
A qPCR Assay for the Quantification of Selected Genotypic Variants of Spodoptera frugiperda Multiple Nucleopolyhedrovirus (Baculoviridae)

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Viral Amplification and DNA Isolation
2.2. Primer Design
2.3. Preparation of Standard Templates for qPCR Calibration
2.4. qPCR Calibration Curves
2.5. Specificity of Variant Primers
2.6. Specificity of Primers against a Heterologous Virus (SeMNPV)
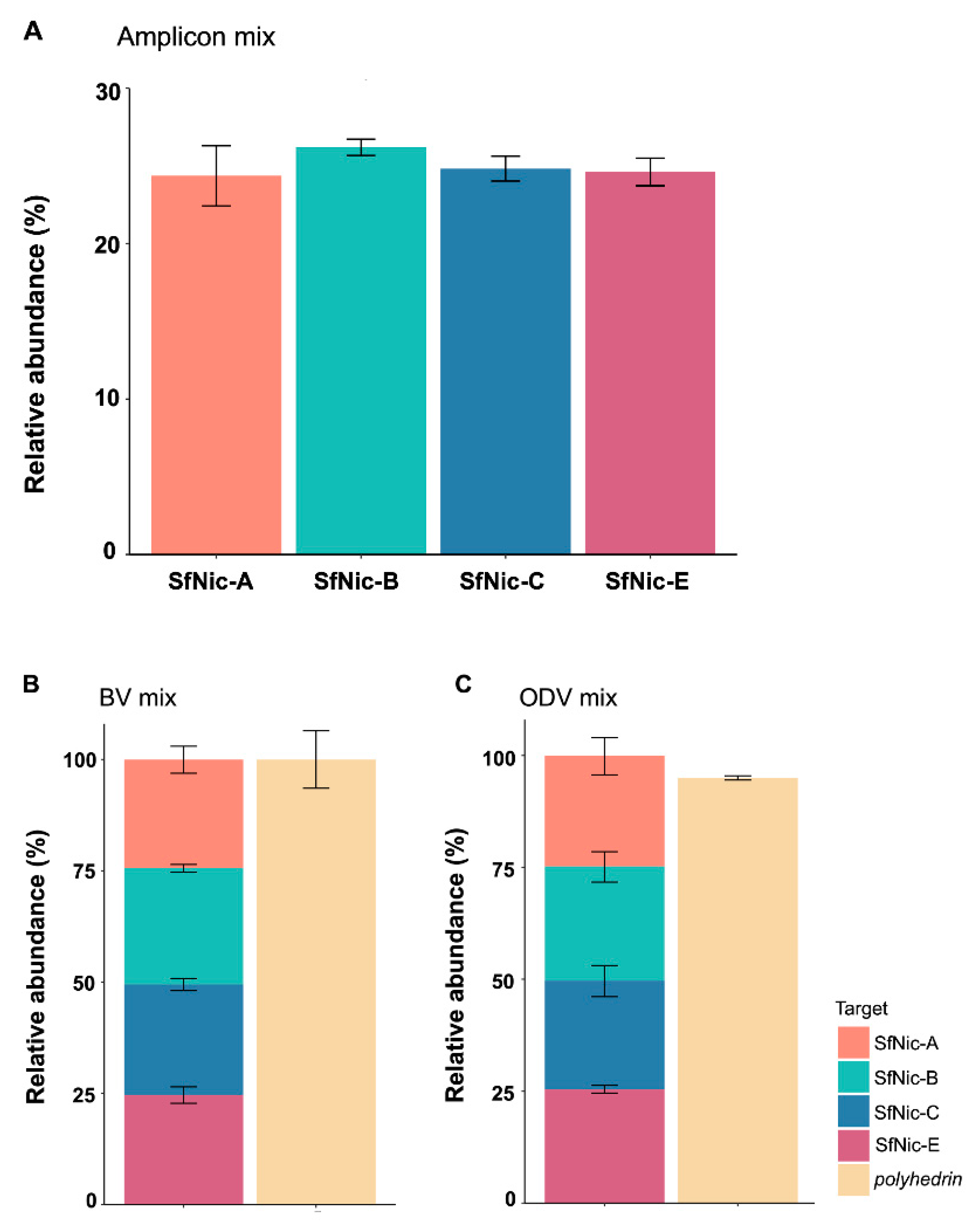
2.7. Quantification of Genotype-Specific Amplicons in a Mixture
2.8. Genotypic Variant Quantification in BV Mixtures
2.9. Genotypic Variant Quantification in Mixtures of DNA Extracted from ODVs
3. Results
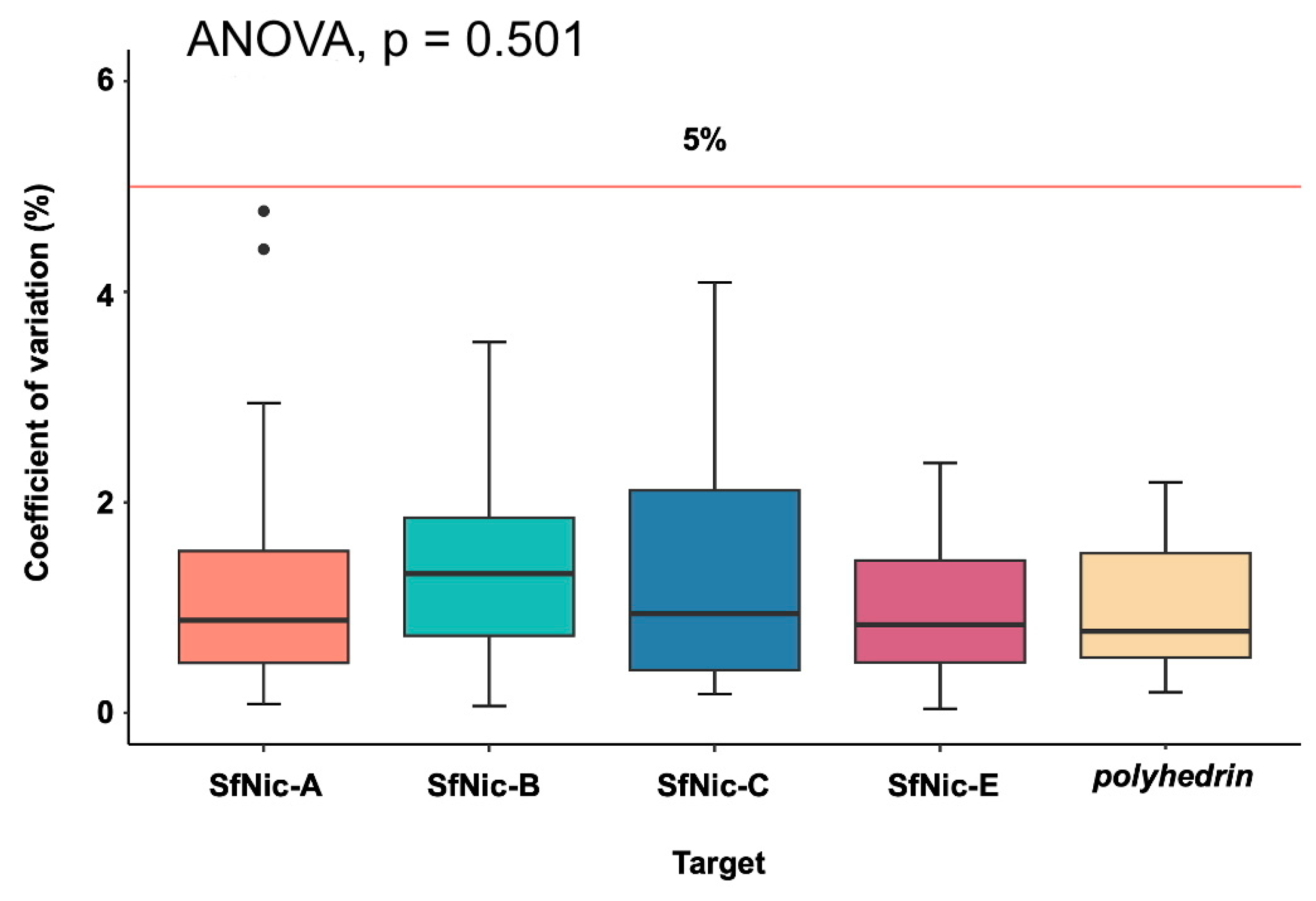
3.1. qPCR Characteristics
3.2. Primer Specificity
3.3. Validation of the Quantification Method in Mixtures of Amplicons, BVs and ODV DNA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harrison, R.L.; Herniou, E.A.; Jehle, J.A.; Theilmann, D.A.; Burand, J.P.; Becnel, J.J.; Krell, P.J.; Van Oers, M.M.; Mowery, J.D.; Bauchan, G.; et al. ICTV Virus Taxonomy Profile: Baculoviridae. J. Gen. Virol. 2018, 99, 1185–1186. [Google Scholar] [CrossRef] [PubMed]
- Rohrmann, G.F. Baculovirus Molecular Biology, 4th ed.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2019. Available online: https://www.ncbi.nlm.nih.gov/books/NBK543458/ (accessed on 17 April 2024).
- Williams, T.; Bergoin, M.; van Oers, M.M. Diversity of large DNA viruses of invertebrates. J. Invertebr. Pathol. 2017, 147, 4–22. [Google Scholar] [CrossRef]
- Moore, S.; Jukes, M. Advances in microbial control in IPM: Entomopathogenic viruses. In Integrated Management of Insect Pests; Kogan, M., Heinrichs, E.A., Eds.; Burleigh Dodds Science Publishing: Cambridge, UK, 2019; pp. 593–648. [Google Scholar] [CrossRef]
- Lacey, L.A.; Grzywacz, D. Basic and Applied Research: Baculovirus. In Microbial Control of Insect and Mite Pests: From Theory to Practice; Academic Press: Amsterdam, The Netherlands, 2017; pp. 27–46. [Google Scholar] [CrossRef]
