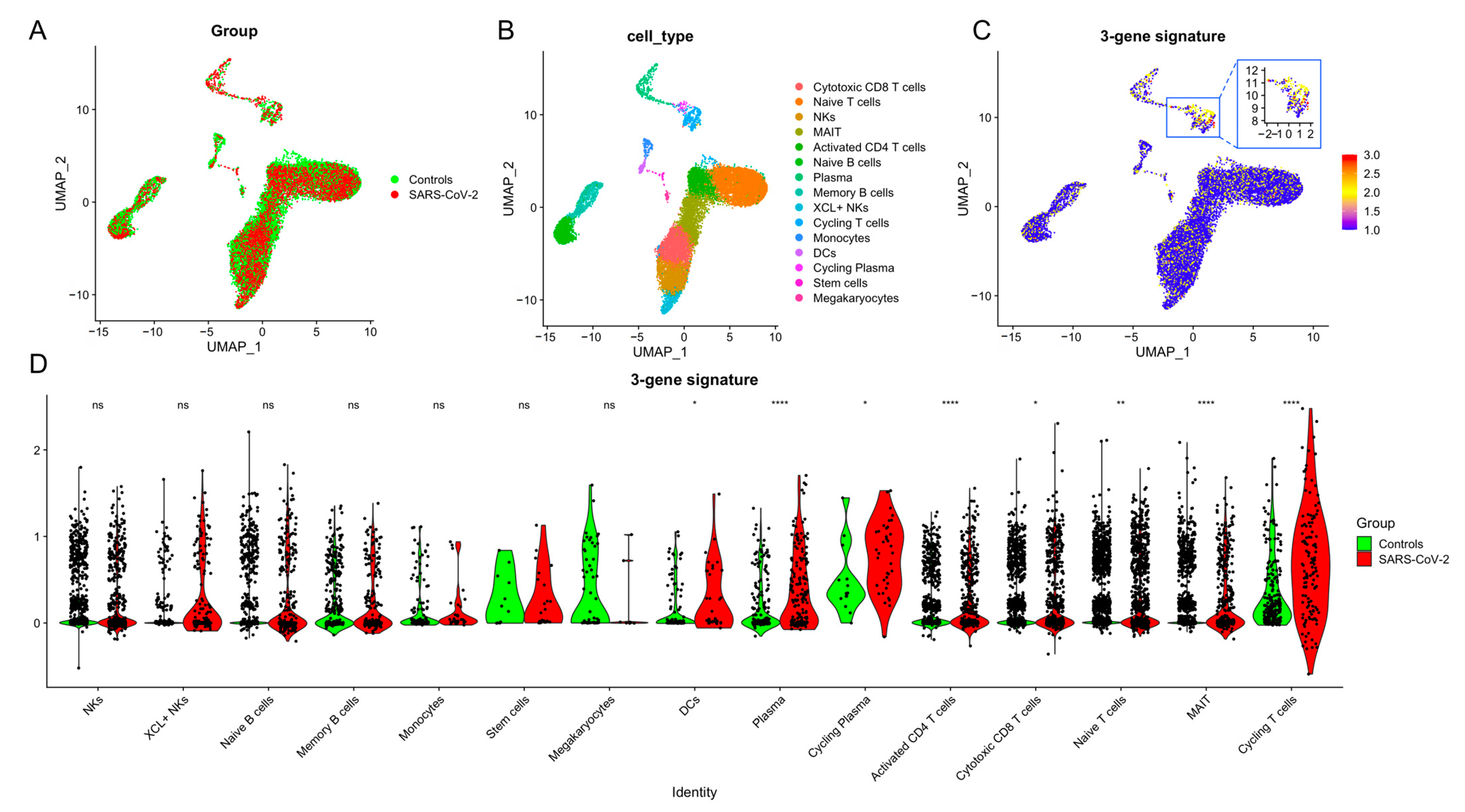
2.12. Single-Cell RNA-Seq Analysis
2.13. Statistical Analysis
3. Results
3.1. Clinical Characteristics of the Included Population
3.2. Host Response Profiling in SARS-CoV-2 and Other Common Respiratory Viral Infections
3.3. Identification of SARS-CoV-2 Specific 3-Gene Signature via Integrative Bioinformatics and Machine Learning Approaches
3.4. T Cells Are the Primary Source of the 3-Gene Signature
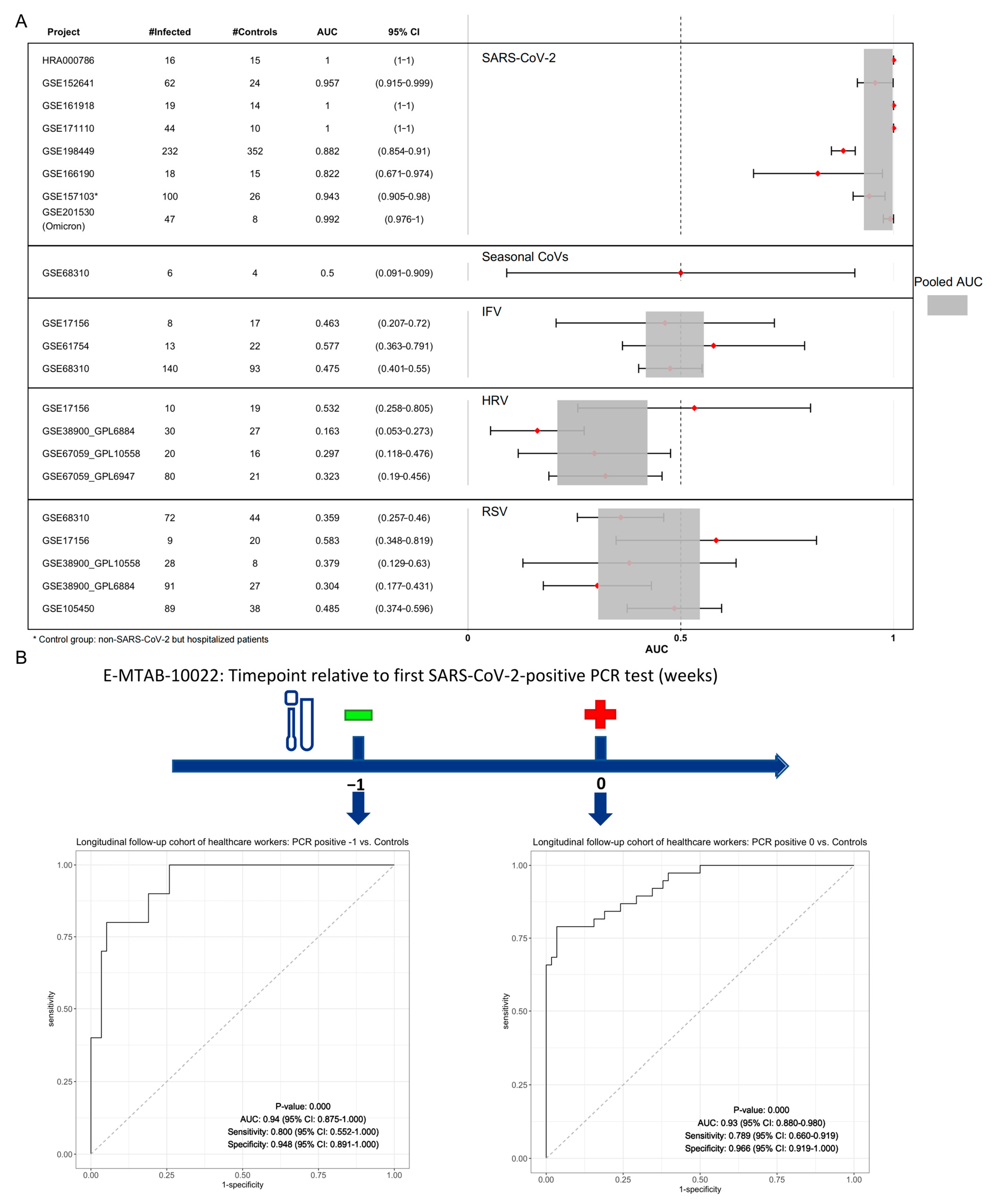
3.5. Validation of a 3-Gene Signature for SARS-CoV-2: Specificity and Predictive Value
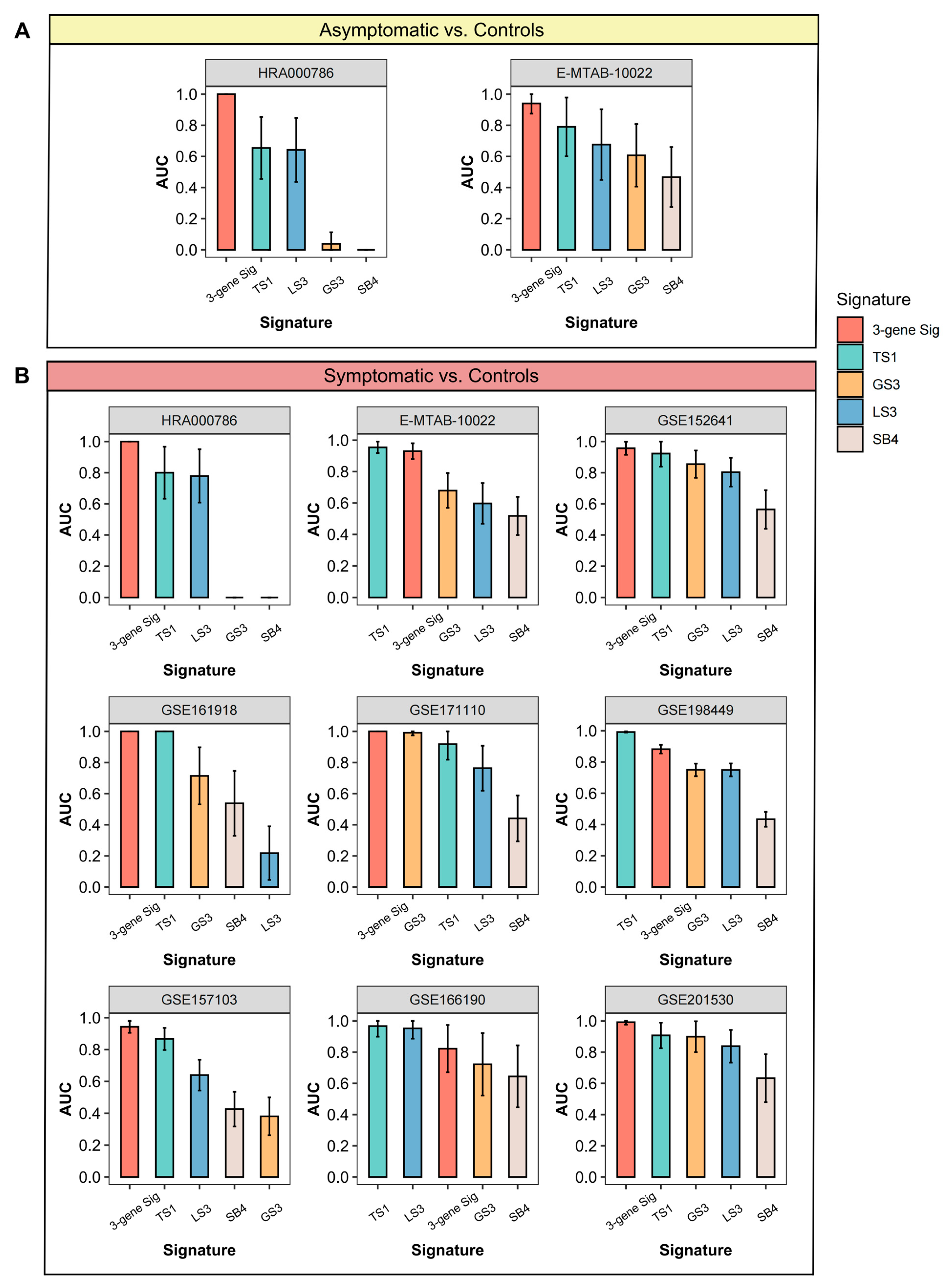
3.6. Comparisons of Gene Expression Signatures
3.7. Query a Gene of Interest via ViRAL Online
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Garrett, N.; Tapley, A.; Andriesen, J.; Seocharan, I.; Fisher, L.H.; Bunts, L.; Espy, N.; Wallis, C.L.; Randhawa, A.K.; Miner, M.D.; et al. High Asymptomatic Carriage with the Omicron Variant in South Africa. Clin. Infect. Dis. 2022, 75, e289–e292. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Liu, J.; Liu, Q.; Kang, L.; Liu, R.; Jing, W.; Wu, Y.; Liu, M. Global Percentage of Asymptomatic SARS-CoV-2 Infections among the Tested Population and Individuals with Confirmed COVID-19 Diagnosis: A Systematic Review and Meta-analysis. JAMA Netw. Open 2021, 4, e2137257. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Hu, Y.; Peng, B.; Tang, X.J.; Wang, W.; Su, K.; Luo, C.; Wu, B.; Zhang, F.; Zhang, Y.; et al. Effective control of SARS-CoV-2 transmission in Wanzhou, China. Nat. Med. 2021, 27, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Sakurai, A.; Suzuki, M.; Komoto, S.; Ide, T.; Ishihara, T.; Doi, Y. Shedding of Viable Virus in Asymptomatic SARS-CoV-2 Carriers. MSphere 2021, 6, e00019-21. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.A.; Quandelacy, T.M.; Kada, S.; Prasad, P.V.; Steele, M.; Brooks, J.T.; Slayton, R.B.; Biggerstaff, M.; Butler, J.C. SARS-CoV-2 Transmission from People without COVID-19 Symptoms. JAMA Netw. Open 2021, 4, e2035057. [Google Scholar] [CrossRef]
- Gao, W.; Lv, J.; Pang, Y.; Li, L.M. Role of asymptomatic and pre-symptomatic infections in COVID-19 pandemic. BMJ 2021, 375, n2342. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.J.; Zhang, H.Y.; Ren, L.L.; Lu, Q.B.; Ren, X.; Zhang, C.H.; Wang, Y.F.; Lin, S.H.; Zhang, X.A.; Li, J.; et al. Etiological and epidemiological features of acute respiratory infections in China. Nat. Commun. 2021, 12, 5026. [Google Scholar] [CrossRef] [PubMed]
- Cilloniz, C.; Luna, C.M.; Hurtado, J.C.; Marcos, M.Á.; Torres, A. Respiratory viruses: Their importance and lessons learned from COVID-19. Eur. Respir. Rev. 2022, 31, 220051. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, K.; Kawasuji, H.; Murai, Y.; Kaneda, M.; Ueno, A.; Miyajima, Y.; Fukui, Y.; Morinaga, Y.; Yamamoto, Y. Circulating Type I Interferon Levels in the Early Phase of COVID-19 Are Associated with the Development of Respiratory Failure. Front. Immunol. 2022, 13, 844304. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, G.; Wang, R.; Ren, L.; Yuan, Z.; Liu, Y.; Wu, Y.; Chen, R.; Chen, Y.; Diao, B. Serum levels of type I interferon (IFN-I) is associated with the severity of COVID-19. J. Med. Microbiol. 2023, 72, 001694. Available online: https://consensus.app/papers/serum-levels-type-interferon-ifni-associated-severity-sun/eb554e01e18855a2a0bf5fb13d420940/ (accessed on 8 April 2024). [CrossRef] [PubMed]
- Sampson, D.L.; Fox, B.A.; Yager, T.D.; Bhide, S.; Cermelli, S.; McHugh, L.C.; Seldon, T.A.; Sullivan, E.; Zimmerman, J.J.; Noursadeghi, M.; et al. A Four-Biomarker Blood Signature Discriminates Systemic Inflammation Due to Viral Infection versus Other Etiologies. Sci. Rep. 2017, 7, 2914. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Carballa, A.; Barral-Arca, R.; Cebey-López, M.; Bello, X.; Pardo-Seco, J.; Martinón-Torres, F.; Salas, A. Identification of a Minimal 3-Transcript Signature to Differentiate Viral from Bacterial Infection from Best Genome-Wide Host RNA Biomarkers: A Multi-Cohort Analysis. Int. J. Mol. Sci. 2021, 22, 3148. [Google Scholar] [CrossRef] [PubMed]
- Li, H.K.; Kaforou, M.; Rodriguez-Manzano, J.; Channon-Wells, S.; Moniri, A.; Habgood-Coote, D.; Gupta, R.K.; Mills, E.A.; Arancon, D.; Lin, J.; et al. Discovery and validation of a three-gene signature to distinguish COVID-19 and other viral infections in emergency infectious disease presentations: A case-control and observational cohort study. Lancet Microbe 2021, 2, e594–e603. [Google Scholar] [CrossRef]