- Sanjuán, R. Collective infectious units in viruses. Trends Microbiol. 2017, 25, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Leeks, A.; Sanjuán, R.; West, S.A. The evolution of collective infectious units in viruses. Virus Res. 2019, 265, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Andreu-Moreno, I.; Sanjuán, R. Collective viral spread mediated by virion aggregates promotes the evolution of defective interfering particles. mBio 2020, 11, e02156-19. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R. The social life of viruses. Annu. Rev. Virol. 2021, 8, 183–199. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Muñoz, S.L.; Sanjuán, R.; West, S. Sociovirology: Conflict, cooperation, and communication among viruses. Cell Host Microbe 2017, 22, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Allman, B.; Koelle, K.; Weissman, D. Heterogeneity in viral populations increases the rate of deleterious mutation accumulation. Genetics 2022, 222, iyac127. [Google Scholar] [CrossRef] [PubMed]
- Pazmiño-Ibarra, V.; Herrero, S.; Sanjuán, R. Spatially segregated transmission of co-occluded baculoviruses limits virus–virus interactions mediated by cellular coinfection during primary infection. Viruses 2022, 14, 1697. [Google Scholar] [CrossRef]
- Erlandson, M. Genetic variation in field populations of baculoviruses: Mechanisms for generating variation and its potential role in baculovirus epizootiology. Virol. Sin. 2009, 24, 458–469. [Google Scholar] [CrossRef]
- Chateigner, A.; Bézier, A.; Labrousse, C.; Jiolle, D.; Barbe, V.; Herniou, E.A. Ultra deep sequencing of a baculovirus population reveals widespreadgenomic variations. Viruses 2015, 7, 3625–3646. [Google Scholar] [CrossRef] [PubMed]
- Loiseau, V.; Herniou, E.A.; Moreau, Y.; Lévêque, N.; Meignin, C.; Daeffler, L.; Federici, B.; Cordaux, R.; Gilbert, C. Wide spectrum and high frequency of genomic structural variation, including transposable elements, in large double-stranded DNA viruses. Virus Evol. 2020, 6, vez060. [Google Scholar] [CrossRef] [PubMed]
- Simón, O.; Williams, T.; López-Ferber, M.; Taulemesse, J.M.; Caballero, P. Population genetic structure determines the speed of kill and occlusion body production in Spodoptera frugiperda multiple nucleopolyhedrovirus. Biol. Control 2008, 44, 321–330. [Google Scholar] [CrossRef]
- Clavijo, G.; Williams, T.; Muñoz, D.; López-Ferber, M.; Caballero, P. Entry into midgut epithelial cells is a key step in the selection of genotypes in a nucleopolyhedrovirus. Virol. Sin. 2009, 24, 350–358. [Google Scholar] [CrossRef]
- Segredo-Otero, E.; Sanjuán, R. The effect of genetic complementation on the fitness and diversity of viruses spreading as collective infectious units. Virus Res. 2019, 267, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Baillie, V.L.; Bouwer, G. High levels of genetic variation within Helicoverpa armigera nucleopolyhedrovirus populations in individual host insects. Arch. Virol. 2012, 157, 2281–2289. [Google Scholar] [CrossRef] [PubMed]
- Niz, J.M.; Salvador, R.; Ferrelli, M.L.; de Cap, A.S.; Romanowski, V.; Berretta, M.F. Genetic Variants in Argentinean isolates of Spodoptera frugiperda multiple nucleopolyhedrovirus. Virus Genes 2020, 56, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Masson, T.; Fabre, M.L.; Pidre, M.L.; Niz, J.M.; Berretta, M.F.; Romanowski, V.; Ferrelli, M.L. Genomic diversity in a population of Spodoptera frugiperda nucleopolyhedrovirus. Infect. Genet. Evol. 2021, 90, 104749. [Google Scholar] [CrossRef]
- Parras-Jurado, A.; Muñoz, D.; Beperet, I.; Williams, T.; Caballero, P. Insecticidal traits of variants in a genotypically diverse natural isolate of Anticarsia gemmatalis multiple nucleopolyhedrovirus (AgMNPV). Viruses 2023, 15, 1526. [Google Scholar] [CrossRef]