- McClain, M.T.; Constantine, F.J.; Nicholson, B.P.; Nichols, M.; Burke, T.W.; Henao, R.; Jones, D.C.; Hudson, L.L.; Jaggers, L.B.; Veldman, T.; et al. A blood-based host gene expression assay for early detection of respiratory viral infection: An index-cluster prospective cohort study. Lancet Infect. Dis. 2021, 21, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.M.; Shojaei, M.; Parnell, G.P.; Huang, S.; Nalos, M.; Teoh, S.; O’Connor, K.; Schibeci, S.; Phu, A.L.; Kumar, A.; et al. A novel immune biomarker IFI27 discriminates between influenza and bacteria in patients with suspected respiratory infection. Eur. Respir. J. 2017, 49, 1602098. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Di, C.; Chen, S.; Guo, M.; Yan, J.; Zhu, Z.; Liu, L.; Feng, R.; Xie, Y.; Zhang, R.; et al. Distinct immune signatures discriminate between asymptomatic and presymptomatic SARS-CoV-2pos subjects. Cell Res. 2021, 31, 1148–1162. [Google Scholar] [CrossRef] [PubMed]
- Zaas, A.K.; Chen, M.; Varkey, J.; Veldman, T.; Hero, A.O., III; Lucas, J.; Huang, Y.; Turner, R.; Gilbert, A.; Lambkin-Williams, R.; et al. Gene Expression Signatures Diagnose Influenza and Other Symptomatic Respiratory Viral Infection in Humans. Cell Host Microbe 2009, 6, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Martins, A.J.; Lau, W.W.; Rachmaninoff, N.; Chen, J.; Imberti, L.; Mostaghimi, D.; Fink, D.L.; Burbelo, P.D.; Dobbs, K.; et al. Time-resolved systems immunology reveals a late juncture linked to fatal COVID-19. Cell 2021, 184, 1836–1857.e22. [Google Scholar] [CrossRef]
- Lévy, Y.; Wiedemann, A.; Hejblum, B.P.; Durand, M.; Lefebvre, C.; Surénaud, M.; Lacabaratz, C.; Perreau, M.; Foucat, E.; Déchenaud, M.; et al. CD177, a specific marker of neutrophil activation, is associated with coronavirus disease 2019 severity and death. iScience 2021, 24, 102711. [Google Scholar] [CrossRef]
- Thair, S.A.; He, Y.D.; Hasin-Brumshtein, Y.; Sakaram, S.; Pandya, R.; Toh, J.; Rawling, D.; Remmel, M.; Coyle, S.; Dalekos, G.N.; et al. Transcriptomic similarities and differences in host response between SARS-CoV-2 and other viral infections. iScience 2021, 24, 101947. [Google Scholar] [CrossRef] [PubMed]