- Escribano, A.; Williams, T.; Goulson, D.; Cave, R.D.; Chapman, J.W.; Caballero, P. Selection of a nucleopolyhedrovirus for control of Spodoptera frugiperda (Lepidoptera: Noctuidae): Structural, genetic and biological comparison of four isolates from the Americas. J. Econ. Entomol. 1999, 92, 1079–1085. [Google Scholar] [CrossRef]
- Simón, O.; Palma, L.; Beperet, I.; Muñoz, D.; López-Ferber, M.; Caballero, P.; Williams, T. Sequence comparison between three geographically distinct Spodoptera frugiperda multiple nucleopolyhedrovirus isolates: Detecting positively selected genes. J. Invertebr. Pathol. 2011, 107, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Simón, O.; Williams, T.; López-Ferber, M.; Caballero, P. Genetic structure of a Spodoptera frugiperda nucleopolyhedrovirus population: High prevalence of deletion genotypes. Appl. Environ. Microbiol. 2004, 70, 5579–5588. [Google Scholar] [CrossRef] [PubMed]
- Simón, O.; Williams, T.; López-Ferber, M.; Caballero, P. Functional importance of deletion mutant genotypes in an insect nucleopolyhedrovirus population. Appl. Environ. Microbiol. 2005, 71, 4254–4262. [Google Scholar] [CrossRef]
- Lopez-Ferber, M.; Simón, O.; Williams, T.; Caballero, P. Defective or effective? Mutualistic interactions between virus genotypes. Proc. R. Soc. B Biol. Sci. 2003, 270, 2249–2255. [Google Scholar] [CrossRef]
- Barrera, G.; Williams, T.; Villamizar, L.; Caballero, P.; Simón, O. Deletion genotypes reduce occlusion body potency but increase occlusion body production in a Colombian Spodoptera frugiperda nucleopolyhedrovirus population. PLoS ONE 2013, 8, e77271. [Google Scholar] [CrossRef]
- Boogaard, B.; Van Oers, M.M.; Van Lent, J.W. An advanced view on baculovirus per os infectivity factors. Insects 2018, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shang, Y.; Chen, C.; Liu, S.; Chang, M.; Zhang, N.; Hu, H.; Zhang, F.; Zhang, T.; Wang, Z.; et al. Baculovirus per os infectivity factor complex: Components and assembly. J. Virol. 2019, 93, e02053-18. [Google Scholar] [CrossRef]
- Simón, O.; Williams, T.; Cerutti, M.; Caballero, P.; López-Ferber, M. Expression of a peroral infection factor determines pathogenicity and population structure in an insect virus. PLoS ONE 2013, 8, e78834. [Google Scholar] [CrossRef] [PubMed]
- Mihm, J.A. Técnicas Eficientes para la Crianza Masiva e Infestacion de Insectos, en la Selección de las Plantas Hospedantes para Resistencia al Gusano Cogollero, Spodoptera frugiperda; Centro Internacional de Mejoramiento de Maiz y Trigo (CIMMYT): El Batán, Mexico, 1984; p. 16. [Google Scholar]
- Williams, T.; Melo-Molina, G.d.C.; Jiménez-Fernández, J.A.; Weissenberger, H.; Gómez-Díaz, J.S.; Navarro-de-la-Fuente, L.; Richards, A.R. Presence of Spodoptera frugiperda multiple nucleopolyhedrovirus (SfMNPV) occlusion bodies in maize field soils of Mesoamerica. Insects 2023, 14, 80. [Google Scholar] [CrossRef]
- Simón, O. (Institute for Multidisciplinary Research in Applied Biology, Universidad Pública de Navarra, Pamplona, Spain). Unpublished genome sequence data from the auxiliary gene rich variable region of SfMNPV-NIC isolate, 2024.
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Thermofisher. Multiple Primer Analyzer. 2024. Available online: https://www.thermofisher.com/mx/es/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/thermo-scientific-web-tools/multiple-primer-analyzer.html (accessed on 10 May 2024).
- Chang, A.; Chau, V.; Landas, J.; Pang, Y. Preparation of calcium competent Escherichia coli and heat-shock transformation. JEMI Meth. 2017, 1, 22–25. [Google Scholar]
- Molina-Ruiz, C.S.; Zamora, J.A.; Williams, T. (Instituto de Ecología AC, Xalapa, Veracruz, Mexico). Partial genome sequences (contigs with unclosed gaps) of variants SfNic-A, SfNic-C and SfNic-E obtained by next generation sequencing, 2024.
- Thermofisher. DNA Copy Number Calculator. Available online: https://www.thermofisher.com/mx/es/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/thermo-scientific-web-tools/dna-copy-number-calculator.html (accessed on 5 April 2024).