- Vono, M.; Huttner, A.; Lemeille, S.; Martinez-Murillo, P.; Meyer, B.; Baggio, S.; Sharma, S.; Thiriard, A.; Marchant, A.; Godeke, G.J.; et al. Robust innate responses to SARS-CoV-2 in children resolve faster than in adults without compromising adaptive immunity. Cell Rep. 2021, 37, 109773. [Google Scholar] [CrossRef] [PubMed]
- Sauerwald, N.; Zhang, Z.; Ramos, I.; Nair, V.D.; Soares-Schanoski, A.; Ge, Y.; Mao, W.; Alshammary, H.; Gonzalez-Reiche, A.S.; van de Guchte, A.; et al. Pre-infection antiviral innate immunity contributes to sex differences in SARS-CoV-2 infection. Cell Syst. 2022, 13, 924–931.e4. [Google Scholar] [CrossRef] [PubMed]
- Overmyer, K.A.; Shishkova, E.; Miller, I.J.; Balnis, J.; Bernstein, M.N.; Peters-Clarke, T.M.; Meyer, J.G.; Quan, Q.; Muehlbauer, L.K.; Trujillo, E.A.; et al. Large-Scale Multi-omic Analysis of COVID-19 Severity. Cell Syst. 2021, 12, 23–40.e7. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Knabl, L.; Walter, M.; Dai, Y.; Füßl, M.; Caf, Y.; Jeller, C.; Knabl, P.; Obermoser, M.; Baurecht, C.; et al. Prior Vaccination Exceeds Prior Infection in Eliciting Innate and Humoral Immune Responses in Omicron Infected Outpatients. Front. Immunol. 2022, 13, 916686. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Rosenheim, J.; Bell, L.C.; Chandran, A.; Guerra-Assuncao, J.A.; Pollara, G.; Whelan, M.; Artico, J.; Joy, G.; Kurdi, H.; et al. Blood transcriptional biomarkers of acute viral infection for detection of pre-symptomatic SARS-CoV-2 infection: A nested, case-control diagnostic accuracy study. Lancet Microbe 2021, 2, e508–e517. [Google Scholar] [CrossRef] [PubMed]
- Mejias, A.; Dimo, B.; Suarez, N.M.; Garcia, C.; Suarez-Arrabal, M.C.; Jartti, T.; Blankenship, D.; Jordan-Villegas, A.; Ardura, M.I.; Xu, Z.; et al. Whole blood gene expression profiles to assess pathogenesis and disease severity in infants with respiratory syncytial virus infection. PLoS Med. 2013, 10, e1001549. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Franco, L.M.; Atmar, R.L.; Quarles, J.M.; Arden, N.; Bucasas, K.L.; Wells, J.M.; Niño, D.; Wang, X.; Zapata, G.E.; et al. Host Transcriptional Response to Influenza and Other Acute Respiratory Viral Infections—A Prospective Cohort Study. PLoS Pathog. 