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Kralik, P.; Ricchi, M. A basic guide to real time PCR in microbial diagnostics: Definitions, parameters, and everything. Front. Microbiol. 2017, 8, 108. [Google Scholar] [CrossRef] [PubMed]
- Presa-Parra, E.; Navarro-de-la-Fuente, L.; Williams, T.; Lasa, R. Can low concentration flufenoxuron treatment increase the pathogenicity or production of nucleopolyhedrovirus occlusion bodies in Spodoptera exigua (Hübner) or Spodoptera frugiperda (J.E. Smith) (Lepidoptera: Noctuidae)? Acta Zool. Mex. 2023, 39, e3912627. [Google Scholar] [CrossRef]
- Stoepler, T.M.; Castillo, J.C.; Lill, J.T.; Eleftherianos, I. A simple protocol for extracting hemocytes from wild caterpillars. J. Vis. Exper. 2012, 69, 4173. [Google Scholar] [CrossRef]
- Wennmann, J.T.; Keilwagen, J.; Jehle, J.A. Baculovirus Kimura two-parameter species demarcation criterion is confirmed by the distances of 38 core gene nucleotide sequences. J. Gen. Virol. 2018, 99, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Hewson, I.; Brown, J.M.; Gitlin, S.A.; Doud, D.F. Nucleopolyhedrovirus detection and distribution in terrestrial, freshwater, and marine habitats of Appledore Island, Gulf of Maine. Microb. Ecol. 2011, 62, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Krokene, P.; Heldal, I.; Fossdal, C.G. Quantifying Neodiprion sertifer nucleopolyhedrovirus DNA from insects, foliage and forest litter using the quantitative real-time polymerase chain reaction. Agric. Forest Entomol. 2012, 15, 120–125. [Google Scholar] [CrossRef]
- Graham, R.; Tummala, Y.; Rhodes, G.; Cory, J.; Shirras, A.; Grzywacz, D.; Wilson, K. Development of a real-time qPCR assay for quantification of covert baculovirus infections in a major African crop pest. Insects 2015, 6, 746–759. [Google Scholar] [CrossRef]
- Ramirez-Arias, F.G.; Lasa, R.; Murillo, R.; Navarro-de-la-Fuente, L.; Mercado, G.; Williams, T. Post-mortem incubation influences occlusion body production in nucleopolyhedrovirus-infected larvae of Spodoptera frugiperda. Biol. Control 2019, 135, 33–40. [Google Scholar] [CrossRef]
- Velasco, E.A.; Molina-Ruíz, C.S.; Gómez-Díaz, J.S.; Williams, T. Properties of nucleopolyhedrovirus occlusion bodies from living and virus-killed larvae of Spodoptera frugiperda (Lepidoptera: Noctuidae). Biol. Control 2022, 174, 105008. [Google Scholar] [CrossRef]
- Zwart, M.P.; van Oers, M.M.; Cory, J.S.; van Lent, J.W.M.; van der Werf, W.; Vlak, J.M. Development of a quantitative real-time PCR for determination of genotype frequencies for studies in baculovirus population biology. J. Virol. Meth. 2008, 148, 146–154. [Google Scholar] [CrossRef] [PubMed]
- dMIQE Group; Huggett, J.F. The digital MIQE guidelines update: Minimum information for publication of quantitative digital PCR experiments for 2020. Clin. Chem. 2020, 66, 1012–1029. [Google Scholar] [CrossRef] [PubMed]
- Kaupp, W.J.; Ebling, P.M. Effect of mechanical processing and long-term storage on biological activity of Virtuss. Can. Entomol. 1993, 125, 975–977. [Google Scholar] [CrossRef]
- Jorio, H.; Tran, R.; Kamen, A. Stability of serum-free and purified baculovirus stocks under various storage conditions. Biotechnol. Prog. 2006, 22, 319–325. [Google Scholar] [CrossRef]