2015, 11, e1004869. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Fernandez, R.; Tapia, L.I.; Yang, C.-F.; Torres, J.P.; Chavez-Bueno, S.; Garcia, C.; Jaramillo, L.M.; Moore-Clingenpeel, M.; Jafri, H.; Peeples, M.E.; et al. Respiratory Syncytial Virus Genotypes, Host Immune Profiles, and Disease Severity in Young Children Hospitalized with Bronchiolitis. J. Infect. Dis. 2017, 217, 24–34. [Google Scholar] [CrossRef]
- Davenport, E.E.; Antrobus, R.D.; Lillie, P.J.; Gilbert, S.; Knight, J.C. Transcriptomic profiling facilitates classification of response to influenza challenge. J. Mol. Med. 2015, 93, 105–114. [Google Scholar] [CrossRef]
- Heinonen, S.; Jartti, T.; Garcia, C.; Oliva, S.; Smitherman, C.; Anguiano, E.; de Steenhuijsen Piters, W.A.; Vuorinen, T.; Ruuskanen, O.; Dimo, B.; et al. Rhinovirus Detection in Symptomatic and Asymptomatic Children: Value of Host Transcriptome Analysis. Am. J. Resp. Crit. Care 2016, 193, 772–782. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Love, M.I.; Wolfgang, H.; Simon, A. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Rao, A.M.; Dermadi, D.; Toh, J.; Jones, L.M.; Donato, M.; Liu, Y.; Su, Y.; Dai, C.L.; Kornilov, S.A.; et al. Multi-cohort analysis of host immune response identifies conserved protective and detrimental modules associated with severity across viruses. Immunity 2021, 54, 753–768.e5. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, T.E.; Wong, H.R.; Khatri, P. Robust classification of bacterial and viral infections via integrated host gene expression diagnostics. Sci. Transl. Med. 2016, 8, 346ra91. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Catalina, M.D.; Bachali, P.; Geraci, N.S.; Grammer, A.C.; Lipsky, P.E. Gene expression analysis delineates the potential roles of multiple interferons in systemic lupus erythematosus. Commun. Biol. 2019, 2, 140. [Google Scholar] [CrossRef]
- Kolde, R.; Laur, S.; Adler, P.; Vilo, J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics 2012, 28, 573–580. [Google Scholar] [CrossRef]
- Su, J.Q.; Liu, J.S. Linear combinations of multiple diagnostic markers. J. Am. Stat. Assoc. 1993, 88, 1350–1355. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, P.; Zhao, Y.; Zhuang, Z.; Wang, Z.; Song, R.; Zhang, J.; Liu, C.; Gao, Q.; Xu, Q.; et al. Single-Cell Sequencing of Peripheral Mononuclear Cells Reveals Distinct Immune Response Landscapes of COVID-19 and Influenza Patients. Immunity 2020, 53, 685–696.e3. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, eabc6027. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; John Wherry, E. T cell responses in patients with COVID-19. Nat. Rev. Immunol. 2020, 20, 529–536. [Google Scholar] [CrossRef]
- Le Bert, N.; Clapham, H.E.; Tan, A.T.; Chia, W.N.; Tham, C.Y.; Lim, J.M.; Kunasegaran, K.; Tan, L.W.L.; Dutertre, C.A.; Shankar, N.; et al. Highly functional virus-specific cellular immune response in asymptomatic SARS-CoV-2 infection. J. Exp. Med. 2021, 218, e20202617. [Google Scholar] [CrossRef]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Strålin, K.; Gorin, J.B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168.e14. [Google Scholar] [CrossRef]
- Mann, E.R.; Menon, M.; Knight, S.B.; Konkel, J.E.; Jagger, C.; Shaw, T.N.; Krishnan, S.; Rattray, M.; Ustianowski, A.; Bakerly, N.D.; et al. Longitudinal immune profiling reveals key myeloid signatures associated with COVID-19. Sci. Immunol. 2020, 5, eabd6197. [Google Scholar] [CrossRef] [PubMed]
- Junqueira, C.; Crespo, Â.; Ranjbar, S.; De Lacerda, L.B.; Lewandrowski, M.; Ingber, J.; Parry, B.; Ravid, S.; Clark, S.; Schrimpf, M.R.; et al. FcγR-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature 2022, 606, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.S.; et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e17. [Google Scholar] [CrossRef] [PubMed]
- Smits, V.A.J.; Cabrera, E.; Freire, R.; Gillespie, D.A. Claspin–checkpoint adaptor and DNA replication factor. FEBS J. 2019, 286, 441–455. [Google Scholar] [CrossRef]