- Lasa, R.; Williams, T.; Caballero, P. Insecticidal properties and microbial contaminants in a Spodoptera exigua multiple nucleopolyhedrovirus (SeMNPV, Baculoviridae) formulation stored at different temperatures. J. Econ. Entomol. 2008, 101, 42–49. [Google Scholar] [CrossRef]
- Beperet, I.; Simón, O.; López-Ferber, M.; van Lent, J.; Williams, T.; Caballero, P. Mixtures of insect pathogenic viruses in a single virion: Towards the development of custom designed insecticides. Appl. Environ. Microbiol. 2021, 87, e02180-20. [Google Scholar] [CrossRef]
- Williams, T.; López-Ferber, M.; Caballero, P. Nucleopolyhedrovirus coocclusion technology: A new concept in the development of biological insecticides. Front. Microbiol. 2022, 12, 810026. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Target | Primer Sequence (5′-3′) | Tm (°C) | Amplicon Size (bp) |
|---|---|---|---|
| SfNic-A | AFw 5′-TCGAGCGTTCGTAACATTGTG-3′ | 60.2 | 113 |
| ARv 5′-GGCCAAATTCAAAACGGAAA-3′ | 56.8 | ||
| SfNic-B | BFw 5′-ACACCACCGAACTGACTTGGAACGA-3′ | 59.9 | 103 |
| BRv 5′-GTTCGTCGGCAGTACATGAATC-3′ | 59.7 | ||
| SfNic-C | CFw 5′-GCCGCGTTTAGTAACAGCAAA-3′ | 60.4 | 150 |
| CRv 5′-TGATTTTCTTCCGTTCTCTGACAC-3′ | 60.2 | ||
| SfNic-E | EFw 5′-TCTTGGTCATGTCCGCAAAA-3′ | 57.1 | 122 |
| ERv 5′-CGCGCTCGATCGTGAGTAT-3′ | 58.6 | ||
| polyhedrin | polhFw 5′-GCCCGTGTACGTAGGAAACA-3′ | 59.3 | 110 |
| polhRv 5′-ACTCTTCGAAGGAGTGCGTG-3′ | 59.1 |
| Target | R2 | Slope | y-Intercept | Efficiency (%) |
|---|---|---|---|---|
| SfNic-A | 0.957 ± 0.040 | −3.354 ± 0.040 | 40.74 ± 0.53 | 98.9 ± 2.1 |
| SfNic-B | 0.993 ± 0.030 | −3.334 ± 0.003 | 38.86 ± 0.70 | 99.4 ± 0.2 |
| SfNic-C | 0.989 ± 0.004 | −3.479 ± 0.200 | 39.78 ± 0.37 | 97.2 ± 2.5 |
| SfNic-E | 0.993 ± 0.005 | −3.335 ± 0.700 | 39.19 ± 0.21 | 98.5 ± 1.4 |
| polyhedrin | 0.998 ± 0.003 | −3.269± 0.050 | 35.92 ± 0.30 | 100.9 ± 1.1 |
| Specificity (%) | ||||
|---|---|---|---|---|
| Sample | SfNic-A | SfNic-B | SfNic-C | SfNic-E |
| SfNic-A | 100 | 0.05 | – | 0.0015 |
| SfNic-B | – | 99.7 | – | – |
| SfNic-C | – | 0.2 | 100 | – |
| SfNic-E | – | 0.05 | – | 99.999 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molina-Ruiz, C.S.; Zamora-Briseño, J.A.; Simón, O.; Lasa, R.; Williams, T. A qPCR Assay for the Quantification of Selected Genotypic Variants of Spodoptera frugiperda Multiple Nucleopolyhedrovirus (Baculoviridae). Viruses 2024, 16, 881. https://doi.org/10.3390/v16060881
Molina-Ruiz CS, Zamora-Briseño JA, Simón O, Lasa R, Williams T. A qPCR Assay for the Quantification of Selected Genotypic Variants of Spodoptera frugiperda Multiple Nucleopolyhedrovirus (Baculoviridae). Viruses. 2024; 16(6):881. https://doi.org/10.3390/v16060881
Chicago/Turabian StyleMolina-Ruiz, Cindy S., Jesús Alejandro Zamora-Briseño, Oihane Simón, Rodrigo Lasa, and Trevor Williams. 2024. "A qPCR Assay for the Quantification of Selected Genotypic Variants of Spodoptera frugiperda Multiple Nucleopolyhedrovirus (Baculoviridae)" Viruses 16, no. 6: 881. https://doi.org/10.3390/v16060881
APA StyleMolina-Ruiz, C. S., Zamora-Briseño, J. A., Simón, O., Lasa, R., & Williams, T. (2024). A qPCR Assay for the Quantification of Selected Genotypic Variants of Spodoptera frugiperda Multiple Nucleopolyhedrovirus (Baculoviridae). Viruses, 16(6), 881. https://doi.org/10.3390/v16060881