- Benevolo, M.; Musio, A.; Vocaturo, A.; Donà, M.G.; Rollo, F.; Terrenato, I.; Carosi, M.; Pescarmona, E.; Vocaturo, G.; Mottolese, M. Claspin as a biomarker of human papillomavirus-related high grade lesions of uterine cervix. J. Transl. Med. 2012, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Motadi, L.R.; Bhoola, K.D.; Dlamini, Z. Expression and function of retinoblastoma binding protein 6 (RBBP6) in human lung cancer. Immunobiology 2011, 216, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Batra, J.; Hultquist, J.F.; Liu, D.; Shtanko, O.; Von Dollen, J.; Satkamp, L.; Jang, G.M.; Luthra, P.; Schwarz, T.M.; Small, G.I.; et al. Protein Interaction Mapping Identifies RBBP6 as a Negative Regulator of Ebola Virus Replication. Cell 2018, 175, 1917–1930.e13. [Google Scholar] [CrossRef]
- Hou, P.; Yang, K.; Jia, P.; Liu, L.; Lin, Y.; Li, Z.; Li, J.; Chen, S.; Guo, S.; Pan, J.A.; et al. A novel selective autophagy receptor, CCDC50, delivers K63 polyubiquitination-activated RIG-I/MDA5 for degradation during viral infection. Cell Res. 2021, 31, 62–79. [Google Scholar] [CrossRef]
- Xu, X.; Wu, Y.; Kummer, A.G.; Zhao, Y.; Hu, Z.; Wang, Y.; Liu, H.; Ajelli, M.; Yu, H. Assessing changes in incubation period, serial interval, and generation time of SARS-CoV-2 variants of concern: A systematic review and meta-analysis. BMC Med. 2023, 21, 374. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Tao, X.; Ye, S.; Tai, Q.; You, Y.-A.; Huang, X.; Liang, M.; Wang, K.; Wen, H.; You, C.; et al. A T-Cell-Derived 3-Gene Signature Distinguishes SARS-CoV-2 from Common Respiratory Viruses. Viruses 2024, 16, 1029. https://doi.org/10.3390/v16071029
Li Y, Tao X, Ye S, Tai Q, You Y-A, Huang X, Liang M, Wang K, Wen H, You C, et al. A T-Cell-Derived 3-Gene Signature Distinguishes SARS-CoV-2 from Common Respiratory Viruses. Viruses. 2024; 16(7):1029. https://doi.org/10.3390/v16071029
Chicago/Turabian StyleLi, Yang, Xinya Tao, Sheng Ye, Qianchen Tai, Yu-Ang You, Xinting Huang, Mifang Liang, Kai Wang, Haiyan Wen, Chong You, and et al. 2024. "A T-Cell-Derived 3-Gene Signature Distinguishes SARS-CoV-2 from Common Respiratory Viruses" Viruses 16, no. 7: 1029. https://doi.org/10.3390/v16071029
APA StyleLi, Y., Tao, X., Ye, S., Tai, Q., You, Y.-A., Huang, X., Liang, M., Wang, K., Wen, H., You, C., Zhang, Y., & Zhou, X. (2024). A T-Cell-Derived 3-Gene Signature Distinguishes SARS-CoV-2 from Common Respiratory Viruses. Viruses, 16(7), 1029. https://doi.org/10.3390/v16071029